Introduction
Sepsis and septic shock within intensive care units
is a major, global healthcare problem, with the mortality rate of
septic shock currently ~25% (1).
The morbidity and mortality of sepsis remain high despite knowledge
of its etiological and pathophysiological mechanisms, and
implementation of guidelines for its prevention, such as those
proposed by the Surviving Sepsis Campaign (1). Sepsis is characterized by increased
vascular permeability, unbalanced inflammation and immune
modulation, which form a complex, crosslinked network of cells,
mediators and signaling pathways. Endothelial cells (ECs) and
endothelial microparticles (EMPs) are known to be vital links in
the network (2).
ECs form an interface between circulating blood and
the rest of the vessel wall, and are involved in inflammation,
coagulation, the immune response, vascular tone and other important
biological processes (3). Hence,
EC dysfunction or damage is associated with a wide range of
conditions, including acute respiratory distress syndrome (ARDS),
sepsis and septic shock (4).
Therefore, it is necessary to discover the pathophysiological
mechanism linking endothelial dysfunction with sepsis.
EMPs are small vesicles generated by activated,
apoptotic or injured ECs, ranging from 0.1-1 µm in size (5). EMP concentrations in circulating
blood are very low under normal conditions, but increases
significantly under pathological conditions (6). EMPs are generated and released when
ECs are activated by proinflammatory, prothrombotic or proapoptotic
factors, or exposed to high shear stress (7). EMPs contain protein, lipid and
nuclear material from their point of origin, but their composition
depends on the stimulus triggering their generation (5). EMPs are associated with functions
including inflammatory and coagulation responses, immune response,
angiogenesis, cell proliferation and migration: As a result, EMPs
are considered useful biomarkers in the evaluation of a wide range
of diseases, including cardiovascular diseases, diabetes,
pre-eclampsia, metabolic syndrome, ARDS and sepsis (8). However, of greatest interest to
intensivists is the involvement of EMPs in acute inflammatory
diseases, such as sepsis and septic shock. Previous research has
reported that EMP levels are elevated in meningococcal sepsis
(9) and the injection of EMPs into
mouse and rat lungs induce acute lung injury (10). A previous study regarding septic
shock patients in intensive care units demonstrated that EMPs are
biomarkers of septic shock-induced disseminated intravascular
coagulopathy, and could be used to evaluate early vascular injury
(11). EMPs also act as vectors in
intercellular information exchange during physiological and
pathological processes, with some of the mechanisms involved being
documented (6). EMPs affect
neighboring and distant cells through the transfer of
membrane-associated receptors, releasing directly active proteins,
exchanging genetic information or inducing the adaptive immune
response (12). EMPs may also
activate target cells, including monocytic cells, and amplify
harmful responses including inflammation, thrombosis and vascular
dysfunction, but research in this area is lacking. A direct
correlation between the proteins that compose EMPs and tumor
necrosis factor-α (TNF-α)-stimulated ECs has been previously
demonstrated (13). The
endothelial proteins transferred by EMPs may be involved in the
interaction between EMPs and their target cells, which may result
in endothelial dysfunction (14).
Despite a relationship between EMP protein
composition and their parental ECs having been demonstrated
(13), to the best of our
knowledge, the effect of EMPs on their parental ECs remains
unknown. Since the major pathological changes during sepsis occur
in the microvascular and pulmonary microvascular endothelial cells,
human pulmonary microvascular endothelial cells (HPMECs) were used
for the present study. As TNF-α is a proinflammatory cytokine
widely used to mimic acute inflammation, TNF-α was used to
stimulate HPMECs to generate EMPs, as previously (14). The primary aim of the current study
was to stimulate normal HPMECs in vitro with EMPs from
TNF-α-activated HPMECs. The cytokine profiles of normal control
HPMECs and EMP-stimulated HPMECs were comprehensively compared
using a proteome profiler array, and the mechanisms of EMP-related
inflammation and immune modulation were explored.
The present study aims to aid understanding of the
function of EMPs and the mechanism underlying endothelial
dysfunction to assess EMPs as a novel and effective target for the
diagnosis and therapy of sepsis.
Materials and methods
HPMEC culture and EMP collection
HPMECs (ScienCell Research Laboratories, Inc.,
Carlsbad, CA, USA) were cultured in endothelial cell culture medium
(ScienCell Research Laboratories, Inc.) consisting of 5% fetal
bovine serum (ScienCell Research Laboratories, Inc.) and
endothelial cell growth supplement (ECGS; ScienCell Research
Laboratories, Inc.) under standard cell culture conditions (37°C
and 5% CO2). HPMECs from passages 4–6 were used for
further experiments, when the cells were ~80–90% confluent. HPMECs
were incubated in serum-free endothelial cell medium with ECGS,
containing 100 ng/ml of TNF-α (PeproTech, Inc., Rocky Hill, NJ,
USA) for 24 h in 25 ml culture flasks as described previously
(14). Following incubation, the
cell-conditioned medium was harvested and centrifuged at 200 × g
for 5 min at room temperature to remove cell debris. The
supernatant was collected and ultracentrifuged at 100,000 × g for 2
h at a temperature of 4°C. The supernatant was then discarded. The
sediment was washed once with phosphate-buffered saline (PBS) and
resuspended in PBS. The EMP pellet was either used immediately to
stimulate HPMECs or stored at ≤-20°C until use. Repeated
freeze-thaw cycles were avoided.
