Introduction
Leukaemia is a malignant and proliferative disease
originating from multipotent haemopoietic stem cells. Although
treatment of leukaemia has greatly improved over the past decades,
conventional combination chemotherapy remains ineffective for
numerous patients. Most therapeutic failures are attributed to
cellular resistance to anti-leukemic therapy. Various factors
contribute to drug resistance, including alteration in drug
transport, dysregulation of DNA replication and repair, and
impaired apoptosis (1).
Recently, abnormal glycometabolism of cancer cells
has become of focus. Previous studies supported the idea that
enhanced glycolysis is associated with decreased sensitivity to
various forms of tumour therapy; glycolytic cancers have been
demonstrated to be highly refractory to chemo- and radiotherapies
(2–4). Over 90 years ago, Warburg observed
that cancer cells exhibited increased glycolysis despite the
presence of ample oxygen, which was termed the ‘Warburg effect’, or
aerobic glycolysis (5). Aerobic
glycolysis inhibition has been demonstrated to increase drug
sensitivity in certain cancer cells. Key proteins in the glycolytic
pathway have been thoroughly investigated, including glucose
transporters (GLUTs), HK, pyruvate kinase (PK) and -LDH (6–9).
However, the effects of glycolytic metabolism on chemo-agent
sensitivity, and the causal association between increased
glycolytic activity and decreased sensitivity to anticancer agents
in refractory tumours and leukemias, remains to be fully
elucidated. It remains unknown whether targeting cancer cell energy
supply via the glycolytic pathway may serve as a potential
therapeutic strategy to overcome MDR. How glycolytic metabolism
affects cell processes, particularly anticancer agent efficiency,
is not fully understood. The present study systematically
investigated the glycolysis-metabolic status, and the association
between drug sensitivity and aerobic glycolysis in sensitive and
MDR leukaemia cells in normoxic conditions. Increased aerobic
glycolysis was demonstrated to be present in leukaemia MDR cells,
and inhibition of glycolysis potently sensitises MDR cells to the
anticancer agent adriamycin (ADM), accompanied by overactivation of
the AKT serine/threonine kinase (AKT)-mechanistic target of
rapamycin (mTOR)-c-Myc pathway.
Materials and methods
Reagents and antibodies
The reagents used in this study included oxamate
(Ox; Alfa-Aesar, Haverhill, MA, USA), 2-deoxyglucose (2-DG; Yuanye
Bio-Technology Co., Ltd., Shanghai, China), ADM (Wanle
Bio-Technology Co., Ltd., Hangzhou, China), glucose, lactate, HK,
PK, and LDH assay kits (Jiancheng Bioengineering Institute,
Nanjing, China) and a phosphofructokinase (PFK) assay kit (Kemin
Industries Co., Ltd., Zhuhai, China). The following primary
antibodies were used: Mouse polyclonal anti-β-actin (Zhongshan
Jinqiao Bio-Technology Co., Ltd., Beijing, China), rabbit
polyclonal anti-GLUT4, anti-HK-II, anti-phosphorylated (p)-AKT
(Thr308), anti-mTOR, anti-p-mTOR (Ser2448) (ImmunoWay
Biotechnology, Plano, TX USA); anti-AKT, anti-p-AKT (Ser473),
anti-c-Myc (Cell Signaling Technology, Inc., Danvers, MA, USA), and
rabbit polyclonal anti-LDHA (Hangzhou HuaAn Biotechnology Co.,
Ltd., Hangzhou, China).
Cell culture
The K562 human leukaemia cell line was purchased
from the American Type Culture Collection (Manassas, VA, USA), and
the K562/ADM ADM-resistant cell line was obtained from the Shanghai
Jiaotong University School of Medicine (Shanghai, China). The cells
were maintained in RPMI-1640 media (HyClone; GE Healthcare Life
Sciences, Inc.) supplemented with 10% foetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.) and cultivated at 37°C in a 5%
CO2 incubator. K562/ADM cells were stimulated with 5
mg/l ADM every 45 days to maintain increased drug resistance, and
then were used after cultured 2 weeks without ADM.
In vitro cytotoxicity assay
For the cytotoxicity assay, a density 1×105 cells/ml
were plated into 96-well plates. Cells were subsequently incubated
with various concentrations of Ox (0, 1, 2.5, 5, 10 and 20 mM) or
2-DG (0, 0.05, 0.25, 0.5, 1 and 2 mM) and ADM (0.01 mg/l for K562
cells, 0.8 mg/l for K562/ADM cells) for 48 h at 37°C. A total of 10
µl MTT solution was then added to each well, followed by incubation
for 4 h at 37°C. 10% SDS was added to each of the wells, and
incubated overnight at 37°C to dissolve the formazan crystals.
Finally, the absorbance values of each well at 570 nm were
quantified using a PowerWave X Plate Reader (BioTek Instruments,
Inc., Winooski, VT, USA). Each dose of the compound was tested in
quadruplicate.
Measurement of glucose concentration
and lactate production
Cells were seeded at a density of 1×105 cells/ml.
Culture media was collected at 48 h after treatment with various
concentrations of Ox (0, 2.5 and 10 mM) or 2-DG (0, 0.5 and 2 mM)
at 37°C and stored at −80°C until assayed. The glucose and lactate
assay kits (Jiancheng Bioengineering Institute, Nanjing, China)
were used to determine the concentrations of glucose and lactate in
the culture media. Experiments were performed in triplicate.
