Introduction
An adequate O2 supply via the vasculature
is important for the normal function of the cardiovascular system
(1). Reduced or impaired blood
flow patterns in the larger conductance and resistance arteries are
a factor in numerous pathological conditions that are associated
with hypoxia-induced injury (2–4). It
is known that acute hypoxia is associated with a number of
cardiovascular diseases, including myocardial infarction (5), pulmonary hypertension (6,7) and
mesenteric ischemia (8) and thus
leads to an increase in blood pressure together with peripheral
vascular resistance due to reduction of tone in certain vascular
beds and critical injury or death (4).
In general, vascular dysfunction and reduction of
vasomotor tone by hypoxia/ischemia are characterized by
abnormalities in intercellular communication, ion transport and the
transport of biologically active substances between adjacent
vascular smooth muscle cells (VSMCs) and vascular endothelial cells
(ECs) (9). The gap junction (GJ)
in the vasculature provides intercellular communication channels
that permit the direct exchange of ions and small signaling
molecules between neighboring cells and thus it is involved in the
regulation of vasomotor tone and the coordination of vascular
function by electrical and chemical coupling formed within and
between the ECs and the VSMCs (9–11).
The roles of the GJ are not confined to providing electrical
coupling between neighboring cells; in various types of cells, the
GJ serves a critical role in deciding cell survival vs. cell death
under hypoxia/ischemia (12).
The GJ results from the docking of two hemichannels,
which are formed by the assembly of six connexins (Cxs) (10,13).
Four isoforms of Cxs (Cx37, Cx40, Cx43 and Cx45) are expressed in
the vasculature of mammals, with the predominant expression of Cx37
and Cx40 in ECs and Cx43 and Cx45 in the VSMCs (9,10).
The present study identified that hypoxia/ischemia preconditioning
in the heart suppresses electrical and chemical gap junctional
intercellular communication (GJIC) of cardiomyocytes (14). The reduction of GJ coupling or GJ
proteins in cardiomyocytes during hypoxia/ischemia is widely
assumed to promote the development of arrhythmias (15). Several studies (13,15–18)
have identified different molecular mechanisms concerning the
effect of hypoxia on GJIC, including a decline in expression in a
time-dependent manner, dephosphorylation and redistribution of
Cx43, the degradation of Cxs and the attenuation of endothelial
adenosine triphosphate (ATP) release by hypoxic exposure. The
present study on the actions of Cxs following exposure to hypoxic
stresses focuses on the myocardium and astrocytes in the brain and
vascular endothelial; however, the association between Cxs
expression and Cxs functional involvement in the vascular smooth
muscle layer of resistance arteries during hypoxia remains to be
elucidated.
To address this question, using a well-defined in
vitro hypoxic model, the present study investigated whether
acute hypoxia suppresses the vasoconstriction of MRA in
Sprague-Dawley (SD) rats and if vasoconstriction suppression is
associated with the reduction of GJIC and Cx43/45 expression. The
present study was performed to produce an improved understanding of
the mechanisms involved in the change of vasomotor responses
following acute hypoxia and to provide a hypothesis for the
inhibition of expression and function of Cxs in MRAs during acute
hypoxia conditions.
Materials and methods
Animals
Female and male 12-week-old SD rats (260–280 g;
n=18) supplied by Beijing Vital River Laboratory Animal Technology
Co., Ltd. (license number: SCXK (BJ) 2012–0001), were used in the
current study. All protocols were approved by the Institutional
Animal Care and Use Committee at the Medical College of Shihezi
University (Xinjiang, China) and were consistent with the
Guidelines for the Care and Use of Laboratory Animals published by
the US National Institutes of Health (Public Health Service Policy
on Humane Care and Use of Animals, DHEW Publication No. 96-01, PHS
Policy revised in 2002). Rats were anesthetized with an
intramuscular injection (1 ml/kg) of a mixture of
ketamine/xylazine/acepromazine (500/20/10 mg in 8.5 ml
H2O) (Sigma-Aldrich, Merck KGaA Millipore, Darmstadt,
Germany) and subsequently rats were euthanized with an overdose of
100 mg/kg sodium pentobarbital (Sigma-Aldrich, Merck kGaA). The
third-order branch of the MRA was harvested from the upper ileum
mesentery and the surrounding connective and adipose tissue were
removed for whole-cell patch clamp recordings, pressure myographic
measurement and western blot analysis.
Hypoxia vascular model
An acute hypoxia model of MRA segments in
vitro was established according to previous studies (4,19,20).
The rats were euthanized and the descending third-order branch of
the MRA (~10 mg) was removed. The MRA segment was rinsed several
times in PBS [composed of the following (g/l): NaCl 8.0, KCl 0.2,
KH2PO4 0.24, Na2HPO4
1.44; pH 7.4]. The connective adipose tissue surrounding the MRA
and the endothelial cells was cleaned and the MRA rings (~0.4 mm
long, 200 mm in outer diameter) were immersed in DMEM/F12 medium
with high glucose (Hyclone; GE Healthcare Bio Sciences, Pittsburgh,
PA, USA) at 37°C for 24 h and then replaced with DMEM/F12 medium of
low glucose (Hyclone; GE Healthcare Bio Sciences). Following
hypoxia treatment for 5 min or 1 h at 37°C in a humidified
atmosphere of <3% O2, 5% CO2 and 95%
N2, the medium was replaced with PBS and the MRA rings
were used in western blot analysis. The vascular normoxic group in
this process was placed in an incubator at 37°C. In the study by
Nuñez et al (19), rings of
pulmonary and systemic arteries obtained from rats were suspended
in an organ bath (37°C) containing Krebs solution and an atmosphere
of 12% O2, 5% CO2 and 83% N2,
which produced an O2 concentration similar to that
present in aortic blood. Hypoxia was induced by flooding with 95%
N2 and 5% CO2, which decreased the
concentration of O2 in the bath to ~5×10−6
mol/l. In the experimental design of the present study, the hypoxia
vascular model was induced by flooding the vessels with 5%
O2, 90% N2 and 5% CO2 or 2.5%
O2, 92.5% N2 and 5% CO2, which was
expected to reduce the concentration of O2 to levels
similar to those reported by Nuñez et al (19). In addition, each experiment based
on the acute hypoxia model of MA segment included three independent
replicates and the preparation of the arteries was performed
according to Lee et al (21).