Human cytokine array analysis
HPMECs (5×106 cells/flask) were cultured in 25 ml
culture flasks and were serum-starved for 2 h. HPMECs were then
evenly divided into a normal control HPMEC group and an
EMP-stimulated HPMEC group. The normal control HPMEC group was
incubated in serum-free endothelial cell medium with ECGS. The
EMP-stimulated HPMEC group was incubated in serum-free endothelial
cell medium with ECGS containing EMPs (10 µg/ml of total protein)
collected as previously described. Following 24 h incubation, cell
culture supernatants from the 2 groups were collected and the
particulates removed by centrifugation at 200 × g for 5 min at room
temperature. The expression of human cytokines in the samples of
the 2 groups was detected using the Proteome Profiler Array Human
Cytokine Array Panel A (R&D Systems, Inc., Minneapolis, MN,
USA) according to the manufacturer's protocol. The films were
scanned using a Tanon 5500 instrument (Tanon Science and Technology
Co., Ltd., Shanghai, China) and subjected to densitometric analysis
using Image J software (version 1.46r; National Institutes of
Health, Bethesda, MD, USA).
ELISA analysis of interferon
gamma-induced protein 10 (IP-10)
HPMECs were cultured in 96-well plates (105
cells/well) and were serum-starved for 2 h. Cells were subsequently
equally divided into 1 normal control HPMEC group and 9
EMP-stimulated HPMEC groups. The normal control HPMEC group was
incubated in serum-free endothelial cell medium with ECGS. The 9
EMP-stimulated HPMEC groups were incubated in serum-free
endothelial cell medium with ECGS containing 2, 5 and 10 µg/ml of
EMP total protein for 24 h and 10 µg/ml of EMP total protein for 1,
3, 6, 18, 24 and 48 h, respectively. Following incubation, cell
culture supernatants were collected from each group and the
particulates were removed by centrifugation at 200 × g for 5 min at
room temperature. Human IP-10 ELISA analysis (cat. no. ELH-IP10;
RayBiotech, Norcross, GA, USA; detection limit: 8 pg/ml) of the
samples was performed according to the manufacturer's protocol to
detect the concentration of IP-10. All standards and samples were
run in duplicate within each plate. The 96-well plate was read at
450 nm using a Epoch Multi-Volume Spectrophotometer System (BioTek
Instruments, Inc., Winooski, VT, USA). The absorbance of samples
was compared against the absorbance of the standards to calculate
the concentration of the samples.
Immunofluorescence analysis of nuclear
factor-κB (NF-κB)
HPMECs (2×105 cells/well) were cultured in Millicell
EZ SLIDE 8-well glass (Merck Millipore, Darmstadt, Germany) and
were serum-starved for 2 h. Cells were equally divided into a
normal control HPMEC group and an EMP-stimulated HPMEC group. The
normal HPMEC group was incubated in serum-free endothelial cell
medium with ECGS. The EMP-stimulated HPMEC group was incubated in
serum-free endothelial cell medium with ECGS containing EMPs (10
µg/ml of total protein) for 24 h. For immunofluorescence microscopy
analysis, the HPMEC monolayers of the 2 groups were washed twice
with 0.01 M PBS at a pH of 7.4 and were fixed with 4% formaldehyde
at room temperature for 10 min. Cells were blocked with 1% fetal
bovine serum in 0.01 M PBS for 30 min at room temperature. Indirect
immunofluorescence was performed by staining the samples with
rabbit NF-κB p65 primary antibody (cat. no. sc-372; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at a dilution of 1:100
overnight at 4°C. Cells were then washed 3 times for 5 min with
0.01 M PBS and stained with an anti-rabbit fluorescein
isothiocyanate-labelled secondary immunoglobulin G antibody (cat.
no. F0382; Sigma-Aldrich; Merck Millipore) at a dilution of 1:500
for 1 h at room temperature in the dark and washed 3 times for 5
min in 0.01 M PBS. Cell nuclei were stained by
4′,6-diamidino-2-phenylindole (Sigma-Aldrich; Merck Millipore) at a
dilution of 1:1,000 for 10 min at room temperature in the dark,
washed 3 times for 3 min in 0.01 M PBS, and then observed by
fluorescence microscopy (Olympus BX61; Olympus Corporation, Tokyo,
Japan). The software used for analysis was Image J software
(version 1.46r; National Institutes of Health).
Western blot analysis of NF-κB
HPMECs (5×106 cells/flask) were cultured in 25 ml
culture flasks and were serum-starved for 2 h. Cells were equally
divided into a normal control HPMEC group and an EMP-stimulated
HPMEC group. The normal control HPMEC group was incubated in
serum-free endothelial cell medium with ECGS. The EMP-stimulated
HPMEC group was incubated in serum-free endothelial cell medium
with ECGS containing EMPs (10 µg/ml of total protein) collected as
previously described. Following 24 h incubation, the culture medium
was aspirated and the HPMEC monolayers washed twice with ice cold
0.01 M PBS. Nucleic protein was extracted using the Nuclear and
Cytoplasmic Protein Extraction kit (Sangon Biotech Co., Ltd.,
Shanghai, China). The protein concentration of the samples was
quantified using a bicinchoninic acid protein assay kit (Pierce;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Protein was
dissolved in Laemmli sample buffer (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and boiled for 5 min at 95°C, then 20 µg was
separated by electrophoresis on a 12.5% sodium dodecyl
sulphate-polyacrylamide gel under non-reducing conditions. Protein
was subsequently electrophoretically transferred onto a
polyvinylidene fluoride membrane and subsequently blocked using 5%
non-fat milk solution for 1 h at room temperature. The membrane was
incubated overnight with rabbit anti-NF-κB p65 primary antibody
(cat. no. sc-372; Santa Cruz Biotechnology, Inc.) at a dilution of
1:500 and a temperature of 4°C. The membrane was then washed 3
times for 10 min with tris-buffered saline with Tween (TBST; 25 mM
Tris, pH 7.2, 150 mM saline, 0.05% Tween) and incubated with
horseradish peroxidase-conjugated mouse anti-rabbit antibody (cat.