Quantification of enzymatic
activity
Cells were seeded at a density of 1×105 cells/ml and
incubated for 48 h at 37°C and were centrifuged at 1,000 × g for 6
min at room temperature and the supernatant was collected. Then the
supernatant was washed with PBS and lysed in ice-cold WIP tissue
and cell lysis solution (Beijing Cellchip Biotechnology Co., Ltd.
Beijing, China) for 5 min. The cell lysates were centrifuged at
12,000 × g for 15 min at 4°C to collect the supernatant. Cell
lysate was stored at −80°C until assayed. HK, PFK, PK and LDH
activity in cell lysates was determined using HK, PK, and LDH assay
kits (Jiancheng Bioengineering Institute, Nanjing, China) and PFK
assay kit (Kemin Industries Co., Ltd., Zhuhai, China). The protein
concentrations in the cell lysates were quantified using a
bicinchoninic acid (BCA) protein assay kit (Beyotime Biotechnology
Co., Ltd., Shanghai, China). Enzymatic activity was normalized to
the quantity of total protein. Experiments were performed in
triplicate.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from the cells was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). cDNA was derived using the total RNA as a
template by a PrimeScript RT reagent kit (Perfect Real Time)
obtained from Takara Bio, Inc. (Otsu, Japan) according to the
manufacturer's protocol. qPCR was conducted using the SYBR Premix
Ex Taq™ II (Tli RNaseH Plus) kit (Takara Bio, Inc.). Gene
expression levels were analysed using a Rotor-Gene 3000 qPCR
amplifier (Corbett Co., Ltd., Australia). The thermocycling
conditions were as follows: 10 sec at 95°C; followed by 40 cycles
of 95°C for 5 sec, 60°C for 30 sec. All samples were analyzed using
β-actin gene expression as an internal control. The relative mRNA
level of GLUT4, LDHA and hypoxia-inducible factor-1α (HIF-1α) was
determined by the 2-ΔΔCq method (10). The primers used for qPCR (Table I) were designed and synthesized by
Takara Biotechnology Co., Ltd. (Dalian, China).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Primer sequence |
|---|
| GLUT4 | F:
GCTGCGAATAAACAGGCAGGA |
|
| R:
CAGCACAGCAGTGATGACAGTGA |
| LDHA | F:
CGTGCATTCCCGATTCCT |
|
| R:
CAACAGCACCAACCCCAAC |
| HIF-1α | F:
TTGCTCATCAGTTGCCACTTCC |
|
| R:
AGCAATTCATCTGTGCTTTCATGTC |
| β-actin | F:
TGGCACCCAGCACAATGAA |
|
| R:
CTAAGTCATAGTCCGCCTAGAAGCA |
Western blotting
Treated cells were washed with PBS and lysed in
ice-cold RIPA lysis buffer (Beijing Solarbio Science &
Technology Co. Ltd., Beijing, China). The cell lysates were
centrifuged at 12,000 × g for 15 min at 4°C to collect the
supernatant. A bicinchoninic acid (BCA) protein assay kit was used
to determine the protein concentration. The proteins (30 µg) were
separated using 10% SDS-PAGE and then transferred onto a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). The membranes were blocked using 5% non-fat milk for 1 h,
followed by overnight incubation with primary antibodies at 4°C.
The primary antibodies used were as follows: β-actin (1:700; cat.
no. sc-1616-R), GLUT4 (1:1,000; cat. no. YT1930), HK-II (1:4,000;
cat. no. YM0350), LDHA (1:1,000; cat. no. 1007-2), AKT (1:1,000;
cat. no. 4691), p-AKT (Ser473) (1:1,000; cat. no. 4060), p-AKT
(Thr308) (1:500; cat. no. YP0007), mTOR (1:1,000; cat. no. YT2915),
p-mTOR (Ser2448) (1:1,000; cat. no. YP0176) and c-Myc (1:500; cat.
no. 13987). Subsequently, the membranes were washed with Tween-20
and PBS and then incubated again for 1 h at room temperature with
horseradish peroxidase conjugated goat anti-rabbit (cat. no.
ZB-2301) or goat anti-mouse (cat. no. ZB-2305) secondary antibody
(1:10,000; Zhongshan Jinqiao Bio-Technology Co., Ltd.). Protein
bands were visualized using enhanced chemiluminescence reagents
(EMD Millipore). β-actin served as an internal control. Band
intensities were determined using ImageJ software version 1.45S
(National Institutes of Health, Bethesda, MD, USA; imagej.nih.gov/ij/).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Two-tailed Student's t-test was used to assess the difference
between two groups. One way analysis of variance followed by
Dunnett's multiple comparisons test was used to determine
differences between groups. P<0.05 was considered to indicate a
statistically significant difference. Statistical analysis was
performed using SPSS software version 13.0 (SPSS, Inc., Chicago,
IL, USA).
Results
K562/ADM leukaemia cells exhibit
increased aerobic glycolytic activity
Our previous study demonstrated that K562/ADM cells
acquired MDR, which was induced by ADM treatment of the K562
parental sensitive cell line and associated with expression levels
of P-glycoprotein (P-gp) (11). In
the present study, the K562 and K562/ADM leukaemic cell lines were
used to assess the association between glycolytic activity and MDR.
