Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
disease, which primarily affects small joints of the hands and
feet. The pathological characteristic of RA includes synovial
hyperplasia and inflammation, pannus formation, cartilage loss and
joint destruction (1).
Fibroblast-like synoviocytes (FLS), a type of synovial lining cell,
is considered as a pivotal effector cell in the inflamed joint. FLS
exhibit high proliferation rates, constitutive expression of
cytokines, and anchorage-independent cell growth (2–5). The
hyperplasia of FLS leads to pannus formation and joint destruction
(6). The production of cytokines,
such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β and
IL-6, causes the erosion of cartilage and contributes to
pathogenesis of RA (7).
Tripartite motif-containing protein 3 (TRIM3)
belongs to TRIM proteins family, which regulates diverse biological
functions including innate immune response, development and
carcinogenesis (8,9). Emerging evidence has suggested that
TRIM3 is a candidate tumor suppressor gene for glioblastomas
(10–12) and colorectal cancer (13). Overexpression of TRIM3 inhibited
cell proliferation of cancer cells (13,14).
However, little is known about the expression and function of TRIM
proteins, including TRIM3 in RA progression.
In the present study, we found that the mRNA and
protein levels of TRIM3 were significantly decreased in RA synovial
tissues. TRIM3 exerted an anti-proliferation role in primary
cultured FLS from RA patients via p38 signaling pathway. Taken
together, results of the current study suggest TRIM3 may be a
candidate therapeutic target for RA.
Materials and methods
Tissue specimens
A total of 30 RA patients and 12 joint trauma
patients (healthy control) were enrolled in this study. RA patients
were diagnosed according to the American College of Rheumatology
Criteria for Classification of RA (15). Written informed consent was
obtained from all patients, and the study was approved by the
Ethics Committee of the First Affiliated Hospital of Soochow
University. Synovial tissue samples were obtained during joint
replacement surgery, snap-frozen and stored at −80°C.
Real-time polymerase chain reaction
(PCR)
Total RNA was isolated from collected synovial
tissue samples using TRIzol (Invitrogen, Carlsbad, CA, USA)
according to the manufacturers protocol. After treating with
RNase-free DNase (Roche Diagnostics, Indianapolis, IN, USA) to
remove any residual genomic DNA, total RNA (2 µg) was
reverse-transcribed using cDNA synthesis kit (Thermo Fisher
Scientific, Inc., Rockford, IL, USA). TRIM3 mRNA levels were
examined by real-time PCR with SYBR-Green qPCR Master Mixes (Thermo
Fisher Scientific Inc.) on an ABI 7300 cycler (Applied Biosystems,
Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) mRNA levels were used for normalization. The primers were
as follows: TRIM3, 5-GAGGGCAA GTTCAAGACCAAG-3 and 5-GGAAGGTAAAGACGC
AGCAAG-3; GAPDH, 5-CACCCACTCCTCCACCTTTG-3 and
5-CCACCACCCTGTTGCTGTAG-3. TRIM3 mRNA expression was calculated
using the CT method as previously described (16).
Western blot analysis
Protein were extracted from frozen tissue samples
and cells by using RIPA buffer in the presence of proteinase
inhibitor cocktail (Sigma, St. Louis, MO, USA). Protein
concentrations were determined using the BCA kit (Thermo Fisher
Scientific, Inc.). Equal amounts of protein from samples were
separated on 10% SDS-PAGE and then transferred to a nitrocellulose
membrane (Millipore Corp., Bedford, MA, USA). For western blot
analysis, the membrane was pre-incubated with 5% skimmed milk in
TBST (Tris-buffered saline containing 0.1% Tween-20) at 25°C for 1
h, and then incubated with primary antibodies overnight at 4°C.
After being washed three times in TBST, horseradish
peroxidase-conjugated secondary antibodies (Beyotime Institute of
Biotechnology, Shanghai, China) were added and incubated for 1 h at
25°C. After three washes in TBST, hybridized bands were detected
using the ECL detection kit (Millipore Corp.). The sources of
primary antibodies were as follows: antibodies against TRIM3,
cyclin D1, and p21 were from Abcam (Cambridge, MA, USA); antibodies
to PCNA, p53, p-p38, p38 and GAPDH were from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Isolation of FLS cell lines from RA
patients
FLS cell lines were prepared from the synovium of RA
patients as previously described (17). In brief, synovial tissues were
minced into 1-mm pieces and treated with 0.4% type 1 collagenase
(Sigma) and DNase in Dulbeccos modified Eagles medium (DMEM;
Hyclone, Logan, UT, USA) for 2 h at 37°C in a 5%
CO2-humidified incubator. Undigested tissues were
removed with a cell strainer.