no. sc-2357; Santa Cruz Biotechnology, Inc.) at a dilution of
1:5,000 for 1 h at room temperature, and the membrane was then
washed 3 times for 10 min in TBST. Proteins were visualized using a
Tanon 5500 instrument (Tanon Science and Technology Co., Ltd.).
Histone H3 (cat. no 4499; Cell Signaling Technology, Inc., Danvers,
MA, USA; 1:2,000) levels were evaluated as a protein loading
control. The incubation condition and the secondary antibody was
the same as for NF-κB p65.
Statistical analysis
Experiments were performed in triplicate. Results
were expressed as the mean ± standard deviation and were analyzed
using one-way analysis of variance tests followed by Tukey's range
tests for multiple comparisons. Comparative statistical analysis
between two groups was performed using Student's t-tests. All
statistical analyses were performed using SPSS 22.0 (IBM SPSS,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Proteome of proinflammatory cytokines
released from normal HPMECs and EMP-stimulated HPMECs
The proteome profiler array (Fig. 1A) of the proinflammatory cytokines
present in the cell supernatants of the normal control HPMEC group
(Fig. 1B) and the EMP-stimulated
HPMEC group (Fig. 1C) revealed
some differences between the groups. The 6 cytokines in present in
both groups were growth regulated oncogene α (GROα), interleukin
(IL-)6, IL-8, monocyte chemoattractant protein 1 (MCP-1),
macrophage migration inhibitory factor (MIF) and serpin E1
(Fig. 1B and C), and 7 cytokines
were revealed to be unique to the EMP-stimulated HPMEC group,
including complement component 5a (C5a), granulocyte colony
stimulating factor (G-CSF), granulocyte macrophage colony
stimulating factor (GM-CSF), soluble intercellular adhesion
molecule-1 (sICAM-1), IP-10, C-C motif chemokine ligand 5 (RANTES)
and TNF-α (Fig. 1C). Therefore,
the cytokines secreted from HPMEC cells differ following exposure
to EMPs from TNF-α activated HPMECs. Different pixel densities and
therefore protein quantities were also observed for 2 of the 6
common cytokines, with IL-6 protein expression levels increased in
the EMP-stimulated HPMEC group compared with the normal control
group (P=0.018; Fig. 2) and IL-8