Metabolic flux in the two cell lines was assessed by glucose
consumption and lactate export. As presented in Fig. 1A, glucose consumption in K562/ADM
cells was increased compared with the sensitive control cells
(P<0.05). A similar trend was observed in lactate export
(Fig. 1B). The lactate export of
K562/ADM cells was increased by 1.34-fold compared with K562 cells
(P<0.001). Increases in glucose consumption and lactate
accumulation indicated that the glycolytic pathway is highly active
in ADM-resistant cells.
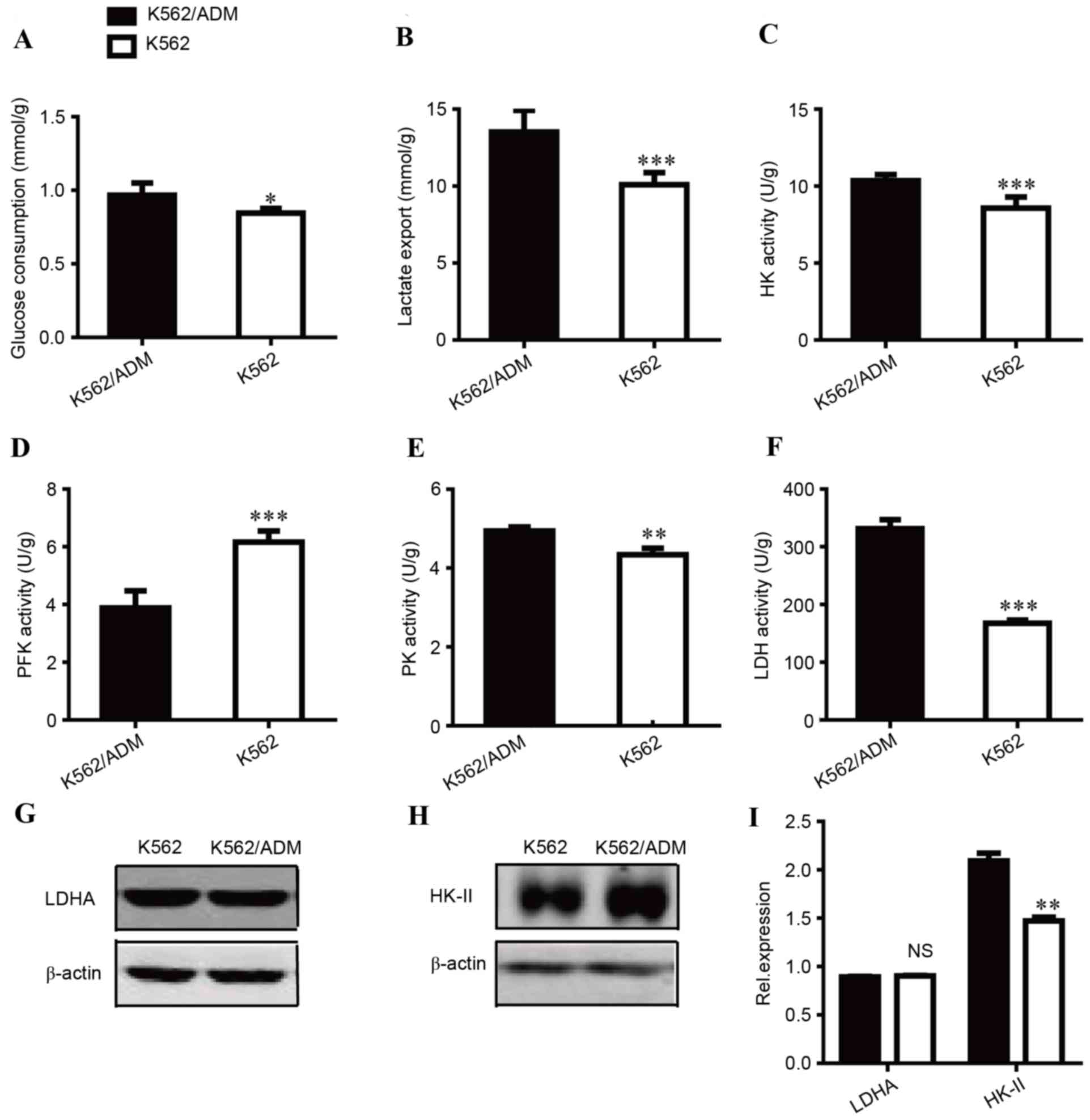 | Figure 1.Comparison of the glycolytic status
between K562 and K562/ADM cells. (A) Glucose consumption, (B)
lactate export, (C) HK activity, (D) PFK activity, (E) PK activity
and (F) LDH activity were measured. Western blot images of (G) LDHA
and (H) HK-II protein expression levels. (I) Quantification of LDHA
and HK-II relative protein expression levels. Data are presented as
the mean ± standard deviation (n=3). *P<0.05, **P<0.01, and
***P<0.001. NS, not significant; ADM, adriamycin; HK,
hexokinase; PFK, phosphofructokinase; PK, pyruvate kinase; LDHA,
lactate dehydrogenase A. |
To investigate the mechanisms underlying these
differences, the enzyme activities of HK (Fig. 1C), PFK (Fig. 1D), PK (Fig. 1E) and LDH (Fig. 1F) were compared between K562 and
K562/ADM due to their key roles in glycolysis. ADM-resistant
K562/ADM cells had increased HK, PK, and LDH activity compared with
their treatment-sensitive counterparts, which may contribute to
increased glycolysis. Notably, LDH activity was markedly increased;
~2-fold greater LDH activity was observed in K562/ADM cells
compared with treatment-sensitive controls, which is consistent
with its increased lactate export. PK activity was slightly
increased, and the increase in HK activity was moderate. In
addition, as presented in Fig. 1D,
the activity levels of PFK, a critical driver of glycolytic flux,
were decreased in K562/ADM cells, which supported the increased
proliferation of K562 cells (data not shown). Thus, LDH and HK were
regarded as two important targets for subsequent experiments.