The dissociated cells were centrifuged at 1,000 rpm
for 10 min, resuspended in DMEM supplemented with 10% fetal bovine
serum (FBS; Gibco, Carlsbad, CA, USA), 100 U/ml penicillin, and 100
µg/ml streptomycin and seeded in 10-cm culture plates. Medium was
replaced every 2–3 days to remove suspended cells, and cells were
sub-cultured when the primary culture reached confluence. After the
third passage, the cells exhibited characteristic morphology of
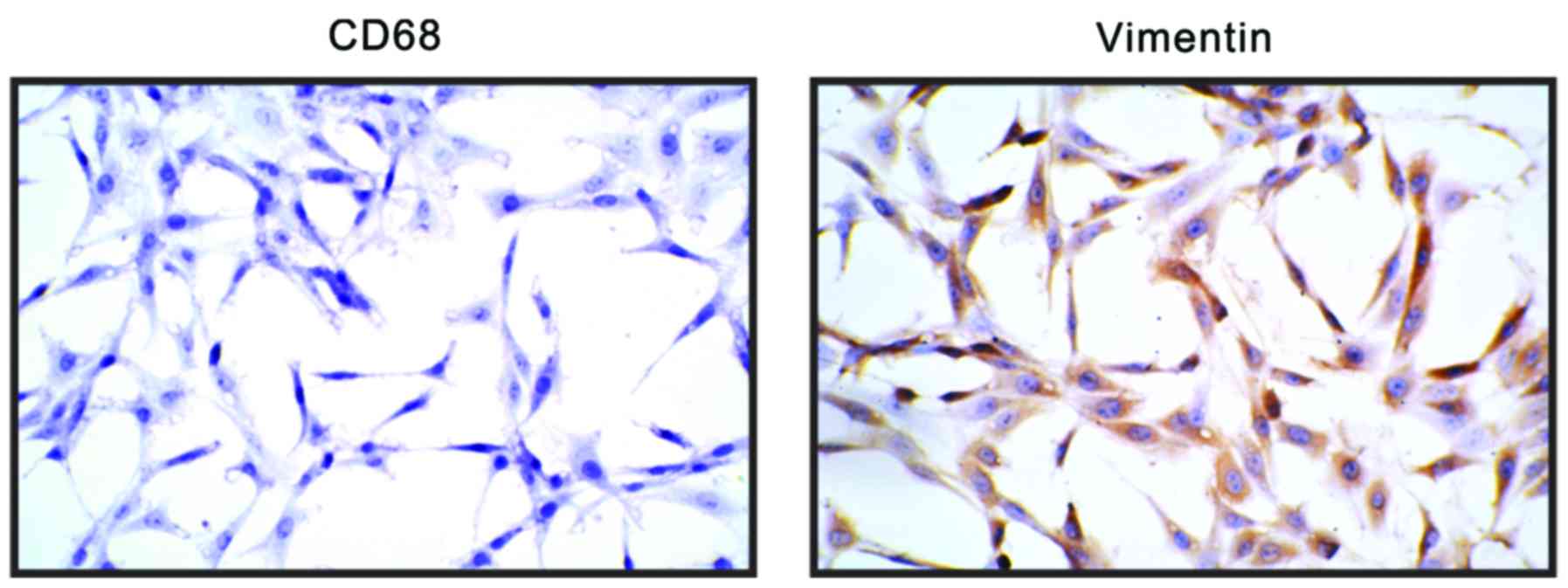
fibroblasts and were verified by immunocytochemistry as a
homogeneous population (>95% vimentin-positive and CD68-negative
(5); Fig. 1). FLS at passages 4–8 were used for
the experiments.
Lentivirus production and RNA
interference
To create TRIM3 lentiviral expression construct, the
full length human TRIM3 cDNA was inserted into pLVX-puro (Clontech
Laboratories, Inc., Palo Alto, CA, USA) and verified by sequencing.
pLVX-puro or pLVX-puro-TRIM3 together with viral packaging plasmids
(psPAX2 and pMD2G) was transfected into HEK293T cells using
Lipofectamine 2000 (Invitrogen) according to the manufacturers
protocol. Vector or TRIM3 expression viral supernatant was
harvested at 48 h after transfection and used to transduce FLS cell
lines.
siRNA targeting human TRIM3 mRNA (5-GCUCACUG
UCACUACCAAA-3) and a non-specific scramble siRNA sequence (siNC)
were synthesized by GenePharma (Shanghai, China) and transfected
into FLS cells using Lipofectamine 2000 (Invitrogen) according to
the manufacturers instructions.
Experimental grouping
To investigate the function of TRIM3 in RA, the FLS
cells were divided into three groups: group 1 (Mock), cells were
cultured without any treatment; group 2, cells were infected with
pLVX-puro vector virus; group 3, cells were infected with
TRIM3-expressing virus. Cell Counting Kit-8 (CCK-8) assays were
carried out at 0, 1, 2 and 3 days after viral infection. Western
blot analysis and enzyme-linked immunosorbent assays were performed
at 48 h after viral infection.
To further explore the involvement of p38 signaling,
the FLS cells were divided into five groups: group 1 (Mock), cells
were cultured without any treatment; group 2, cells were pretreated
with DMSO for 1 h and then transfected with control siRNA (siNC);
group 3, cells were pretreated with 10 µM p38 inhibitor (SB203580;
Selleck Chemicals, Houston, TX, USA) for 1 h and then transfected
with siNC; group 4, cells were pretreated with DMSO for 1 h and
then transfected with TRIM3 siRNA (siTRIM3); group 5, cells were
pre-treated with 10 µM SB203580 for 1 h and then transfected with
siTRIM3. CCK-8 assays were carried out at 0, 1, 2 and 3 days after
siRNA transfection. Western blot analysis was performed at 48 h
after siRNA transfection.