protein expression levels decreased in the EMP-stimulated HPMEC
group compared with the normal control group (P=0.029, Fig. 2). EMP exposure therefore alters the
quantity of cytokines secreted from HPMEC cells.
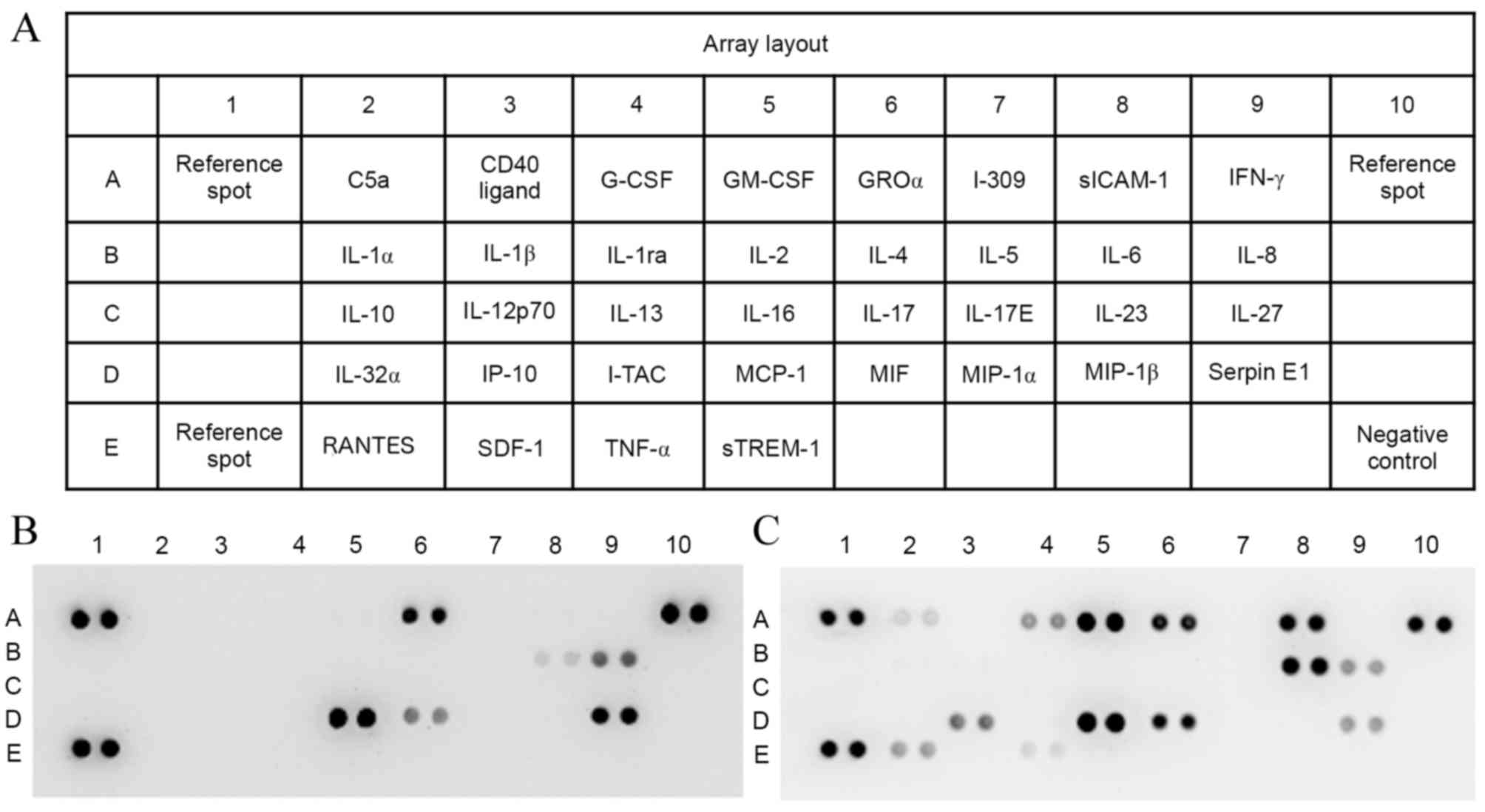 | Figure 1.Proteome array of proinflammatory
cytokines. (A) Array layout. Cytokines present in cell culture
supernatants of (B) normal control HPMECs and (C) endothelial
microparticle-stimulated HPMECs. The intensity of each protein spot
represents the quantity of the protein. HPMEC, human pulmonary
microvascular endothelial cell; C5a, complement component 5a; CD40,
cluster of differentiation 40; G-CSF, granulocyte colony
stimulating factor; GM-CSF, granulocyte macrophage colony
stimulating factor; GROα, growth regulated oncogene α; I-309,
inflammatory cytokine I-309; sICAM-1, soluble intercellular
adhesion molecule-1; IFN-γ, interferon γ; IL, interleukin; IP-10,
interferon γ-induced protein 10; I-TAC, C-X-C motif chemokine 11;
MCP-1, monocyte chemoattractant protein 1; MIF, macrophage
migration inhibitory factor; MIP, macrophage inflammatory protein;
RANTES, C-C motif chemokine ligand 5; SDF-1, stromal cell-derived
factor-1; TNF-α, tumor necrosis factor-α; sTREM-1, soluble
triggering receptor expressed on myeloid cells 1. |
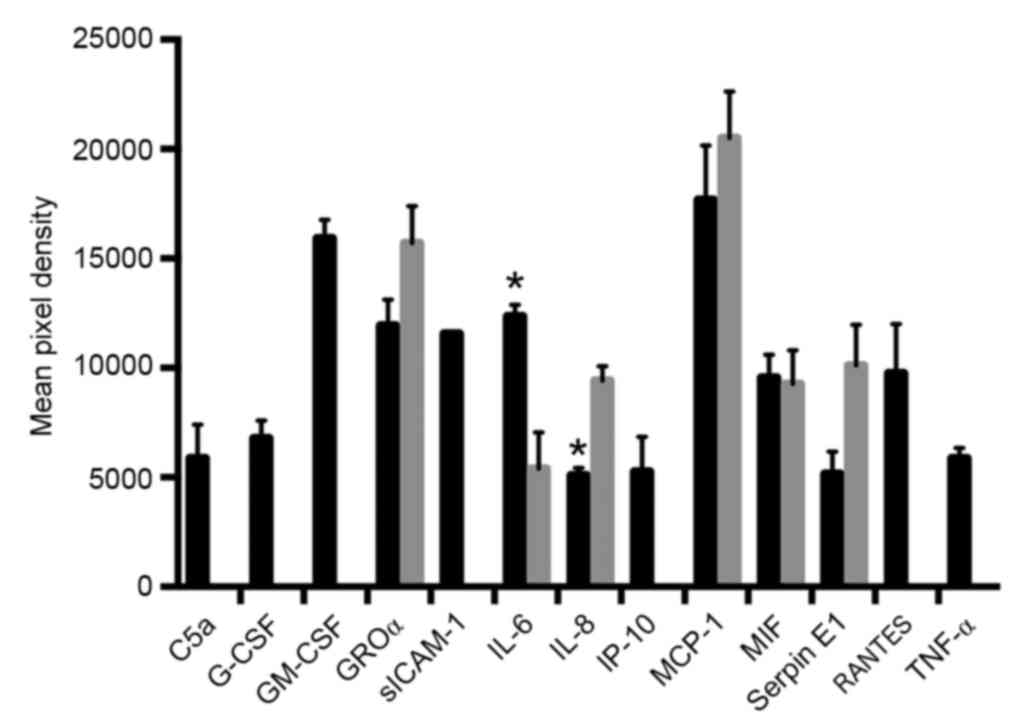 | Figure 2.Mean pixel density of the proteome
array protein spots in normal control HPMECs (grey bars) and
EMP-stimulated HPMECs (black bars). *P<0.05 vs. normal control
HPMECs. HPMEC, human pulmonary microvascular endothelial cells;
EMP, endothelial microparticle; C5a, complement component 5a;
G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte
macrophage colony stimulating factor; GROα, growth regulated
oncogene α; siCAM-1, soluble intercellular adhesion molecule-1; IL,
interleukin; IP-10, interferon γ-induced protein 10; MCP-1,
monocyte chemoattractant protein 1; MIF, macrophage migration
inhibitory factor; RANTES, C-C motif chemokine ligand; TNF-α, tumor
necrosis factor-α. |
The concentration of IP-10 released by
HPMECs in response to EMP stimulation is dose and time
dependent
The IP-10 concentration in the normal control group
was very low (24.55±1.32 pg/ml; Fig.