Tight-seal whole-cell patch clamp
recording
As described in a previous study (22), 0.4 mm is the standard length of MRA
for tight-seal whole-cell patch clamp recording. In general, the
shorter the blood vessel, the smaller the adverse effects for the
clamping space. In addition, an average length of 0.4 mm for the
MRA was selected in order to maintain the voltage stability of
patch clamp and thus ensuring the accuracy of results. Following
exposure to 5 min acute hypoxia the MRA was transferred to a
glass-bottomed Petri dish filled with an aerated external solution
composed of (mM): NaCl 138, KCl 5, CaCl2 1.6,
MgCl2 1.2, Na-HEPES 5, HEPES 6 and glucose 7.5. The
preparation was secured at the bottom of the dish using the weight
of a platinum strip at each end and digested with collagenase A (1
mg/ml) dissolved in the external solution at 37°C for 15 min. The
collagenase A digestion was used only for the patch clamp recording
and was to expose the smooth muscle cells and avoid the adverse
effect of adventitial connective tissue on membrane current. In
addition, the two ends of MRA segment were secured at the bottom of
the dish using a platinum strip and thus making it difficult for
the collagenase is to enter into the vascular lumen. The enzyme was
washed away gently twice with external solution and the MRA segment
was further cleaned to remove the adventitial tissue. The Petri
dish was then placed onto the stage of an inverted microscope
equipped with micromanipulators. The specimen was continuously
superfused with the external solution (0.2 ml/min) at room
temperature (22–25°C).
Conventional whole-cell recordings were performed
using an Axon 700B amplifier (Axon Instruments; Molecular Devices,
LLC, Sunnyvale, CA, USA) as described previously (23). Recording pipettes were pulled from
borosilicate glass capillaries with filaments using a P-97 puller
(Sutter Instrument, Novato, CA, USA). The pipette typically had a
resistance of ~5 MΩ subsequent to being filled with internal
solution containing the following (mM): K-gluconate 130, NaCl 10,
CaCl2 2, MgCl2 1.2, HEPES 10, ethylene
glycol-bis (b-aminoethylether) N,N',N'-tetraacetic acid 5 and
glucose 7.5. The membrane current or voltage signal was low-pass
filtered at 10 kHz; the data were recorded on a PC equipped with a
Digidata 1440A AD-interface and pClamp 10.2 software (Axon
Instruments; Molecular Devices) at a sampling interval of 10, 20 or
100 msec. A Minidigi digitizer and Axoscope 10.2 software (Axon
Instruments; Molecular Devices) were used to simultaneously perform
gap-free recording at a sampling interval of 50 msec.
The seal resistance usually reached 1–20 GΩ prior to
the rupture of the membrane. Membrane rupture was achieved by a
high-frequency buzz current and/or suction pressure from the
pipette. The transient current over the membrane input capacitance
(Cinput) was routinely uncompensated to monitor and
calculate the access resistance (Ra) and the membrane
parameters on- or off-line.
Pressure myographic
The isolated MRA vascular segments were rapidly
removed and pinned in a dissecting dish filled with aerated 4°C
physiological salt solution (PSS) containing (mM): NaCl 118.9, KCl
4.7, MgSO4 1.2, KH2PO4 1.2,
CaCl2 2.5, NaHCO3 25 and glucose 11. The
arteries were cannulated at their ends with glass micropipettes
(1.2 mm; World Precision Instruments, LLC, Sarasota, FL, USA),
which were pulled to 300 to 400 µm in diameter for the MRA and
secured using a 11–0 nylon monofilament suture. At all times, care
was taken to avoid excessive tension or stretching of the vascular
tissues during dissection. Once the tissues were mounted, the
vessel and the perfusion chamber were transferred to an inverted
trinocular microscope equipped with an analog video camera and
computer-assisted image capture system (Pressure Myograph System;
Danish Myo Technology A/S, Aarhus, Denmark) (24) to continuously record the outer
diameter of the MRA. The bath temperature was held constant at 37°C
and was continuously monitored with a thermal microprobe placed
immediately adjacent to the mounted vessel. The chamber was
superfused at 1.5 ml/min with warmed and aerated physiologic salt
solution (pH 7.4, aerated with 95% O2 and 5%
CO2 for the control group and 5% O2 and 95%
N2 for the hypoxiagroup) and maintained at 37°C. The MRA
segments were pressurized with a stepwise increase in transmural
pressure that was applied in 10 mmHg increments up to the
appropriate working pressure (60 mmHg) with 5 min of equilibration
at each pressure, according to requirement. All experiments were
performed under conditions of zero intraluminal flow. The bath was
changed to a recirculating buffer circuit (total volume 20 ml) for
treatment with various drugs. The diameter was continuously
determined by a video dimension analyzer and recorded using a DMT
Vessel Acquisition Suite (Danish Myo Technology A/S). Cumulative
dose-response curves to phenylephrine (PE; 0.1–100 µM) were
generated (24).
Western blot analysis
The MRA segments were homogenized in RIPA buffer (at
a ratio of 10 mg of tissue to 100 µl of RIPA buffer) with freshly
added protease inhibitor phenylmethylsulfonyl fluoride. The
homogenates were incubated at 4°C for 30 min and centrifuged at
12,000 × g for 15 min at 4°C. The supernatant was collected and the
protein concentration in the supernatant was determined. Protein
aliquots (40 mg) were subjected to 4–15% tris-glycine denaturing
gradient gel electrophoresis. The proteins were then transferred
onto a PVDF membrane (EMD Millipore, Billerica, MA, USA). The
membrane was hybridized with specific primary antibodies against
Cxs (1:1,000) at 4°C overnight. Subsequently, the membrane was
incubated with appropriated horseradish peroxidase-conjugated
secondary antibodies (1:10,000) (Beijing Zhongshan Jinqiao
Biotechnology Co., Ltd., Beijing, China) at room temperature for 2
h. Immunoreactive bands were detected using the ECL
chemiluminescence reagent (GE Healthcare Life Sciences, Chalfont,
UK). The membrane was stripped following the manufacturer's
protocol and labeled with β-actin antibody (catalog no. ab8226;
1:1,000; Abcam, Cambridge, MA, USA) as an internal control. The
intensities of the protein bands were analyzed using Quantity One
software (Bio-Rad, Hercules, CA, USA).
Sources of reagents
Cx45 and Cx43 primary antibodies were obtained from
Abcam (catalog no. ab79010 and ab78408 for anti-Cx43 antibody and
anti-Cx45 antibody, respectively), the horseradish
peroxidase-conjugated secondary antibody was obtained from Beijing
Zhongshan Jinqiao Biotechnology Co., Ltd. The bicinchoninic acid
protein assay kit was purchased from Pierce (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). RIPA buffer, PE and
2-aminoethoxydiphenyl borate (2-APB; a GJ inhibitor) were purchased
from Sigma-Aldrich (Merck KgaA). 2-APB was dissolved in dimethyl
sulfoxide as a stock solution prior to being further diluted with
the external solution to achieve the final concentrations. The
final dimethyl sulfoxide concentration in solution was ≤0.1%, which
had no detectable effect on vasomotor activity.