The expression levels of key enzymes are
additionally responsible for metabolic alterations; previous
research has suggested that LDH and HK are associated with tumour
drug resistance (4,12). Therefore, the expression levels of
LDHA (Fig. 1G) and HK-II (Fig. 1H) were measured, which are isoforms
of LDH and HK, respectively. No significant differences in LDHA
expression levels were identified between K562 and K562/ADM cells;
however, HK-II expression levels were significantly increased in
K562/ADM cells compared with K562 cells (P<0.01). These data
implied that K562/ADM cells have increased glycolysis compared with
K562 cells, primarily due to increased HK-II expression levels
and/or HK and LDH activity.
K562/ADM leukaemia cells have
increased sensitivity to glycolytic inhibitors
MTT assays were performed to investigate the effects
of Ox and 2-DG on cell viability in the two cell lines. Ox is an
established inhibitor of LDHA with a potent inhibitory effect on
glycolysis. LDHA catalyses the conversion of pyruvate to lactate,
the last step in the glycolytic pathway. This is a key step that
influences the quantity of pyruvates that enter glycolysis.
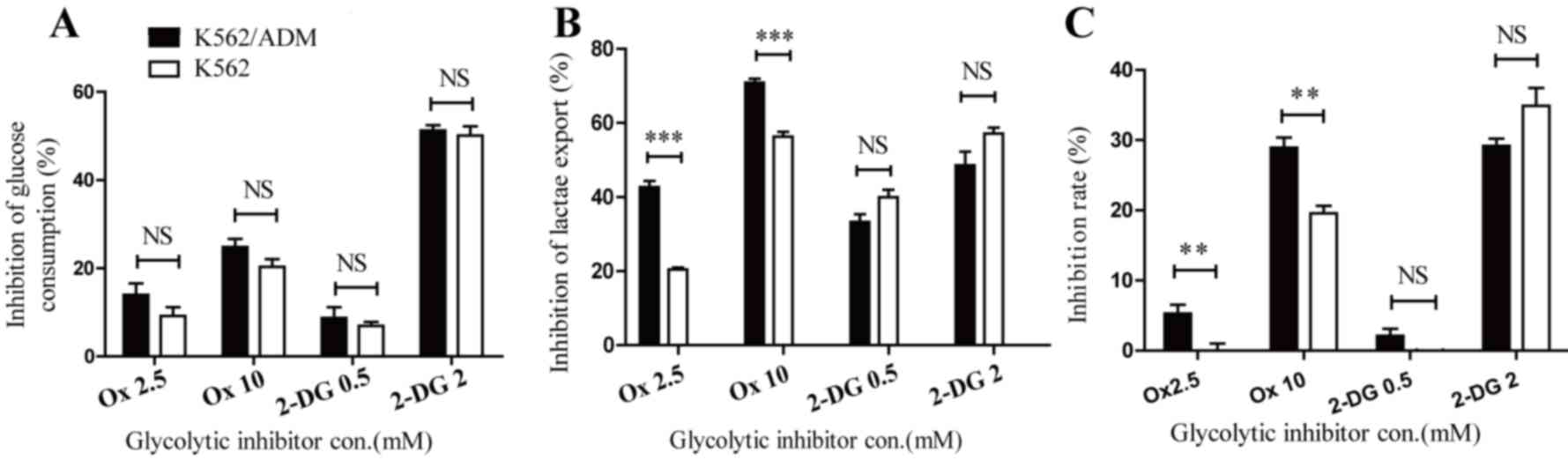
Incubation of K562 and K562/ADM with Ox caused glucose consumption
and lactate export to decrease, confirming its ability to block
energy metabolism. No significant differences in glucose
consumption inhibition were observed between K562 and K562/ADM
cells (Fig. 2A). However, lactate
export inhibition was increased in K562/ADM cells compared with
K562 cells following the administration of Ox (P<0.001; Fig. 2B). Furthermore, as presented in
Fig. 2C, dose-dependent
cytotoxicity was revealed in the two cell lines, with ADM-resistant
cells exhibiting increased sensitivity to the LDHA inhibitor
compared with control cells. The inhibition rates of Ox at 2.5 and
10 mM were 0±1.93 and 19.60±2.09% (for K562 cells) and 5.25±2.52
and 28.91±2.97% (for K562/ADM cells), respectively. The increase in
lactate export inhibition in K562/ADM cells following treatment
with Ox paralleled its increased inhibition ratio, which revealed a
significant association between drug resistance and enhanced
lactate accumulation. The glycolytic inhibitor 2-DG was
additionally used in this study, which is a compound known to
inhibit the first phase of glycolysis catalysed by HK. Following
treatment with 2-DG, glycolytic flux was additionally negatively
affected in both cell lines. Notably, glucose consumption (Fig. 2A) and lactate export inhibition
(Fig. 2B) were similar between
K562 and K562/ADM cells, and no significant differences were
observed in cytotoxicity levels between the two cell lines
(Fig. 2C), in contrast with the
results with Ox treatment. The inhibition of 2-DG 0.5 mM to K562
cells was undetectable in the present study. These data provided
further evidence to confirm the important role of lactic acid
accumulation in drug-resistant leukaemia cells.