3) and the concentration increased progressively with the
increase of dose and the duration of exposure to EMPs (Fig. 3). The concentration of IP-10
released from 7 out of the 8 EMP-stimulated HPMEC conditions was
significantly increased compared with the normal control HPMEC
group (Fig. 3). The HPMECs
stimulated with 10 µg/ml EMP for 24 h generated the highest
concentration of IP-10 (1970.50±68.84 pg/ml; P=0.0003; Fig. 3).
NF-κB expression differs in HPMECs
following exposure to EMPs
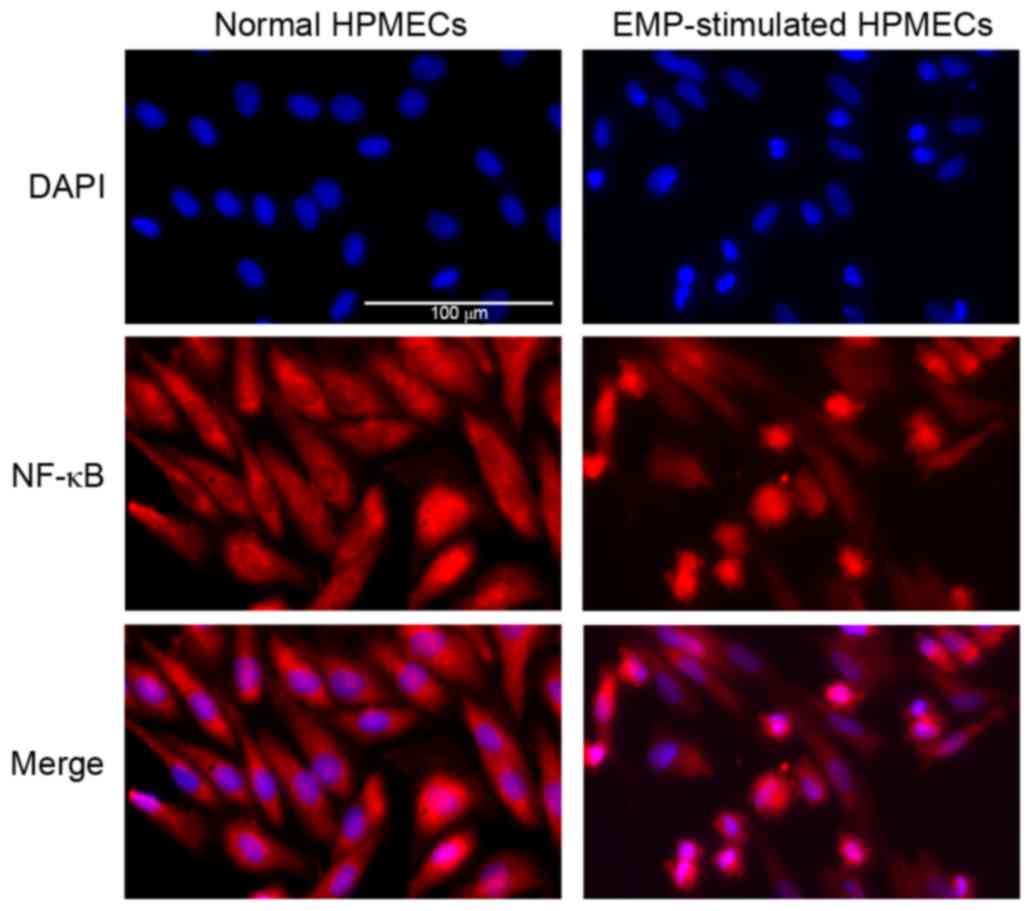
Immunofluorescence analysis revealed increased red
staining of NF-κB in the nucleus when HPMECs were stimulated by
EMPs (Fig. 4), suggesting that
NF-κB was inactive in the cytoplasm of normal control HPMECs, and
was translocated to the nucleus following exposure to EMPs
(Fig. 4). The translocation of
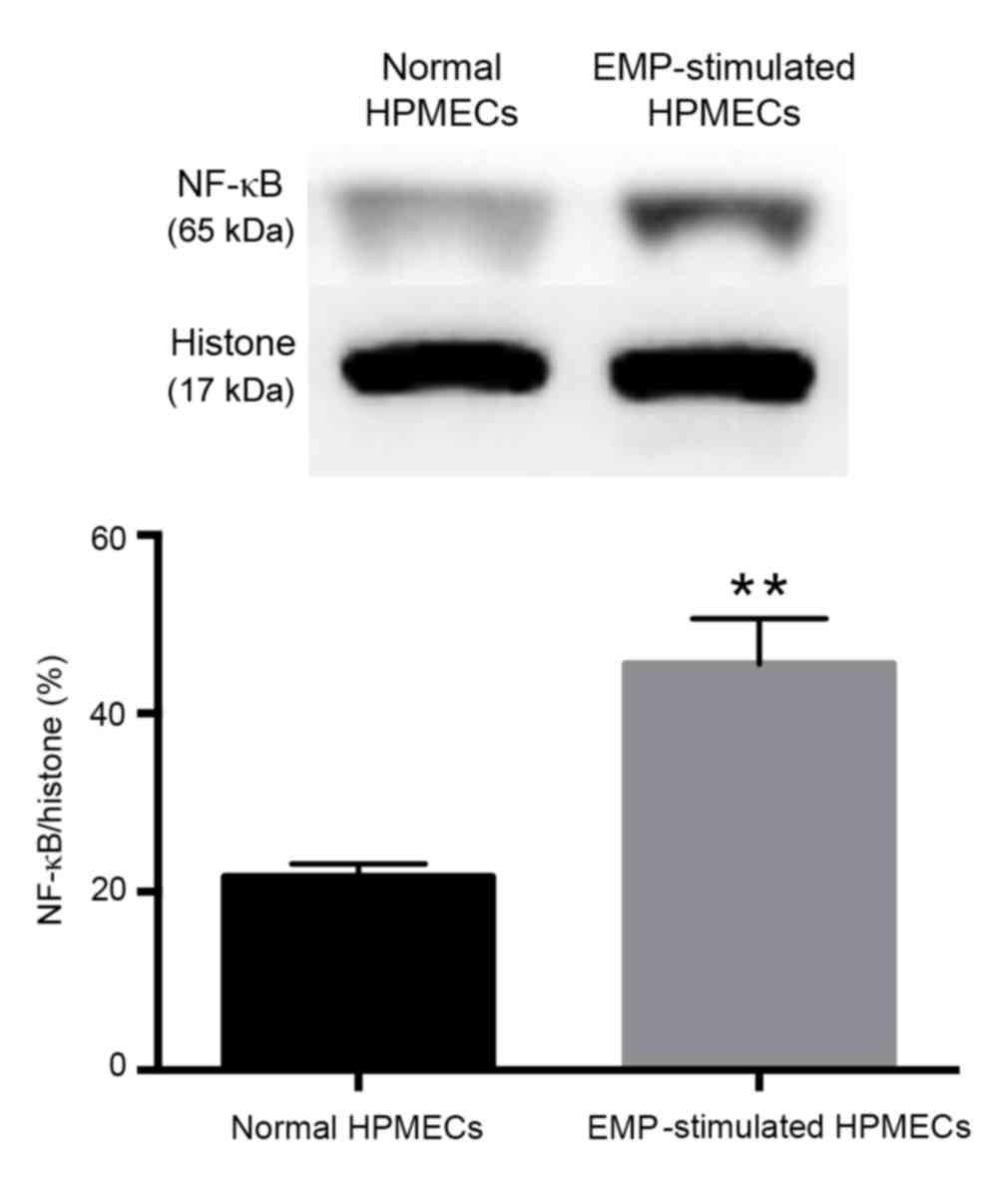
NF-κB in EMP-stimulated HPMECs was further confirmed by western
blot analysis (Fig. 5). The
relative amount of NF-κB in the nucleus protein was significantly
increased in EMP-stimulated cells compared with normal control
cells (P=0.0004; Fig. 5).
Discussion
The present study demonstrated that TNF-α-derived
EMPs activate HPMECs and induce the production of proinflammatory
cytokines, thereby facilitating the inflammatory response. Notably,
IP-10 is critically involved in the activation process and
modulation of inflammation. In addition, the NF-κB pathway was
revealed to be associated with the stimulation of HPMECs by