Statistical analysis
The rats were age matched to minimize individual
differences. The results are expressed at the mean ± standard error
of the mean. For PE-induced vascular reactivity experiments,
vasoconstriction effects were calculated using the following
equation: [vasoconstriction
effect=(DPSS-DPE)/DPSS ×100%].
DPSS, the constant vessel diameter in PSS;
DPE, the constant vessel diameter following treatment of
PE with different concentrations. Statistical analysis was
performed using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA).
The primary statistical analyses were performed using a two-tailed
Student's t-test or where appropriate by analysis of variance.
P<0.05 was considered to indicate a statistically significant
difference.
Results
GJs between MRA VSMCs are suppressed
following exposure to acute hypoxia
Compared to constant oxygen perfusion of the VSMCs
(normoxia group), acute hypoxia 5 min (a duration of hypoxia >5
min would result in the decline of cell activity) significantly
decreased the membrane conductance (Ginput) and membrane
capacitance (Cinput). However, a marked increase in the
membrane resistance (Rinput) and the absolute value of
rest potential was observed in the VSMCs of the acute hypoxia
compared with the normoxia group (n=13; Table I).
 | Table I.Changes of membrane properties of
vascular smooth muscle cells in situ in mesenteric
resistance artery following hypoxia. |
Table I.
Changes of membrane properties of
vascular smooth muscle cells in situ in mesenteric
resistance artery following hypoxia.
| Group |
Rinput (MΩ) |
Ginput (nS) |
Cinput (pF) | RP (mV) |
|---|
| Control | 397±68 | 2.27±0.43 | 201.6±83.0 | −22.1±1.5 |
| Hypoxia |
2,365±340a |
0.49±0.07a |
19.3±3.4b |
−44.5±2.9a |
| Reoxygenation | 566±124 | 2.71±0.46 | 167.4±96.1 | −25.8±1.1 |
Acute hypoxia attenuates
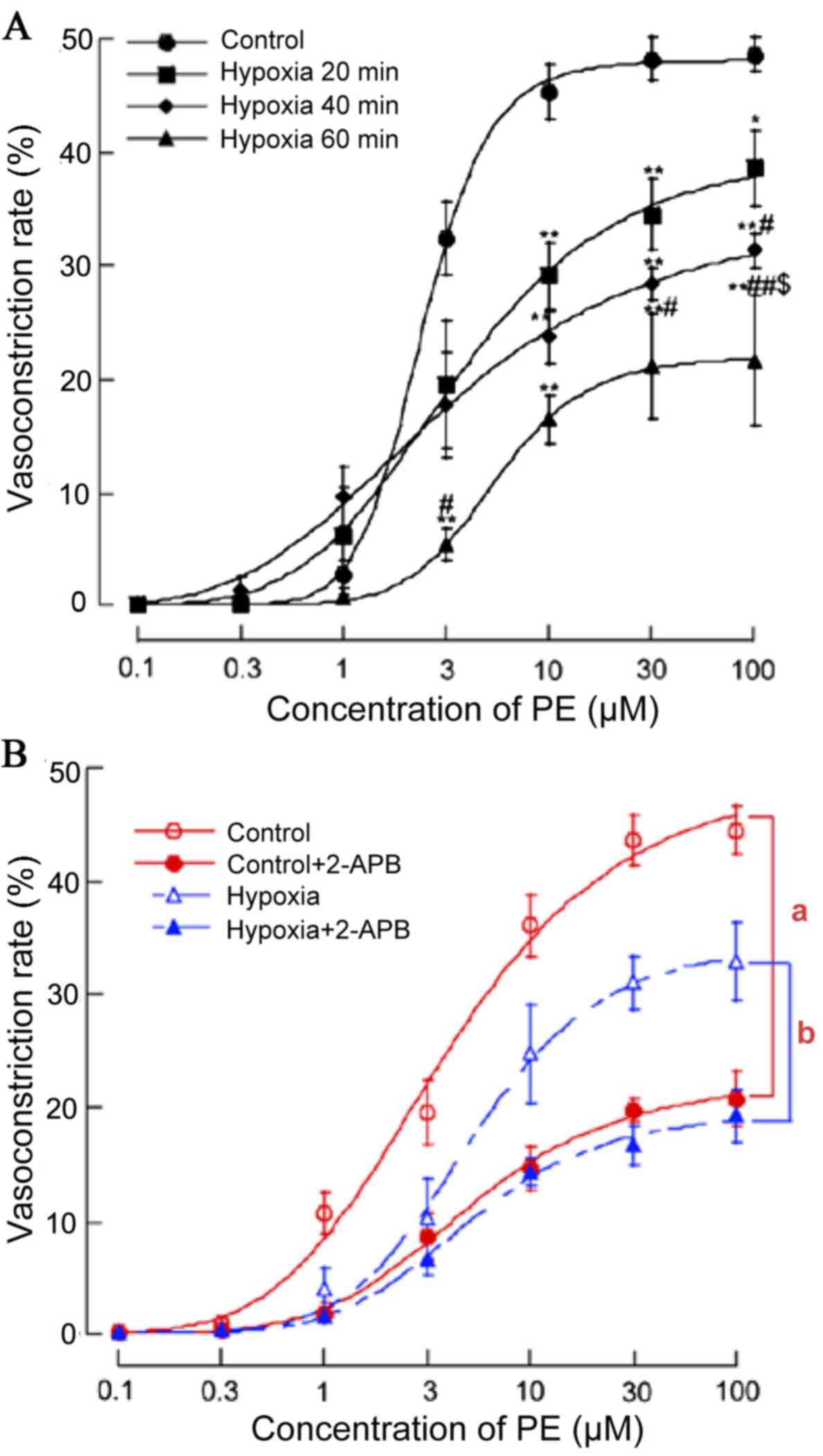
vasoconstriction of MRAs via decreased GJ communication
Vasoconstrictor responses are Cx dependent,
particularly in non-denuded preparations (11). To examine the effects of acute
hypoxia on the smooth muscle dependent vasomotor responses, PE was
applied to the MRAs of acute hypoxia and normoxia rats. PE (0.1–100
µM) initiated concentration-dependent vasoconstriction of the MRAs
of normoxia rats (EC50=3.77 µM) and acute hypoxia rats
(EC50=4.11 µM for 20 min, EC50=3.99 µM for 40
min, EC50=4.48 µM for 60 min). PE (1–100 µM) induced
more pronounced vasoconstriction in normoxia vs. acute hypoxia
rats. (n=7; P<0.01; Fig.
1A).
In addition, to confirm whether the GJ was involved
in the process that acute hypoxia suppressed PE (0.1–100 µM)
initiated concentration-dependent vasoconstriction, the effect of
GJ inhibitor 2-APB (100 µM for 20 min) on PE-induced
vasoconstriction in MRA under normoxia and acute hypoxia was also
investigated. Pre-incubation with 2-APB shifted the
concentration-response curve of PE-induced vasoconstriction
downward (Fig. 1B). The inhibitory
effects of 2-APB on the vasoconstriction induced by PE (1–100 µM)
were greater in the normoxia compared with the acute hypoxia rats
(n=9; P<0.05).