Cell proliferation assay
FLS proliferation was determined with the CCK-8 kit
(Signalway Antibody LLC (SAB), College Park, MD, USA). FLS cells
(3×103/well) were plated into 96-well plates. After
cultured overnight, the cells were treated as indicated and
cultured at 37°C for 0, 1, 2 and 3 days. After the incubation
period, the cultured media were replaced with 10% CCK-8 reagent (in
DMEM), and the cells were incubated at 37°C for another 1 h. The
absorbance at 450 nm was read with a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Immunoassays of IL-1β, IL-6 and
TNF-α
The amounts of secreted cytokines in culture media
were measured by ELISA. Assays were performed following the
manufacturers instructions (Bio-Swamp Life Science, Shanghai,
China). The absorbance was read at 450 nm and recorded on a
microplate reader (Bio-Rad Laboratories, Inc.).
Statistical analysis
The results are presented as the mean ± SD. One-way
analysis of variance followed by Dunnetts multiple comparisons was
used for statistical analysis by GraphPad Prism 6.0 software
(GraphPad, San Diego, CA, USA). P<0.05 was considered
significant.
Results
TRIM3 is downregulated in synovial
tissue samples of RA patients
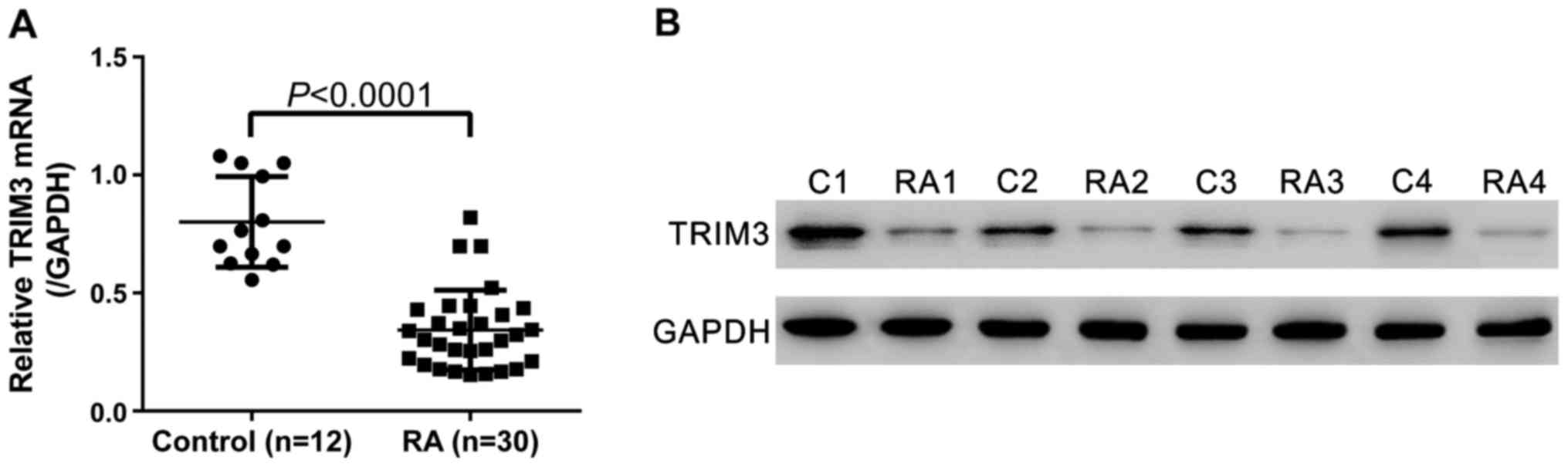
To determine TRIM3 expression in synovial tissues,
synovial tissue specimens were collected from 30 RA patients and 12
joint trauma patients (healthy control). As indicated by real-time
PCR analysis, compared with healthy control, the TRIM3 mRNA in
synovial tissues of RA patients decreased by 57.2% (Fig. 2A, P<0.0001). Western blot
analysis also showed the downregulation of TRIM3 in RA tissues at
translational level (Fig. 2B).
Inhibition of RA FLS cell
proliferation by TRIM3 overexpression
Synoviocyte hyperplasia is the most prominent
characteristic of RA (6). To
investigate the function of TRIM3 in RA, FLS cells were isolated
from the synovium of RA patients and TRIM3 was overexpressed in RA
FLS cells by lentiviral transduction. CCK-8 assays showed that
TRIM3 overexpressing virus (pLVX-puro-TRIM3) significantly
inhibited the proliferation of RA FLS cells at 1, 2 and 3 days as
compared to untreated cells (Mock) and cells transduced with vector
virus (pLVX-puro) (Fig. 3A). The
overexpression of TRIM3 in virally transduced cells was confirmed
by western blot analysis at 48 h after lentiviral transduction
(Fig. 3B).