EMPs.
During sepsis-induced microvascular injury, ECs and
circulating cells release large quantities of microparticles (MPs)
(5). MPs are considered biomarkers
of endothelial function, but also mediate the exchange of
intercellular information (6). MPs
exert proinflammatory, prothrombotic and immunosuppressive effects
on ECs and circulating cells, inducing or promoting inflammation,
disseminated intravascular coagulopathy, immunosuppression and
microvascular injury (15).
Sabatier et al (16)
demonstrated that EMPs bind to monocytic THP-1 cells and induce a
tissue factor-dependent procoagulant cellular response. Wang et
al (17) observed that MPs
from lipopolysaccharide (LPS)-treated THP-1 monocytic cells, but
not untreated cells, bind to and activate endothelial cells in an
IL-1β-dependent manner. These authors also found that monocytic MPs
induced cytokine expression, the activation of NF-κB pathway and
phosphorylation of ERK1/2. However, Wen et al (18) observed different results, in which
LPS-induced monocytic MPs contained proinflammatory cytokines that
had contrasting effects on endothelial cells, acting in a
proinflammatory and procoagulatory manner, but also protecting the
function of the endothelium. These findings elucidate some of the
effects and mechanisms of EMPs on circulating cells and monocytic
MPs on ECs. However, to the best of our knowledge, no previous
research has focused on the effect of EMPs on their parental ECs.