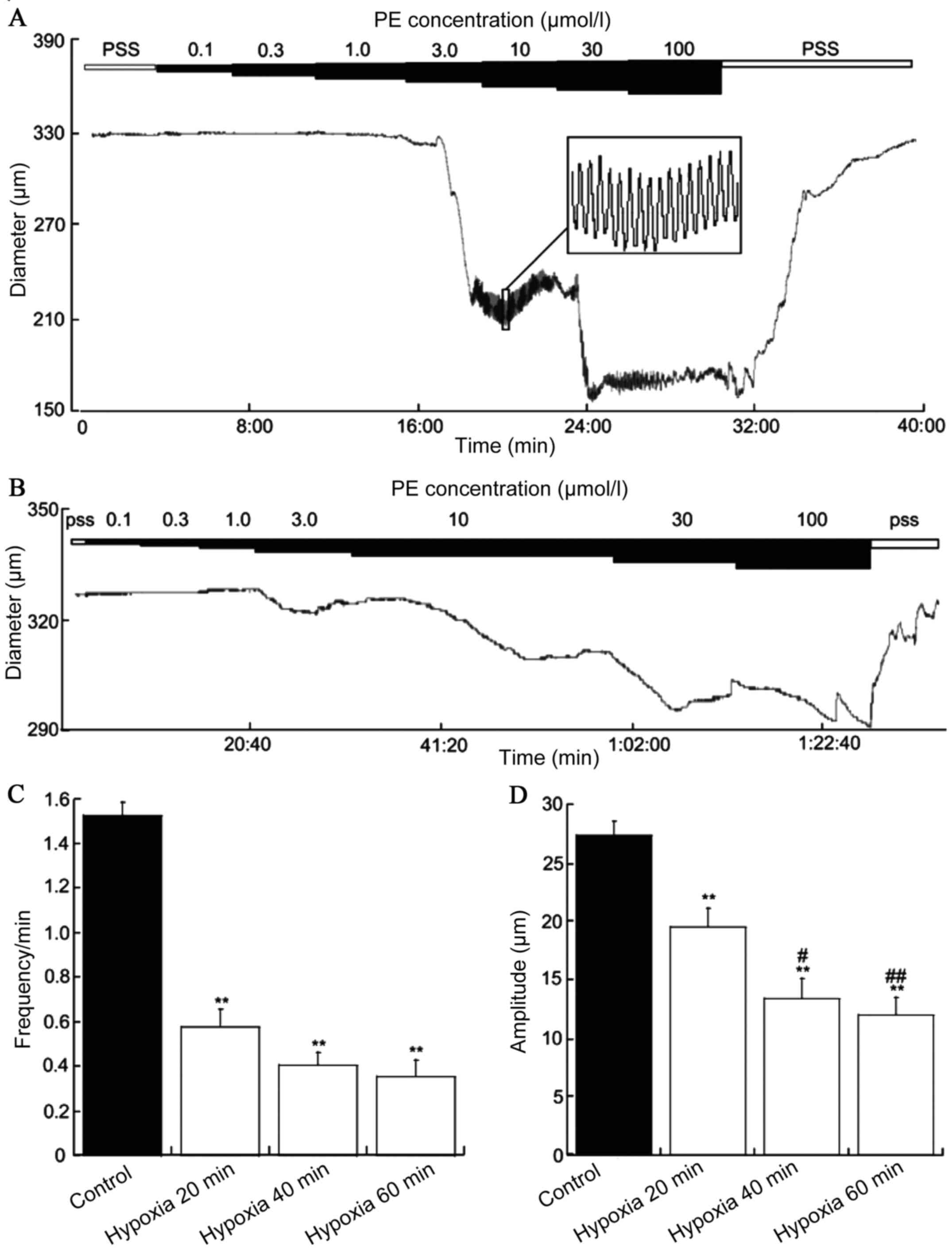
Acute hypoxia for 20, 40 and 60 min
inhibited the frequency of contraction
The diameter of the MRAs was 366.6±11.5 µm (n=16)
and no difference was observed between the MRA of acute hypoxia for
20, 40 and 60 min. The vasoconstrictor PE (0.1–100 µM) was applied
to the MRAs of SD rats and constricted the blood vessels to a
stable state exhibiting spontaneously vascular vasomotor activity.
The frequency was 1.52±0.11 per min (n=16) and the amplitude
27.35±1.21 per min (n=18). Acute hypoxia for 20, 40 and 60 min
PE-induced vasomotion frequencies were 0.57±0.08 (n=6), 0.40±0.05
(n=4) and 0.35±0.07 (n=4), and the amplitudes were 19.52±1.29
(n=6), 13.36±1.71 (n=4) and 12.00±1.46 (n=4), respectively. The
frequency and amplitude were significantly reduced in acute hypoxia
for 20, 40 and 60 min vs. the control group (P<0.01; Fig. 2).