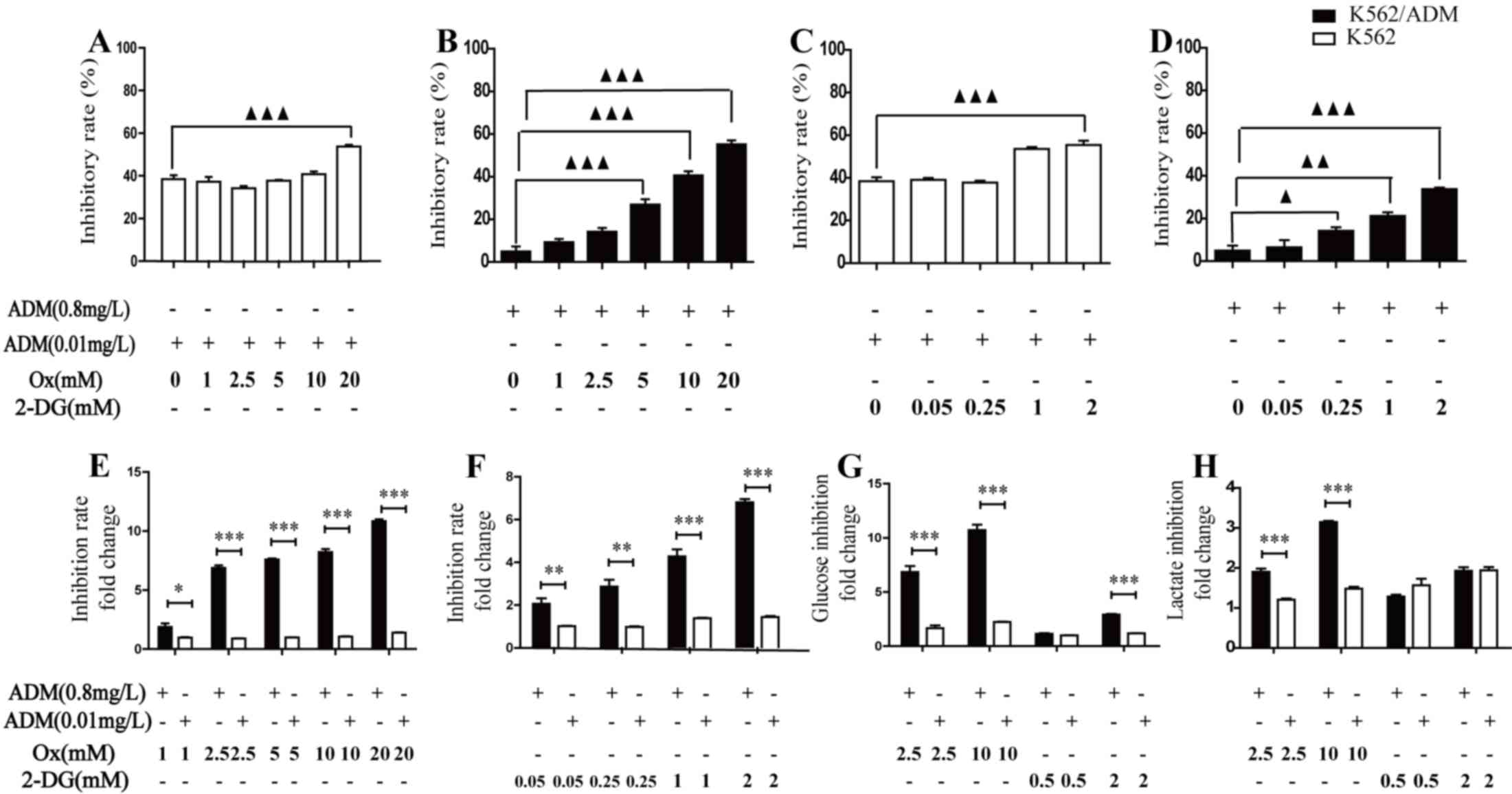
Inhibition of glycolysis effectively
restores the sensitivity of K562/ADM cells to ADM treatment
The above observations suggested a novel strategy
for effectively killing cancer cells and overcoming drug
resistance. One approach is to inhibit glycolysis and thus decrease
lactic acid production during chemotherapy. The cytotoxicity of ADM
was markedly increased following treatment with Ox in K562
(Fig. 3A) and K562/ADM (Fig. 3B) cells, and 2-DG in K562 (Fig. 3C) and K562/ADM (Fig. 3D) cells. In K562/ADM cells, this
inhibition efficacy was significant when Ox and 2-DG concentrations
were >5 mM or >0.25 mM, respectively. In K562 cells,
significance was observed at 20 mM or 2 mM for Ox and 2-DG,
respectively. Following treatment with Ox (Fig. 3E) and 2-DG (Fig. 3F), increased inhibition rates were
observed in K562/ADM cells compared with K562 cells. These data
indicated that while drug-resistant K562/ADM cells were less
sensitive to chemotherapy compared with K562 cells, they
demonstrated increased chemotherapeutic efficacy when combined with
increasing concentrations of a glycolytic inhibitor. To further
confirm the glycolysis inhibition effect and understand alterations
in energy metabolism following combination treatment, glucose
consumption and lactate production as glycolysis biochemical
indicators were measured in the two cell lines. Following treatment
with ADM plus the glycolysis inhibitor for 48 h, glucose
consumption and lactate production were revealed to be decreased in