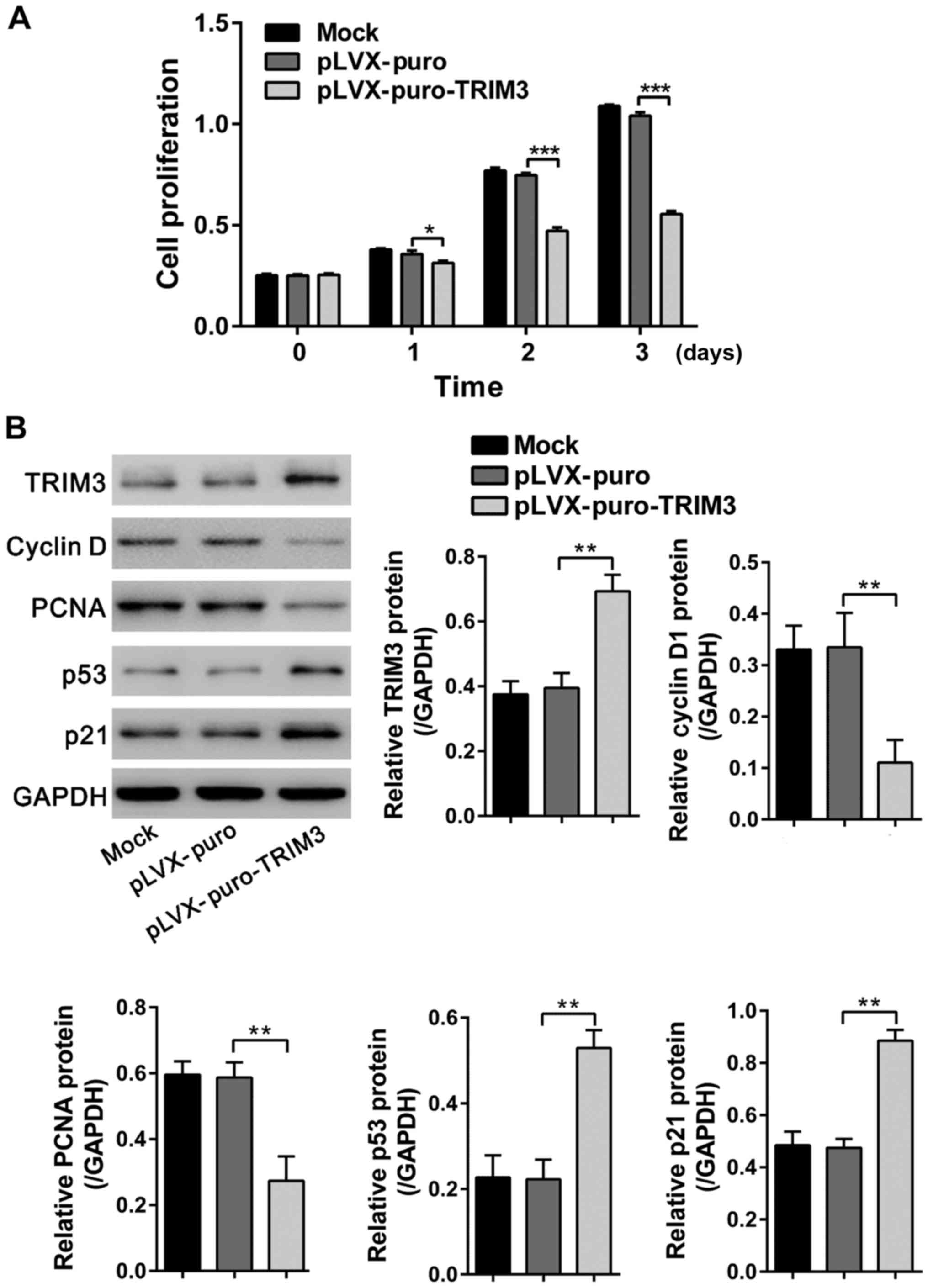 | Figure 3.Inhibition of RA FLS cell
proliferation by TRIM3 overexpression. (A) Results of CCK-8 assays
following infection of pLVX-puro or pLVX-puro-TRIM3 lentivirus into
RA FLS cells. Proliferation was significantly suppressed by
pLVX-puro-TRIM3 viral transduction. (B) Expression of TRIM3, cyclin
D1, PCNA, p53 and p21 was evaluated by western blot analysis at 48
h after viral transduction. Data are based on three independent
experiments, and shown as mean ± SD. *P<0.05, **P<0.01,
***P<0.001. RA, rheumatoid arthritis; FLS, fibroblast-like
synoviocytes; TRIM3, tripartite motif-containing protein 3; CCK-8,
Cell Counting Kit-8. |
The expression levels of cell proliferation
regulators, cyclin D1, PCNA, p53 and p21, were also evaluated by
western blot analysis. The levels of PCNA (a marker of cell
proliferation) (18) and cyclin D1
(a cell cycle-promoting protein) (19) were downregulated, while the levels
of p53 (a growth suppressor protein) (20) and its downstream target gene, p21,
was upregulated by TRIM3 overexpression (Fig. 3B). These results suggested that
TRIM3 inhibited the proliferation of RA FLS cells.