The present study demonstrated that TNF-α-derived EMPs stimulated
resting HPMECs to become active. Compared to resting, normal
control HPMECs, EMP-activated HPMECs released a greater range and
altered quantities of proinflammatory factors including C5a,
sICAM-1, IL-6 and IP-10, which are associated with endothelial
dysfunction. EMPs derived from TNF-α-treated HPMECs induced HPMECs
to produce proinflammatory cytokines and thus facilitated the
inflammatory response. Notably, the quantity of IL-8, an important
proinflammatory factor, decreased in EMP-activated HPMECs. It is
possible that IL-8 might be associated with the protective function
of EMPs.
In order to further confirm the effect of EMPs on
ECs, expression of IP-10, one of the cytokines largely secreted by
EMP-stimulated HPMECs, was quantitatively measured. IP-10, also
known as C-X-C motif chemokine 10 (CXCL10), is a chemokine produced
by monocytes and endothelial cells following interferon-γ
stimulation that acts as a potent T cell chemoattractant (19). IP-10 facilitates the recruitment of
Th1-type leukocytes to inflammatory sites, and is considered as an
important mediator of immune and inflammatory responses (20). In the present study, IP-10
concentration analysis confirmed the findings of the proteome
array: IP-10 concentration was very low in normal HPMECs and
significantly increased in EMP-stimulated HPMECs. It was also
revealed that the production of IP-10 in EMP-stimulated HPMECs was
time and concentration dependent. The highest level of IP-10
expression was observed when cells were treated with 10 µg/ml EMPs
for 24 h, but the concentration of IP-10 decreased when cells were
treated with EMPs for 48 h. IP-10 may be involved in EMP-induced
activation of HPMECs, and EMP-activated HPMECs may interact with
downstream cells to promote inflammation and induce immune disorder
through the modulation of IP-10. Fang et al (21) demonstrated that TNF-α-activated ECs
were an important source of IP-10, and the TNF-α pathway was
involved in the regulation of IP-10 production during monocyte-EC
interactions. Thus, EMP-activated ECs may be a source of IP-10, and
IP-10 may regulate the cellular interactions of the ECs it
originates from.
The mechanism of EMP generation requires the
transcription of NF-κB (22,23),
but it is unknown whether NF-κB is crucial to EMP function. Wang
et al (17) demonstrated
that MPs from LPS-treated THP-1 monocytic cells activated
intracellular signaling pathways in human endothelial cells,
including the NF-κB pathway. In addition, Bardelli et al
(24) demonstrated that
monocyte-derived MPs induce NF-κB activation in monocytes. The
present study revealed that, similar to monocytic MPs, EMPs
activate the NF-κB pathway in HPMECs. Immunofluorescence and
western blot analysis of NF-κB revealed that NF-κB was distributed
in the cytoplasm of inactive ECs, and translocated to the nucleus
following exposure to EMPs. Therefore, EMPs may activate ECs by
inducing the activation of the NF-κB pathway, resulting in the
release of a number of cytokines in order to initiate and amplify
the inflammatory response. Stimulation of HPECs with 10 µg/ml EMPs
for 24 h simultaneously resulted in increased IP-10 expression and
increased NF-κB translocation. Harris et al (25) previously demonstrated that
symmetrical dimethylation of NF-κB p65 by protein arginine
methyltransferase 5 enhanced IP-10 induction in response to TNF-α.
Therefore, the NF-κB pathway may contribute to the expression of
IP-10 in EMP-stimulated ECs.
There are some limitations in the present study.
Although EMPs activated their parental ECs and the related
mechanisms were proposed, the mechanisms behind the packaging of
secreted cytokines into EMPs and the key components of NF-κB
activation when EMPs activate ECs remain unknown. It was previously
demonstrated that TNF-α-derived EMPs transfer endothelial proteins,
which could affect the interaction between EMPs and target cells
through proteomic methods (14).
Future studies will focus on discovering key mediators contributing
to the effects of EMPs on ECs, and futher studying the involved
mechanisms in order to identify novel targets for the treatment of
endothelial dysfunction.
In conclusion, EMPs activate the inflammatory
response in ECs. EMP-stimulated ECs release a number of
proinflammatory cytokines including IP-10, which is an important
mediator of inflammation and the immune response. The mechanism
underlying the process of EMPs activating ECs may be the NF-κB
pathway. However, the key components packaged within EMPs which
contribute to the activation of ECs remain unknown, and future
studies will investigate these questions. The results of the
present study open a new line of investigation in the research of
EMPs and ECs, and EMPs could serve as potential therapeutic targets
for the treatment of sepsis and other diseases associated with
endothelial dysfunction.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81201451).
References
|
1
|
Dellinger RP, Levy MM, Rhodes A, Annane D,
Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke
R, et al: Surviving sepsis campaign: International guidelines for
management of severe sepsis and septic shock: 2012. Crit Care Med.
41:580–637. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reid VL and Webster NR: Role of
microparticles in sepsis. Br J Anaesth. 109:503–513. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rizvi AA: Cytokine biomarkers, endothelial
inflammation, and atherosclerosis in the metabolic syndrome:
Emerging concepts. Am J Med Sci. 338:310–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vassiliou AG, Mastora Z, Orfanos SE, Jahaj
E, Maniatis NA, Koutsoukou A, Armaganidis A and Kotanidou A:
Elevated biomarkers of endothelial dysfunction/activation at ICU
admission are associated with sepsis development. Cytokine.