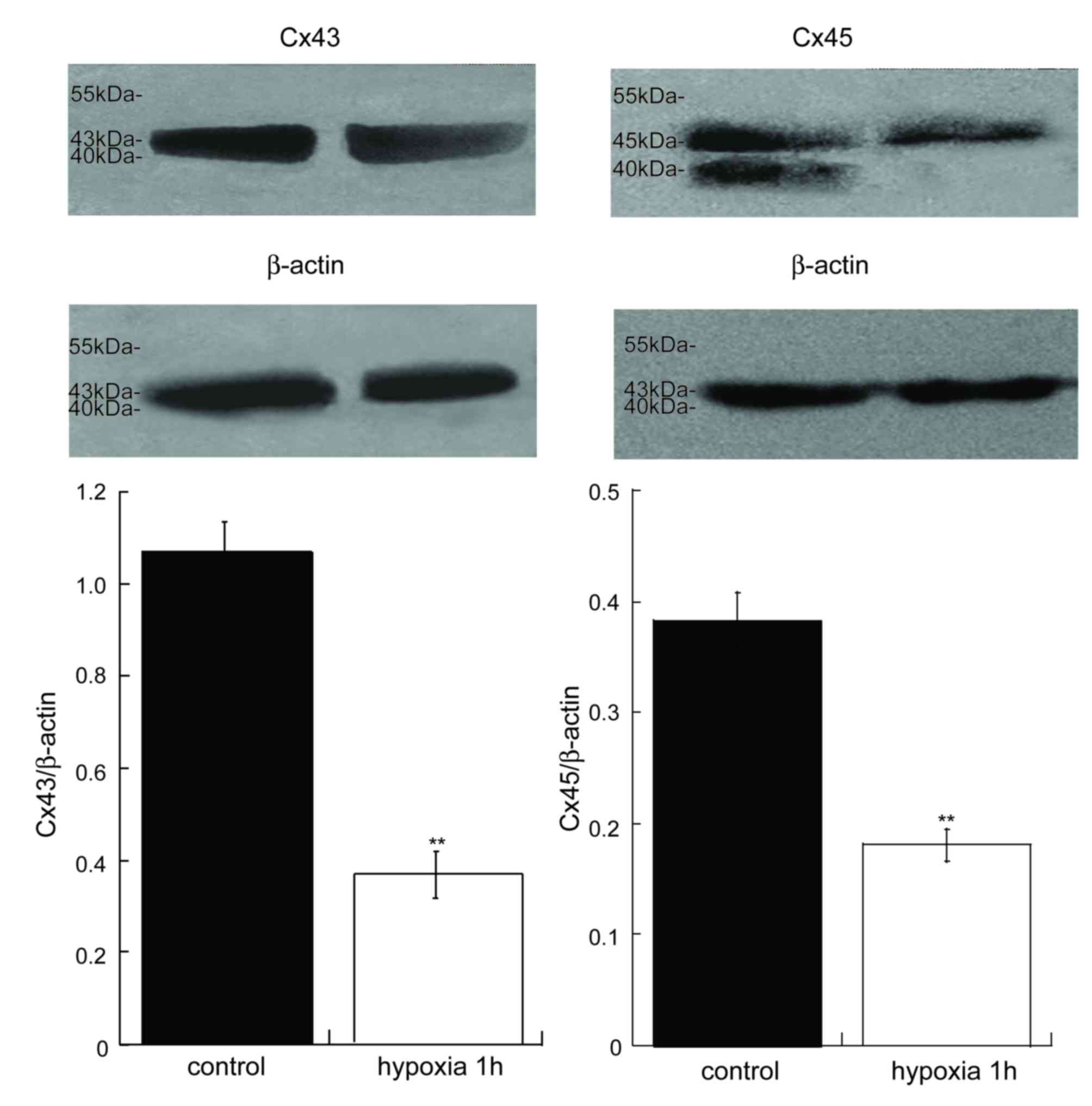
Inhibited vascular Cx43 and Cx45
expression in the MRA of acute hypoxia in vessels in vitro
It has been reported that the expression of Cx43 is
decreased in myocardial cells (25) and periodontalligament cells
(26). In the present study, the
protein expression of Cx43 and Cx45 in the MRAs of acute hypoxia 1
h was notable lower compared with control MRAs (Fig. 3). Therefore, acute hypoxia may
inhibit the expression of Cxs and the performance of GJ
communication, thus resulting in vasomotor dysfunction.
Discussion
The present study investigated the association
between changes in Cx43/45 expression and vasomotor function, and
Cx43/45-mediated GJIC under acute hypoxic conditions in SD rat
MRAs. The key finding of the present study is that the down
regulation of Cx43 and Cx45 expression had a positive correlation
with the reduction of GJIC and vasomotor tone during acute hypoxia,
which was reflected in the capacitance/conductance and the
frequency/amplitude of vasomotion in vitro,
respectively.
Several reports (27–30)
have demonstrated that GJ permeability in the cardiomyocytes and
astrocytes is reduced by hypoxia/ischemia. A 77% reduction and
90–95% reduction in GJIC was observed in astrocytes following 15
min and 30 min of hypoxia, respectively (29). A previous study identified that
acute hypoxia caused vascular hyperpolarization and vasodilation in
the guinea-pig anterior inferior cerebellar artery by increasing
outward current and decreasing the GJs of the VSMCs (31). The reduction in the number of GJs
or the reduction of conductance of each GJ may inhibit overall GJIC
and the two appear to be involved in hypoxia/ischemia-induced
inhibition of GJIC (14). In the
present study, whole-cell recordings from the VSMCs embedded in a
segment of the MRA were used to detect GJ communication in normoxia
and acute hypoxia vessels. This novel technique was practicable for
determining the function of GJs between the VSMCs in a more
physiologically relevant state (compared with dispersed VSMCs)
(32). The results indicated that
whole-cell recording from embedded VSMCs in arteriole segments is
an accessible approach that can be used to study various arteriolar
preparations electrophysiologically (33,34).
In Table I, the membrane
capacitance and conductance of the VSMCs of the MRAs in acute
hypoxia rats were significantly reduced compared with normoxia
rats, which is in agreement with previous studies in cardiomyocytes
and astrocytes, namely that hypoxia may suppress GJIC. The
reduction of the electrical coupling of GJs is often associated
with dephosphorylation and phosphorylation of Cxs (12,14,35).
In the normal cardiovascular system, the majority of the Cxs are
phosphorylated (15,17). Phosphorylation of Cxs serves
important functions in GJ assembly, channel gating and degradation.
There are certain phosphorylation sites where phosphorylated Cxs
may increase GJ assembly whereas others may inhibit formation of
GJs, or reduce the channel opening time (36). For example, acute hypoxia (5–40
min) can induce Cx43 dephosphorylation and reduce junctional
uncoupling in astrocytes and hearts (15,37),
while increasing Cx43-serine 368 phosphorylation status (16,38)
has been demonstrated to change the status of Cx43 channels from
open to closed (16). The change
of phosphorylation status may affect the number of GJs and
conductance of GJs (39,40). In support of this, a possible
explanation is that hypoxia suppresses electrical coupling of
adjacent VSMCs in MRA, perhaps through dephosphorylation or serine
368 phosphorylation of Cxs. The present study hypothesizes that the
phosphorylation state of Cx43 is altered by hypoxia and thus
affects channel opening. It is aimed that future studies will
investigate which specific phosphorylation sites of Cx43 are
affected by acute hypoxia in MRA using mass spectrometry. The
expression or localization of nonphosphorylated and phosphorylated
Cxs during acute hypoxia exposure by western blot analysis or
immunofluorescent staining will be analyzed as the subject of a
future study.
The phosphorylation state of Cxs affects their
half-lives. Previous studies have suggested that the
phosphorylation of specific serine sites on Cx43 and Cx45 result in
Cx degradation by complex mechanisms (41–43).
Phosphorylation of Cx43 on serine 255 by p34cdc2 kinase in Rat1
cells promotes the degradation of Cx43 (42). Thus, the phosphorylation state of
serine residues may also alter Cx stability and/or target the
protein for degradation (41). In
the current study, the expression of Cx43 and Cx45 were
significantly reduced by acute hypoxia for 1 h; this may result
from the alteration of the phosphorylation state of Cx43 and Cx45
promoting their degradation.