Suppression effect of TRIM3 on the
secretion of cytokines from RA FLS
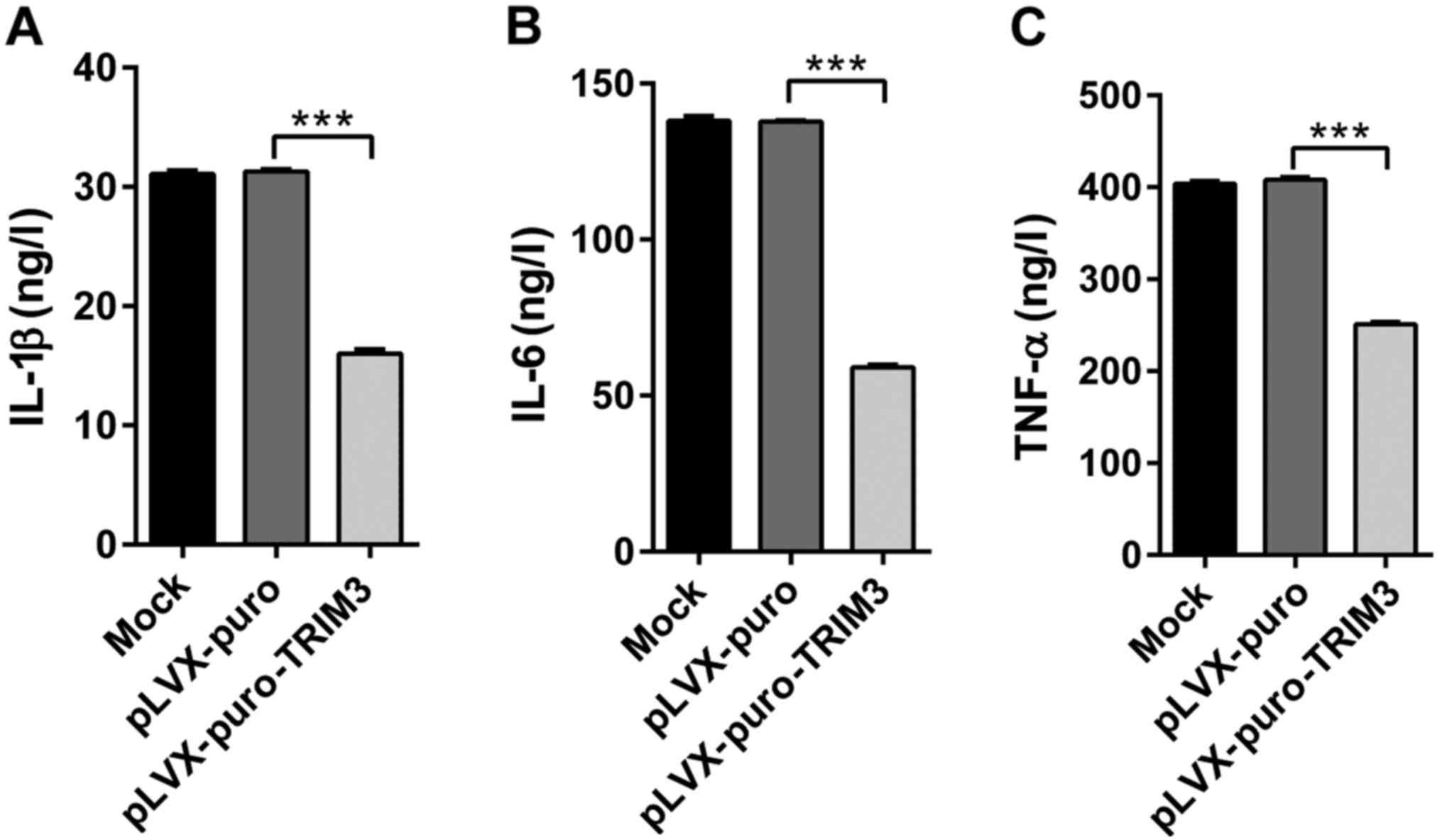
Cytokines, such as TNF-α, IL-1β and IL-6, contribute
to the pathogenesis of RA (7). We
next measured the changes of three cytokines in culture medium
conditioned with RA FLS after overexpression of TRIM3 (Fig. 4). All cytokine levels were
significantly decreased in the presence of TRIM3
overexpression.
TRIM3 inhibits the proliferation of RA
FLS via p38 signaling
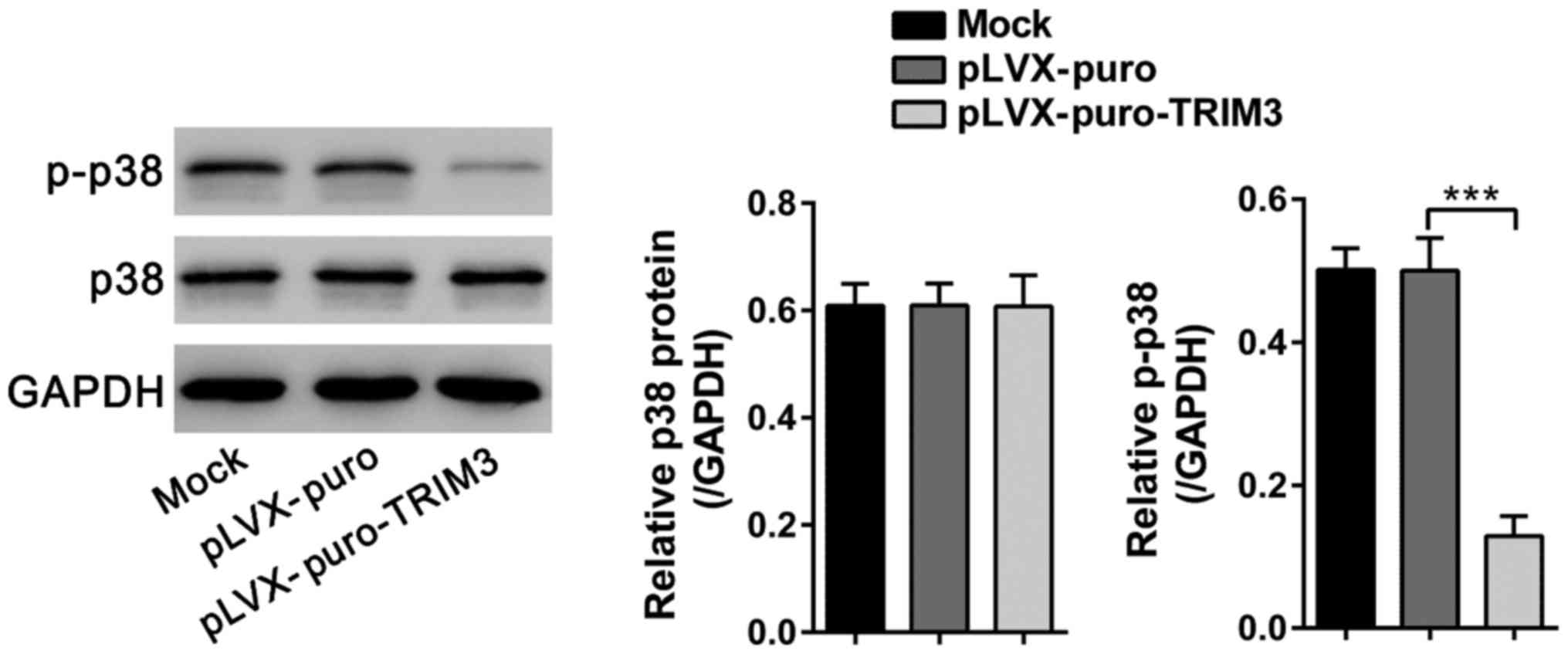
p38 is a key signaling component of inflammatory
disease including RA (21). To
further investigate the molecular mechanism through which TRIM3
inhibited the growth of RA FLS, western blot analyses were
performed to study changes in p38 signaling. As shown in Fig. 5, p-p38 levels were significantly
decreased at 48 h after TRIM3 expressing viral infection, while
total p38 levels were unchanged.
To further explore the involvement of p38 signaling
in suppression effect of TRIM3 on the proliferation of RA FLS, the
p38-specific inhibitor SB203580 was added before TRIM3 siRNA
transfection. Cell proliferation was repressed by SB203580
treatment, but promoted by TRIM3 knockdown. Additionally, SB203580
treatment reduced the promotion effects of TRIM3 on FLS
proliferation (Fig. 6A).
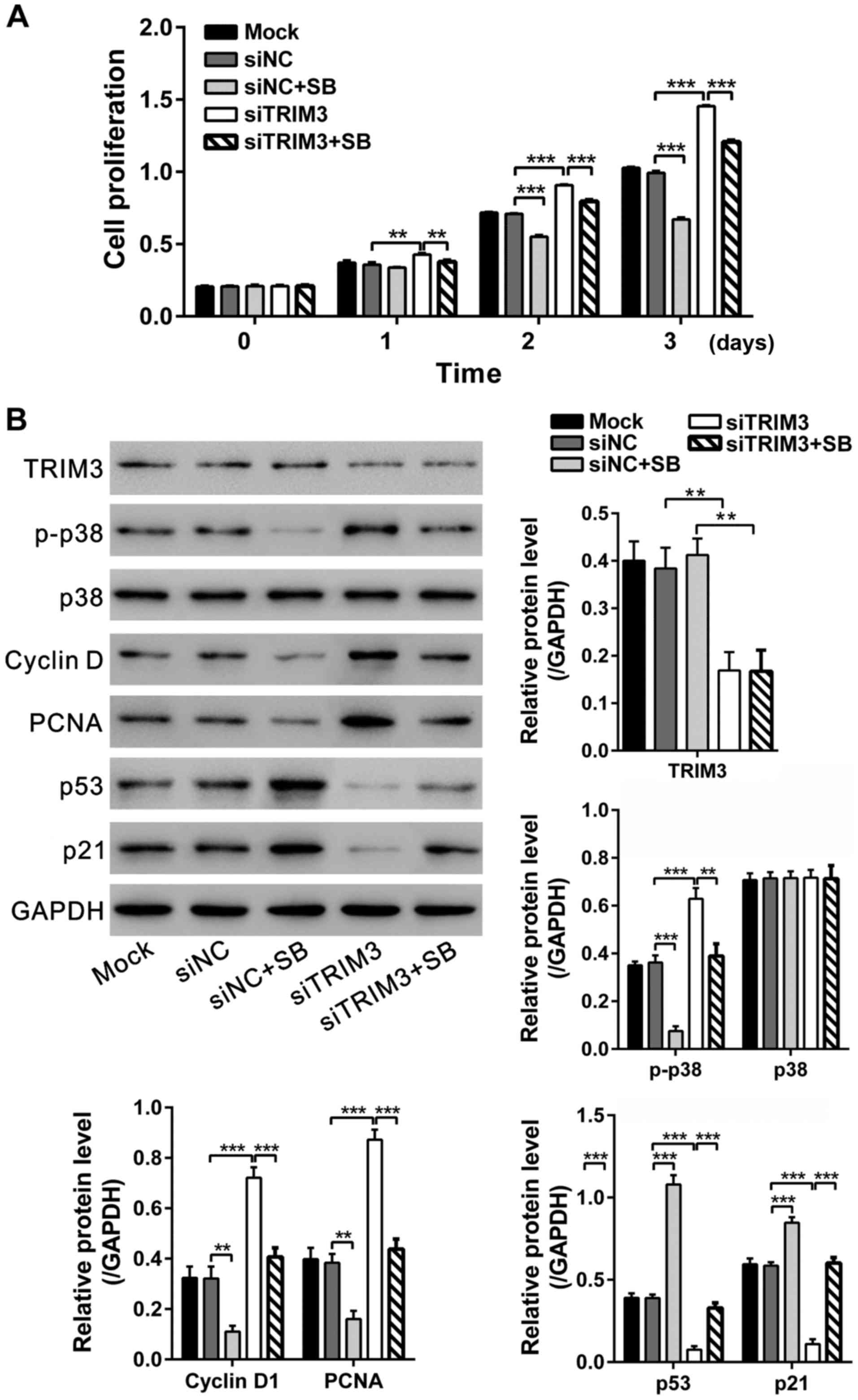 | Figure 6.TRIM3 inhibits the proliferation of RA
FLS via p38 signaling. The FLS cells were pre-treated with DMSO or
10 µM SB203580 for 1 h and then transfected with control siRNA
(siNC) or TRIM3 siRNA (siTRIM3). Cells cultured without any
treatment were set as control (Mock). (A) CCK-8 assays were
performed at 0, 1, 2 and 3 days after siRNA transfection. (B)
Expression of TRIM3, p-p38, p38, cyclin D1, PCNA, p53 and p21 was
evaluated by western blot analysis at 48 h after treatment. Data
are based on three independent experiments, and shown as mean ± SD.