the two cell types, compared with ADM treatment alone. In
accordance with the varying effect on cell viability of combination
treatment of K562 and K562/ADM cells, drug-resistant cells
exhibited increased glycolysis inhibition efficacy compared with
drug-sensitive cells (Fig. 3G and
H). Following treatment with 10 mM Ox or 2 mM 2-DG, the glucose
consumption was decreased by 10.71±1.01 (Ox) and 2.91±0.13 (2-DG)
fold change for K562/ADM cells, and 2.23±0.04 (Ox) and 1.21±0.05
(2-DG) fold change for K562 cells; the lactate production was
decreased by 3.15±0.06 (Ox) and 1.93±0.18 (2-DG) fold change for
K562/ADM cells, 1.48±0.09 (Ox) and 1.94±0.17 (2-DG) fold change for
K562 cells, respectively, relative to ADM treated alone.
These results demonstrated that inhibition of
glycolysis caused glucose consumption and lactate production to
decrease, leading to increased sensitivity to chemotherapy in
leukaemia cells. Notably, the results additionally revealed that
inhibition of glycolysis may condition K562/ADM cells to respond
more efficiently to chemotherapy compared with K562 cells.
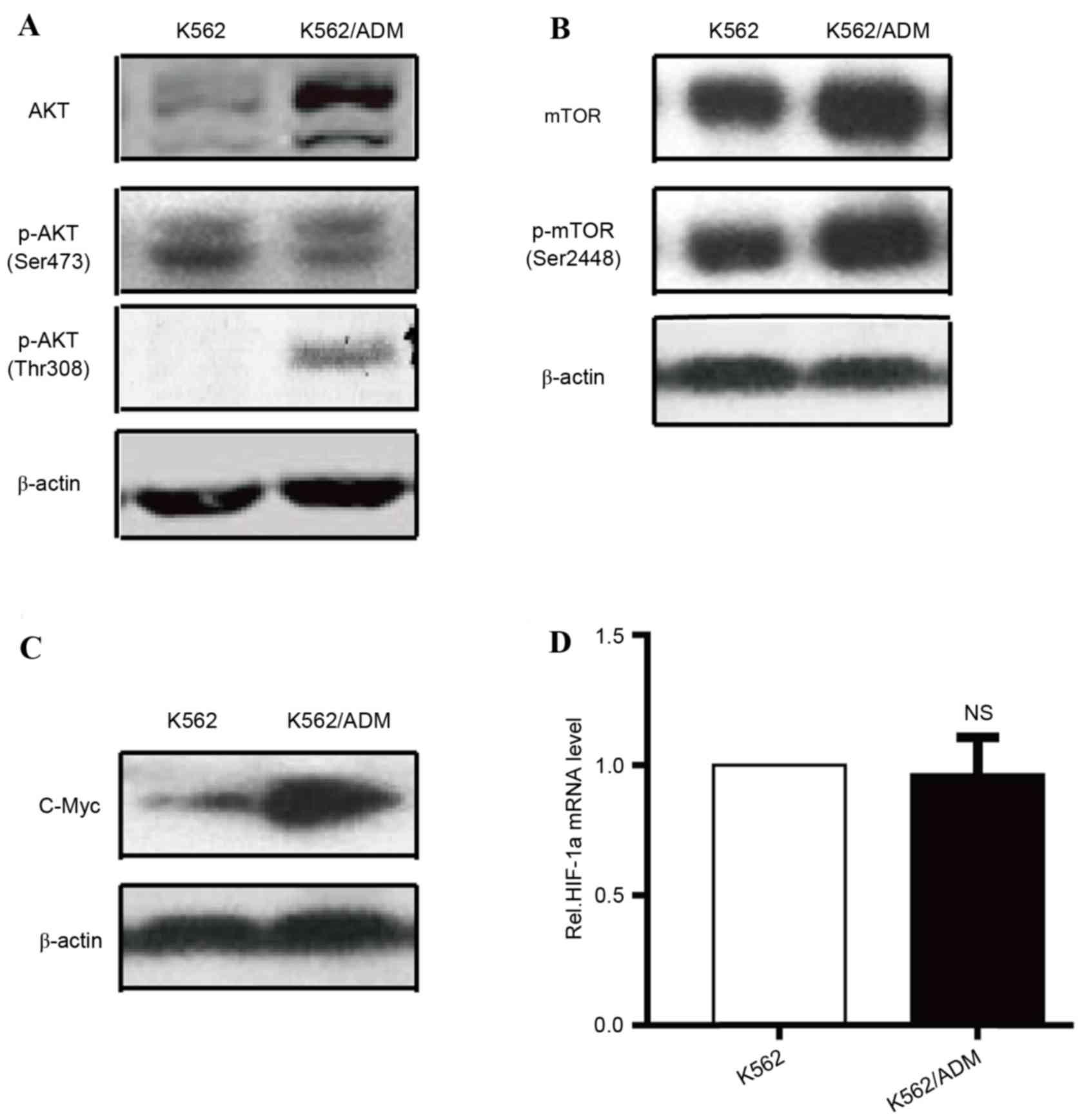
AKT-mTOR pathway over-activation and
increased glycolysis are observed in K562/ADM cells
Previous studies indicated that certain factors are
associated with the metabolic dysregulation of cancer cells,
including c-Myc, the phosphatidylinositol-4,5-bisphosphate/AKT-mTOR
signalling pathway, HIF-1α, AMP-activated protein kinase and p53
(5,13–17).