69:240–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dignat-George F and Boulanger CM: The many
faces of endothelial microparticles. Arterioscler Thromb Vasc Biol.
31:27–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chironi GN, Boulanger CM, Simon A,
Dignat-George F, Freyssinet JM and Tedgui A: Endothelial
microparticles in diseases. Cell Tissue Res. 335:143–151. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mause SF and Weber C: Microparticles:
Protagonists of a novel communication network for intercellular
information exchange. Circ Res. 107:1047–1057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herring JM, McMichael MA and Smith SA:
Microparticles in health and disease. J Vet Intern Med.
27:1020–1033. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nieuwland R, Berckmans RJ, McGregor S,
Böing AN, Romijn FP, Westendorp RG, Hack CE and Sturk A: Cellular
origin and procoagulant properties of microparticles in
meningococcal sepsis. Blood. 95:930–935. 2000.PubMed/NCBI
|
|
10
|
Densmore JC, Signorino PR, Ou J, Hatoum
OA, Rowe JJ, Shi Y, Kaul S, Jones DW, Sabina RE, Pritchard KA Jr,
et al: Endothelium-derived microparticles induce endothelial
dysfunction and acute lung injury. Shock. 26:464–471. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delabranche X, Boisramé-Helms J, Asfar P,
Berger A, Mootien Y, Lavigne T, Grunebaum L, Lanza F, Gachet C,
Freyssinet JM, et al: Microparticles are new biomarkers of septic
shock-induced disseminated intravascular coagulopathy. Intensive
Care Med. 39:1695–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morel O, Morel N, Jesel L, Freyssinet JM
and Toti F: Microparticles: A critical component in the nexus
between inflammation, immunity, and thrombosis. Semin Immunopathol.
33:469–486. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peterson DB, Sander T, Kaul S, Wakim BT,
Halligan B, Twigger S, Pritchard KA Jr, Oldham KT and Ou JS:
Comparative proteomic analysis of PAI-1 and TNF-alpha-derived
endothelial microparticles. Proteomics. 8:2430–2446. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Huang W, Zhang R, Wu J, Li L and
Tang Y: Proteomic analysis of TNF-α-activated endothelial cells and
endothelial microparticles. Mol Med Rep. 7:318–326. 2013.PubMed/NCBI
|
|
15
|
Souza AC, Yuen PS and Star RA:
Microparticles: Markers and mediators of sepsis-induced
microvascular dysfunction, immunosuppression, and AKI. Kidney Int.
87:1100–1108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sabatier F, Roux V, Anfosso F, Camoin L,
Sampol J and Dignat-George F: Interaction of endothelial
microparticles with monocytic cells in vitro induces tissue
factor-dependent procoagulant activity. Blood. 99:3962–3970. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang JG, Williams JC, Davis BK, Jacobson
K, Doerschuk CM, Ting JP and Mackman N: Monocytic microparticles
activate endothelial cells in an IL-1β-dependent manner. Blood.
118:2366–2374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wen B, Combes V, Bonhoure A, Weksler BB,
Couraud PO and Grau GE: Endotoxin-induced monocytic microparticles
have contrasting effects on endothelial inflammatory responses.
PLoS One. 9:e915972014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luster AD, Unkeless JC and Ravetch JV:
Gamma-interferon transcriptionally regulates an early-response gene
containing homology to platelet proteins. Nature. 315:672–676.
1985. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Antonelli A, Ferrari SM, Giuggioli D,
Ferrannini E, Ferri C and Fallahi P: Chemokine (C-X-C motif) ligand
(CXCL)10 in autoimmune diseases. Autoimmun Rev. 13:272–380. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang YS, Zhu LM, Sun ZG, Yu LZ and Xu H:
Tumor necrosis factor-α pathway plays a critical role in regulating
interferon-γ induced protein-10 production in initial allogeneic
human monocyte-endothelial cell interactions. Transplant Proc.
44:993–995. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sapet C, Simoncini S, Loriod B, Puthier D,
Sampol J, Nguyen C, Dignat-George F and Anfosso F: Thrombin-induced
endothelial microparticle generation: Identification of a novel
pathway involving ROCK-II activation by caspase-2. Blood.
108:1868–1876. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leroyer AS, Anfosso F, Lacroix R, Sabatier
F, Simoncini S, Njock SM, Jourde N, Brunet P, Camoin-Jau L, Sampol
J and Dignat-George F: Endothelial-derived microparticles:
Biological conveyors at the crossroad of inflammation, thrombosis
and angiogenesis. Thromb Haemost. 104:456–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bardelli C, Amoruso A, Canova Federici D,
Fresu L, Balbo P, Neri T, Celi A and Brunelleschi S: Autocrine
activation of human monocyte/macrophages by monocyte-derived
microparticles and modulation of PPARg ligands. Br J Pharmacol.
165:716–728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harris DP, Bandyopadhyay S, Maxwell TJ,
Willard B and DiCorleto PE: Tumor necrosis factor (TNF)-α induction
of CXCL10 in endothelial cells requires protein arginine
methyltransferase 5 (PRMT5)-mediated nuclear factor (NF)-κB p65
methylation. J Biol Chem. 289:15328–15339. 2014. View Article : Google Scholar : PubMed/NCBI
|