**P<0.01; ***P<0.001. TRIM3, tripartite motif-containing
protein 3; RA, rheumatoid arthritis; FLS, fibroblast-like
synoviocytes; siTRIM3, TRIM3 siRNA; CCK-8, Cell Counting Kit-8. |
The protein levels of TRIM3, p-p38, p38, cyclin D1,
PCNA, p53 and p21 were then assessed by western blot analysis. As
shown in Fig. 6B, SB203580 blocked
the TRIM3 knockdown mediated increase of p-p38, cyclin D1 and PCNA,
and rescued the TRIM3 knockdown mediated decrease of p53 and p21.
Collectively, these results support the hypothesis that the
suppression effects of TRIM3 on RA FLS proliferation may be
achieved via inactivating p38 signaling.
Discussion
In the present study, we show that the expression of
TRIM3 was significantly lower in RA synovial tissues than in
control tissues. Overexpression of TRIM3 in RA FLS led to decreased
cell proliferation and reduced production of TNF-α, IL-1β and IL-6.
The suppression effects of cell proliferation by TRIM3 were
partially mediated via impaired p38 signaling.
Pathologic expression of TRIM3 has been identified
in glioblastomas (10–12), colorectal cancer (13) and hepatocellular carcinoma, where
its expression levels are lower than in normal tissues. TRIM3
functions as a tumor suppressor (10–13).
Studies of cancer cells have also shown that TRIM3 overexpression
can inhibit cell proliferation (13,14).
The present results of CCK-8 and western blot (PCNA and cyclin D1)
analyses in RA FLS were in accordance with previous findings.
It has been reported that RA synovium expressed many
cytokines (22). Among these
cytokines, IL-1β, IL-6 and TNF-α are critical in the evolution of
RA, partly because anti-cytokine strategies have been successfully
applied for patients (23,24). Several members of TRIMs, such as
TRIM30α, TRIM8 and TRIM21, are involved in the innate immunity
response (8). The current study