The AKT/mTOR signalling pathway appears to be a crucial controller
of metabolic homeostasis via regulation of the expression or
translocation of metabolic genes associated with glycometabolism,
including LDHA, HK-II and GLUTs (18). As dysfunction in these genes has
previously been associated with an enhanced Warburg effect in
K562/ADM cells, the present study compared the expression levels of
AKT, p-AKT (Fig. 4A), mTOR, p-mTOR
(Fig. 4B), HIF-1α, and c-Myc
(Fig. 4C) in the K562/ADM and K562
cell lines. AKT, p-AKT (Thr308), mTOR, p-mTOR (Ser2448), and c-Myc
protein expression levels were upregulated in K562/ADM cells
compared with parental controls, whereas no significant differences
were observed in HIF-1α mRNA (Fig.
4D) and p-AKT (Ser473) protein expression levels between the
two cell types. Previous research has indicated that the
AKT-mTOR-c-Myc signalling pathway has multiple roles in stimulating
glucose consumption and metabolism by regulating GLUTs, HK-II and
LDHA (19). Therefore, the present
study investigated increased glycolytic activity in K562/ADM cells,
which may be in part caused by increased key enzyme activity and/or
overexpression, which was primarily attributable to over-activation
of the AKT-mTOR-c-Myc signalling pathway.
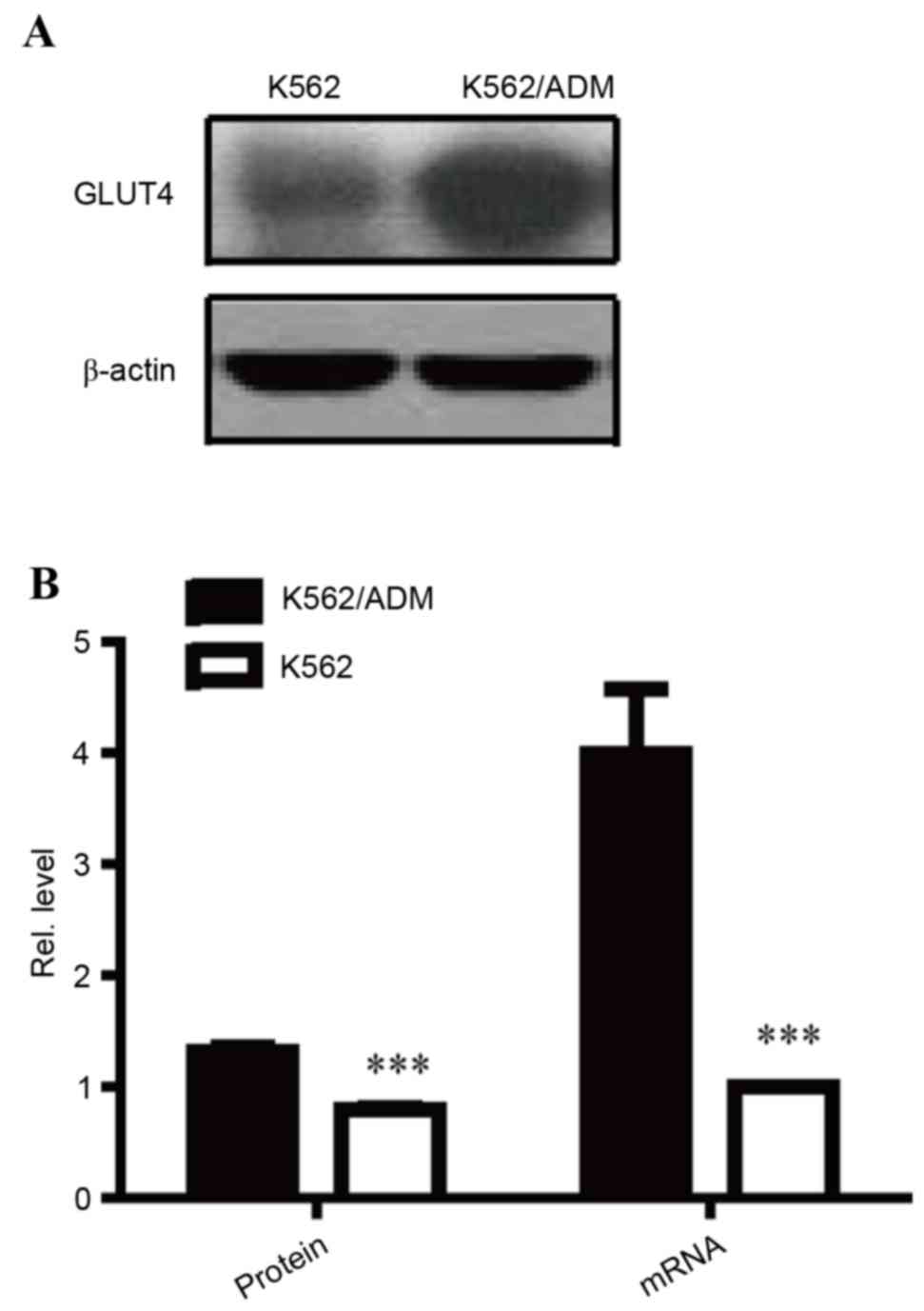
GLUT4 is overexpressed in K562/ADM
cells
AKT has previously been reported to be an important
mediator of the glucose consumption signalling pathway via its
effect on GLUT translocation and activation. Notably, as a
substrate of the AKT-mTOR-c-Myc signalling pathway, GLUT4 was
determined to have increased protein (P<0.001; Fig. 5A and B) and mRNA (P<0.001;
Fig. 5B) expression levels in
K562/ADM cells compared with K562 cells, further confirming the
over-activity of the AKT-mTOR-c-Myc signalling pathway in
ADM-resistant cells. Overexpression of GLUT4 may to contribute to
increased glucose consumption, resulting in aerobic glycolysis.
Discussion
A previous study demonstrated that tumour cells have
substantially different glycometabolism compared with healthy cells
or tissues (7), and have increased
dependency on the glycolytic pathway, rather than mitochondrial
oxidative phosphorylation (5).
Compared with healthy cells, cancer cells are characterised by
increased glucose intake and lactic acid production, and cancer
types with increased glycolysis levels are frequently insensitive
to chemo- and radiotherapy. It is generally recognised that
mitochondrial metabolic defects, aberrant expression levels and/or
activity of glycolytic enzymes, and a hypoxic microenvironment are
primary factors that contribute to the Warburg effect.
How glycolytic metabolism affects drug resistance in