Oxygen deprivation during hypoxic exposure causes
the intracellular accumulation of toxic metabolic products and ATP
depletion, which can lead to cell injury or death (44); GJ and hemi-channels serve a role in
the communication of cell death signals between cells (45). Faigle et al (16) demonstrated that ATP release was
significantly attenuated (2% oxygen, 22±3% after 48 h) by the
selective repression of Cx43 transcription and time-dependent Cx43
total and surface protein repression during hypoxia. The GJs may
transmit death signals between injured and intact cells or lead to
ATP loss and cell death via the opening of GJ or Cx hemi-channels.
The uncoupling of GJ and the closing of the GJ channel during
hypoxia may promote cell survival and thus limit cell necrosis.
Electrical coupling of GJs in the VSMCs serves a
crucial role in the synchronization and coordination of vasomotor
tone (9). Ischemia or hypoxia can
cause systemic vascular system relaxation with the exception of the
pulmonary artery (6,46). Following exposure to hypoxic
conditions, the cardiomyocytes shorten to reduce contraction and
increase the resistance of the whole tissue (17,47).
Following these studies, the present study ascertained the effect
of acute hypoxia on the vasoconstriction of the MRAs and the role
of GJs in the regulation of vasomotor tone of MRAs during hypoxia.
In agreement with previous data, the PE-induced maximal
vasoconstrictive responses in MRAs were less sensitive in the acute
hypoxia compared with the normoxia group (Fig. 1A). As the experiments in the
current study were designed to study the role of GJs in the
regulation of vasomotor tone of MRAs during hypoxia, a
pharmacological procedure with GJ blocker 2-APB was used to inhibit
GJ. 2-APB is a membrane permeable modulator of myo-inositol
1,4,5-triphosphate (IP3) receptors and has been of
widespread recent use in the inhibition or activation of transient
receptor potential (TRP) channels and the blocking of GJ channels
(48). 2-APB was previously
reported to exert opposite effects on members of TRP channels,
inhibiting activity of TRP5 and TRP6 at 20 µM and activating TRP
cation channel subfamily V member (TRPV)-1, TRPV2 and TRPV3 at
higher concentrations (48,49).
Pan et al (50), in the
screening of GJ antagonists, identified that 2-APB was one of the
most effective antagonists, completely blocking A-type horizontal
cell coupling in rabbit retina. 2-APB at higher concentrations (100
µM) was used as a specific GJ channel blocker in critical control
experiments demonstrating peptide permeation through GJs (51) and at 10 µM to disrupt GJIC in the
vascular wall (52). Although
2-APB blocks slow Ca2+ waves in the process of
blockading the IP3 receptor, it should be noted that the
results above may be a result of GJ channel blocking. For example,
a previous study (53)
demonstrated the blockade of urinary bladder smooth muscle calcium
waves by 2-APB and other GJ blockers, however no intracellular
Ca2+ release effect was observed when using thapsigargin
and xestospongin. In addition, the results of a study by Li et
al (54) also demonstrated
that 2-APB may mimic the effects of inhibition of gap communication
by Gap 27 and lead to a significant inhibition of lucifer yellow
uptake and the attenuation of calcium transients in ventricular
myocytes. In the present study, the inhibitory effects of 2-APB on
PE-induced (1–100 µM) vasoconstriction were greater in normoxia
compared with acute hypoxia rats (Fig.
1B). It is therefore hypothesized that a blocking of gap
communication by2-APB may be expected as a consequence of GJ
channel blockade. The changes in contractile response in the MRA
were directly associated with the uncoupling of GJs and the
reduction in electrophysiological properties of the VSMCs in MRA
under hypoxic conditions and suggest that GJ intercellular
communication was inhibited by acute hypoxia and thus led to the
reduction of vasoconstriction in response to PE. The data from the
present study indicated that the inhibitory effect of 2-APB on
PE-induced vasoconstriction was markedly less apparent in acute
hypoxia rats compared with the normoxia control (Fig. 1B). These changes in contractile
response in MRAs were directly associated with the uncoupling of
GJs and the reduction in the electrophysiological properties of the
VSMCs in MRAs under hypoxic conditions.
These results, together with the reduction of
electrical coupling in MRAs, demonstrated that the regulation of GJ
coupling is involved in the vasodilatation improvement in response
to hypoxia conditions. Furthermore, previous studies have
demonstrated that GJs have a crucial role in coordinating the
synchronization of vasomotor tone by synchronizing the changes in
cytoplasmic Ca2+ between VSMCs (55). A plausible explanation for the
reduction of vasoconstriction in MRAs during the condition of acute
hypoxia is that inhibited GJs reduce intercellular Ca2+
wave propagation between the VSMCs of MRAs, which leads to the
response of vasomotion being less in acute hypoxia rats compared
with the normoxia controls.
Subsequent experiments indicated that decreased
vasoconstriction of MRA during acute hypoxic exposure was
associated with the reduction of Cx43 and Cx45 expression. In the
heart, the expression of Cx43 depends on the duration of hypoxia
(18). Wu et al (18) demonstrated that cultured atrial
cells in hypoxia for 6 and 12 h experienced a decrease in the Cx43
expression by ~30–50%. In the present study, exposure to acute
hypoxia for only 1 h cause a significant decrease in Cx43 and Cx45
expression. A reduction in Cx43 and Cx45 expression may lead to the
reduction of GJIC in VSMCs, thereby influencing the electrical
uncoupling of MRA in a low-oxygen environment. The role of Cx43
channels in hypoxia-induced cell injury or death has been studied
in several organs, including the heart and the brain (12,44).
Cx43 in cardiomyocyte mitochondria functions as a key regulator of
cell protection against hypoxia induced cell apoptosis (12). In previous study, Martins-Marques
et al (13) demonstrated
that ischemia results in Cx43 ubiquitin-dependent degradation
localized at the intercalated discs, which may be a novel
regulatory pathway in GJ remodeling associated with
hypoxia/ischemia injury. A similar mechanism of ubiquitin-dependent
degradation may occur in the present study and thus resulting in
the down regulation of Cx43 or Cx45 expression, although the
degradation of Cx43 and Cx45 during acute hypoxia exposure was not
assessed. In contrast with Cx43, the physiological functions of
Cx45 remain unclear, although its expression has been demonstrated
in vascular smooth muscle (56,57).
Whether Cx45 hemichannels and/or GJs are important for
hypoxia-mediated vascular dysfunction remains to be elucidated. The
majority of current studies have failed to establish the functional
associations between Cx45 and hypoxic exposure. In this context,
the data from the current study provides evidence that acute
hypoxia inhibited the level of Cx45, which may be responsible for
the coordinated role of vasomotor tone in mediating cell protection
exposure to hypoxia.
Together, the results reported from the present
study provide the first evidence that acute hypoxia negatively
regulates the vasomotor tone of the vascular smooth muscle layer of
the MRA, possibly through downregulation of Cx43 and Cx45
expression concurrently with the suppression of Cx43- and
Cx45-mediated intercellular communications. In summary, the present
study defines an important contribution of GJ in regulating
vasomotor function during limited oxygen availability, one that may
be essential for the adaptation of vasomotor tone in response to
acute hypoxia.