revealed that TRIM3 overexpression can inhibit the secretion of
IL-1β, IL-6 and TNF-α, indicating the role of TRIM3 in inflammatory
response and the potential clinical application of TRIM3 in RA.
Overexpression of TRIM3 in colorectal cancer cells
is able to upregulate p53 with concomitant induction of downstream
target genes, p21 and GADD45 (13). p38, a cell proliferation regulator,
is a key signaling component of inflammatory disease including RA
(21). Chemotherapeutic agents can
activate p53 via p38 signaling (25). Here, TRIM3 overexpression decreased
the expression of p53, p21 and p-p38 in RA FLS. By using the
p38-specific inhibitor SB203580, we found that the effects of TRIM3
on p53 was dependent on p38 activity. SB203580 blocked the effects
of TRIM3 knockdown that increased cell proliferation, and enhanced
the expression of cyclin D1 and PCNA. Therefore, the collective
evidence indicated that TRIM3 may inactivate p38/p53 signaling
pathway and could serve as a potential therapeutic target.
In conclusion, our findings demonstrated that TRIM3
expression was downregulated in RA synovium. It inhibited RA FLS
proliferation via p38 signaling pathway, and suppressed cytokine
secretion. Thus, TRIM3 plays a potentially protective role for FLS
hyperplasia and inflammation.
References
|
1
|
Majithia V and Geraci SA: Rheumatoid
arthritis: Diagnosis and management. Am J Med. 120:936–939. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ritchlin C, Dwyer E, Bucala R and
Winchester R: Sustained and distinctive patterns of gene activation
in synovial fibroblasts and whole synovial tissue obtained from
inflammatory synovitis. Scand J Immunol. 40:292–298. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bucala R, Ritchlin C, Winchester R and
Cerami A: Constitutive production of inflammatory and mitogenic
cytokines by rheumatoid synovial fibroblasts. J Exp Med.
173:569–574. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lafyatis R, Remmers EF, Roberts AB, Yocum
DE, Sporn MB and Wilder RL: Anchorage-independent growth of
synoviocytes from arthritic and normal joints. Stimulation by
exogenous platelet-derived growth factor and inhibition by
transforming growth factor-beta and retinoids. J Clin Invest.
83:1267–1276. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartok B and Firestein GS: Fibroblast-like
synoviocytes: Key effector cells in rheumatoid arthritis. Immunol
Rev. 233:233–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wicks I, Cooley H and Szer J: Autologous
hemopoietic stem cell transplantation: A possible cure for
rheumatoid arthritis? Arthritis Rheum. 40:1005–1011. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Firestein GS: Invasive fibroblast-like
synoviocytes in rheumatoid arthritis. Passive responders or
transformed aggressors? Arthritis Rheum. 39:1781–1790. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ozato K, Shin DM, Chang TH and Morse HC
3rd: TRIM family proteins and their emerging roles in innate
immunity. Nat Rev Immunol. 8:849–860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hatakeyama S: TRIM proteins and cancer.
Nat Rev Cancer. 11:792–804. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Raheja R, Yeh N, Ciznadija D,
Pedraza AM, Ozawa T, Hukkelhoven E, Erdjument-Bromage H, Tempst P,
Gauthier NP, et al: TRIM3, a tumor suppressor linked to regulation
of p21Waf1/Cip1. Oncogene. 33:308–315. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen G, Kong J, Tucker-Burden C, Anand M,
Rong Y, Rahman F, Moreno CS, Van Meir EG, Hadjipanayis CG and Brat
DJ: Human Brat ortholog TRIM3 is a tumor suppressor that regulates
asymmetric cell division in glioblastoma. Cancer Res. 74:4536–4548.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boulay JL, Stiefel U, Taylor E, Dolder B,
Merlo A and Hirth F: Loss of heterozygosity of TRIM3 in malignant
gliomas. BMC Cancer. 9:712009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Piao MY, Cao HL, He NN, Xu MQ, Dong WX,
Wang WQ, Wang BM and Zhou B: Potential role of TRIM3 as a novel
tumour suppressor in colorectal cancer (CRC) development. Scand J
Gastroenterol. 51:572–582. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Raheja R, Liu Y, Hukkelhoven E, Yeh N and
Koff A: The ability of TRIM3 to induce growth arrest depends on
RING-dependent E3 ligase activity. Biochem J. 458:537–545. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cohen S and Emery P: The American College
of Rheumatology/European League Against Rheumatism criteria for the
classification of rheumatoid arthritis: A game changer. Arthritis
Rheum. 62:2592–2594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Saha SK, Roy S and Khuda-Bukhsh AR:
Ultra-highly diluted plant extracts of Hydrastis canadensis and
Marsdenia condurango induce epigenetic modifications and alter gene
expression profiles in HeLa cells in vitro. J Integr Med.
13:400–411. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nishida K, Komiyama T, Miyazawa S, Shen
ZN, Furumatsu T, Doi H, Yoshida A, Yamana J, Yamamura M, Ninomiya
Y, et al: Histone deacetylase inhibitor suppression of
autoantibody-mediated arthritis in mice via regulation of p16INK4a
and p21WAF1/Cip1 expression. Arthritis Rheum. 50:3365–3376. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Robbins BA, de la Vega D, Ogata K, Tan EM
and Nakamura RM: Immunohistochemical detection of proliferating
cell nuclear antigen in solid human malignancies. Arch Pathol Lab
Med. 111:841–845. 1987.PubMed/NCBI
|
|
19
|
Fu M, Wang C, Li Z, Sakamaki T and Pestell
RG: Minireview: Cyclin D1: Normal and abnormal functions.
Endocrinology. 145:5439–5447. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Montenarh M: Functional implications of
the growth-suppressor oncoprotein p53 (Review). Int J Oncol.
1:37–45. 1992.PubMed/NCBI
|
|
21
|
Pargellis C and Regan J: Inhibitors of p38
mitogen-activated protein kinase for the treatment of rheumatoid
arthritis (Review). Curr Opin Investig Drugs. 4:566–571.
2003.PubMed/NCBI
|
|
22
|
McInnes IB and Schett G: Cytokines in the
pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 7:429–442.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishimoto N, Hashimoto J, Miyasaka N,
Yamamoto K, Kawai S, Takeuchi T, Murata N, van der Heijde D and
Kishimoto T: Study of active controlled monotherapy used for
rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): Evidence of
clinical and radiographic benefit from an × ray reader-blinded
randomised controlled trial of tocilizumab. Ann Rheum Dis.
66:1162–1167. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maini RN and Taylor PC: Anti-cytokine
therapy for rheumatoid arthritis. Annu Rev Med. 51:207–229. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sanchez-Prieto R, Rojas JM, Taya Y and
Gutkind JS: A role for the p38 mitogen-acitvated protein kinase
pathway in the transcriptional activation of p53 on genotoxic
stress by chemotherapeutic agents. Cancer Res. 60:2464–2472.
2000.PubMed/NCBI
|