Acknowledgements
The current study was supported by the National
Natural Science Foundation of China (grant nos. 31260247 and
31460264 to Professor Ke-Tao Ma, grant no. 81460098 to Dr Xin-Zhi
Li and grant no. 81260159 to Dr LiLi).
References
|
1
|
Wanandi SI, Reni P and Syarifah D:
Relative expression of HIF-1α mRNA in rat heart, brain and blood
during induced systemic hypoxia. Makara Seri Sains. 13:185–188.
2009.
|
|
2
|
Lohman AW, Billaud M and Isakson BE:
Mechanisms of ATP release and signalling in the blood vessel wall.
Cardiovasc Res. 95:269–280. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ainslie PN, Ogoh S, Burgess K, Celi L,
McGrattan K, Peebles K, Murrell C, Subedi P and Burgess KR:
Differential effects of acute hypoxia and high altitude on cerebral
blood flow velocity and dynamic cerebral auto regulation:
alterations with hyperoxia. J Appl Physiol. 104:490–498. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tawaa M, Shimosato T, Geddawy A, Imamura T
and Okamura T: Influence of hypoxia on endothelium-derived
no-mediated relaxation in rat carotid, mesenteric and iliac
arteries. Pharmacology. 91:322–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhdanov GG and Sokolov IM: Tissue hypoxia
in acute myocardial infarction and possible approaches to its
correction. Anesteziol Reanimatol. 51–53. 2001.(In Russian).
PubMed/NCBI
|
|
6
|
Ariyaratnam P, Loubani M and Morice AH:
Hypoxic pulmonary vasoconstriction in humans. Biomed Res Int.
2013:6236842013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wan J, Yamamura A, Zimnicka AM, Voiriot G,
Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, et al:
Chronic hypoxia selectively enhances L- and T-type
voltage-dependent Ca2+ channel activity in pulmonary artery by
upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol.
305:L154–L164. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Shraim MM, Zafer MH and Rahman GA:
Acute occlusive mesenteric ischemia in high altitude of
southwestern region of Saudi Arabia. Ann Afr Med. 11:5–10. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Figueroa XF and Duling BR: Gap junctions
in the control of vascular function. Antioxid Redox Signal.
11:251–266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rummery NM and Hill CE: Vascular gap
junctions and implications for hypertension. Clin Exp Pharmacol
Physiol. 31:659–667. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang L, Yin J, Nickles HT, Ranke H,
Tabuchi A, Hoffmann J, Tabeling C, Barbosa-Sicard E, Chanson M,
Kwak BR, et al: Hypoxic pulmonary vasoconstriction requires
connexin 40-mediated endothelial signal conduction. J Clin Invest.
122:4218–4230. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Waza AA, Andrabi K and Hussain MU: Protein
kinase C (PKC) mediated interaction between conexin43 (Cx43) and
K(+) (ATP) channel subunit (Kir6.1) in cardiomyocyte mitochondria:
Implications in cytoprotection against hypoxia induced cell
apoptosis. Cell Signal. 26:1909–1917. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martins-Marques T, Catarino S, Marques C,
Matafome P, Ribeiro-Rodrigues T, Baptista R, Pereira P and Girão H:
Heart ischemia results in connexin43 ubiquitination localized at
the intercalated discs. Biochimie. 112:196–201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miura T, Miki T and Yano T: Role of the
gap junction in ischemic preconditioning in the heart. Am J Physiol
Heart Circ Physiol. 298:H1115–H1125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Turner MS, Haywood GA, Andreka P, You L,
Martin PE, Evans WH, Webster KA and Bishopric NH: Reversible
connexin 43 dephosphorylation during hypoxia and reoxygenation is
linked to cellular ATP levels. Circ Res. 95:726–733. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Faigle M, Seessle J, Zug S, El Kasmi KC
and Eltzschig HK: ATP release from vascular endothelia occurs
across Cx43 hemichannels and is attenuated during hypoxia. PLoS
One. 3:e28012008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsushita S, Kurihara H, Watanabe M,
Okada T, Sakai T and Amano A: Alterations of phosphorylation state
of connexin 43 during hypoxia and reoxygenation are associated with
cardiac function. J Histochem Cytochem. 54:343–353. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu X, Huang W, Luo G and Alain LA: Hypoxia
induces connexin 43 dysregulation by modulating matrix
metalloproteinases via MAPK signaling. Mol Cell Biochem.
384:155–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nuñez C, Victor VM, Martí M and D'Ocon P:
Role of endothelial nitric oxide in pulmonary and systemic arteries
during hypoxia. Nitric Oxide. 15:17–27. 2014. View Article : Google Scholar
|
|
20
|
Ibe JC, Zhou Q, Chen T, Tang H, Yuan JX,
Raj JU and Zhou G: Adenosine monophosphate-activated protein kinase
is required for pulmonary artery smooth muscle cell survival and
the development of hypoxic pulmonary hypertension. Am J Respir Cell
Mol Biol. 49:609–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee YH, Seo JH and Kang BS: Effects of
hypoxia on pulmonary vascular contractility. Yonsei Med J.
39:261–267. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma KT, Li XZ, Li L, Jiang XW, Chen XY, Liu
WD, Zhao L, Zhang ZS and Si JQ: Role of gap junctions in the
contractile response to agonists in the mesenteric artery of
spontaneously hypertensive rats. Hypertens Res. 37:110–115. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma KT, Guan BC, Yang YQ, Nuttall AL and
Jiang ZG: 2-Aminoethoxydiphenyl borate blocks electrical coupling
and inhibits voltage-gated K+ channels in guinea pig arteriole
cells. Am J Physiol Heart Circ Physiol. 300:H335–H346. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li L, Wang R, Ma KT, Li XZ, Zhang CL, Liu
WD, Zhao L and Si JQ: Differential effect of calcium-activated
potassium and chloride channels on rat basilar artery vasomotion. J
Huazhong Univ Sci Technolog Med Sci. 34:482–490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang R, Zhang C, Ruan Y, Liu N and Wang L:
Change in phosphorylation of connexin43 during acute hypoxia and
effects of antiarrhythmic peptide on the phosphorylation. J
Huazhong Univ Sci Technolog Med Sci. 27:241–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kato R, Ishihara Y, Kawanabe N, Sumiyoshi
K, Yoshikawa Y, Nakamura M, Imai Y, Yanagita T, Fukushima H,
Kamioka H, et al: Gap-junction-mediated communication in human
periodontal ligament cells. J Dent Res. 92:635–640. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Naitoh K, Yano T, Miura T, Itoh T, Miki T,
Tanno M, Sato T, Hotta H, Terashima Y and Shimamoto K: Roles of
Cx43-associated protein kinases in suppression of gap
junction-mediated chemical coupling by ischemic preconditioning. Am
J Physiol Heart Circ Physiol. 296:H396–H403. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sato M, Jiao Q, Honda T, Kurotani R,
Toyota E, Okumura S, Takeya T, Minamisawa S, Lanier SM and Ishikawa
Y: Activator of G protein signaling 8 (AGS8) is required for
hypoxia-induced apoptosis of cardiomyocytes: Role of G betagamma
and connexin 43 (Cx43). J Biol Chem. 284:31431–31440. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li WE and Nagy JI: Connexin43
phosphorylation state and intercellular communication in cultured
astrocytes following hypoxia and protein phosphatase inhibition.
Eur J Neurosci. 12:2644–2650. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li W, Hertzberg EL and Spray DC:
Regulation of connexin43-protein binding in astrocytes in response
to chemical ischemia/hypoxia. J Biol Chem. 280:7941–7948. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li XZ, Si JQ, Zhang ZS, Zhao L, Li L and
Ma KT: Acute hypoxia increases outward current and decreases gap
junction of VSMCs in guinea-pig anterior inferior cerebellar
artery. Sheng Li Xue Bao. 63:533–539. 2011.(In Chinese). PubMed/NCBI
|
|
32
|
Guan BC, Si JQ and Jiang ZG: Blockade of
gap junction coupling by glycyrrhetinic acids in guinea pig
cochlear artery: A whole-cell voltage- and current-clamp study. Br
J Pharmacol. 151:1049–1060. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Quinn K and Beech DJ: A method for direct
patch-clamp recording from smooth muscle cells embedded in
functional brain microvessels. Pflugers Arch. 435:564–569. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamamoto Y, Fukuta H, Nakahira Y and
Suzuki H: Blockade by 18beta-glycyrrhetinic acid of intercellular
electrical coupling in guinea-pig arterioles. J Physiol.
511:501–508. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Beardslee MA, Lerner DL, Tadros PN, Laing
JG, Beyer EC, Yamada KA, Kléber AG, Schuessler RB and Saffitz JE:
Dephosphorylation and intracellular redistribution of ventricular
connexin43 during electrical uncoupling induced by ischemia. Circ
Res. 87:656–662. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Solan JL and Lampe PD: Connexin43
phosphorylation: Structural changes and biological effects. Biochem
J. 419:261–272. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Le HT, Sin WC, Lozinsky S, Bechberger J,
Vega JL, Guo XQ, Sáez JC and Naus CC: Gap junction intercellular
communication mediated by connexin43 in astrocytes is essential for
their resistance to oxidative stress. J Biol Chem. 289:1345–1354.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Srisakuldee W, Makazan Z, Nickel BE, Zhang
F, Thliveris JA, Pasumarthi KB and Kardami E: The FGF-2-triggered
protection of cardiac subsarcolemmal mitochondria from calcium
overload is mitochondrial connexin 43-dependent. Cardiovasc Res.
103:72–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shimizu K and Stopfer M: Gap junctions.
Curr Biol. 23:R1026–R1031. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Goodenough DA and Paul DL: Gap Junctions.
Cold Spring Harb Perspect Biol. 1:a0025762009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saffitz JE, Laing JG and Yamada KA:
Connexin expression and turnover: Implications for cardiac
excitability. Circ Res. 86:723–728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lampe PD, Kurata WE, Warn-Cramer BJ and
Lau AF: Formation of a distinct connexin43 phosphoisoform in
mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci.
111:833–841. 1998.PubMed/NCBI
|
|
43
|
Hertlein B, Butterweck A, Haubrich S,
Willecke K and Traub O: Phosphorylated carboxy terminal serine
residues stabilize the mouse gap junction protein connexin45
against degradation. J Membr Biol. 162:247–257. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang X, Ma A, Zhu W, Zhu L, Zhao Y, Xi J,
Zhang X and Zhao B: The role of connexin 43 and hemichannels
correlated with the astrocytic death following ischemia/reperfusion
insult. Cell Mol Neurobiol. 33:401–410. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Krysko DV, Leybaert L, Vandenabeele P and
D'Herde K: Gap junctions and the propagation of cell survival and
cell death signals. Apoptosis. 10:459–469. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Madden JA, Vadula MS and Kurup VP: Effects
of hypoxia and other vasoactive agents on pulmonary and cerebral
artery smooth muscle cells. Am J Physiol. 263:L384–L393.
1992.PubMed/NCBI
|
|
47
|
Jain SK, Schuessler RB and Saffitz JE:
Mechanisms of delayed electrical uncoupling induced by ischemic
preconditioning. Circ Res. 92:1138–1144. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kovacs G, Montalbetti N, Simonin A, Danko
T, Balazs B, Zsembery A and Hediger MA: Inhibition of the human
epithelial calcium channel TRPV6 by 2-aminoethoxydiphenyl borate
(2-APB). Cell Calcium. 52:468–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Colton CK and Zhu MX:
2-Aminoethoxydiphenyl borate as a common activator of TRPV1, TRPV2,
and TRPV3 channels. Handb Exp Pharmacol. 173–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pan F, Mills SL and Massey SC: Screening
of gap junction antagonists on dye coupling in the rabbit retina.
Vis Neurosci. 24:609–618. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Neijssen J, Herberts C, Drijfhout JW,
Reits E, Janssen L and Neefjes J: Cross-presentation by
intercellular peptide transfer through gap junctions. Nature.
434:83–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Griffith TM, Chaytor AT, Bakker LM and
Edwards DH: 5-Methyltetrahydrofolate and tetrahydrobiopterin can
modulate electrotonically mediated endothelium-dependent vascular
relaxation. Proc Natl Acad Sci USA. 102:7008–7013. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hashitani H, Yanai Y and Suzuki H: Role of
interstitial cells and gap junctions in the transmission of
spontaneous Ca2+ signals in detrusor smooth muscles of the
guinea-pig urinary bladder. J Physiol. 559:567–581. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li C, Meng Q, Yu X, Jing X, Xu P and Luo
D: Regulatory effect of connexin 43 on basal Ca2+ signaling in rat
ventricular myocytes. PLoS One. 7:e361652012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Halidi N, Alonso F, Burt JM, Bény JL,
Haefliger JA and Meister JJ: Intercellular calcium waves in primary
cultured rat mesenteric smooth muscle cells are mediated by
Connexin43. Cell Commun Adhes. 19:25–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kurtz A: Renal connexins and blood
pressure. Biochim Biophys Acta. 1818:1903–1908. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schmidt VJ, Jobs A, von Maltzahn J,
Wörsdörfer P, Willecke K and de Wit C: Connexin45 is expressed in
vascular smooth muscle but its function remains elusive. PLos One.
7:e422872012. View Article : Google Scholar : PubMed/NCBI
|