cancer and leukaemia cells remains to be fully elucidated. The
present study used K562 and K562/ADM leukaemia cell lines to
investigate the effects of glycolytic metabolism on MDR of
leukaemia cells in normoxic conditions. Increased glucose
consumption and lactate export suggested that in K562/ADM cells,
the ADM-resistant MDR cell line exhibited an enhanced anaerobic
metabolic phenotype compared with ADM-sensitive K562 cells.
Therefore, the present study focused on the expression levels and
activity of glycolytic enzymes or GLUTs. The detection of key
glycolytic proteins indicated that K562/ADM cells exhibited
increased HK-II and GLUT4 expression levels, and increased LDH, HK
and PK activities, compared with ADM-sensitive cells. These data
suggested that MDR required greater glycolytic metabolic
adaptation, which may regulate the MDR phenotype of chemoresistant
leukaemia cells.
The cells, which were heavily dependent on
glycolysis in a normoxic environment, were potentially sensitive to
glycolytic inhibition. Therefore, 2-DG (a HK inhibitor) and Ox (an
LDHA inhibitor) were used as pharmacological tools to inhibit
glycolysis by blocking the first and the last step of glycolysis,
respectively. The present study demonstrated that Ox and 2-DG
treatment reduced glucose consumption and lactate export in the two
cell lines, and although glucose consumption inhibition did not
differ between the two cell types, lactate export inhibition was
increased in K562/ADM cells treated with Ox, in contrast with the
results of inhibition by 2-DG. Notably, Ox and 2-DG treatment
inhibited proliferation in the two cell types, and inhibition of
aerobic glycolysis and of LDHA activity by Ox were more effective
in killing K562/ADM cells, which cells showed an MDR phenotype,
whereas no significant inhibition of 2-DG was observed between the
two cell lines (Fig. 2C). One
logical conclusion is that inhibition of LDHA prevented the Warburg
effect, and increased growth inhibition in K562/ADM cells was
caused by increased inhibition of lactate production, which
contributed to increased K562/ADM cell sensitivity to Ox.
Therefore, the present study hypothesised that LDH has an important
role in leukaemia drug resistance, and K562/ADM exhibited ‘lactate
addiction’. Further experiments demonstrated that glycolytic
inhibition improved the therapeutic effect of ADM in the two cell
lines and re-sensitised ADM-resistant cells, with Ox exhibiting
stronger effects on chemotherapy than 2-DG. It is understood that
ATP-binding cassette transporters of resistant cells require ATP as
the energy source to pump anticancer drugs out of the cells to
avoid their lethal effects (20).
Our previous study revealed that drug-resistant K562/ADM cells
overexpressed P-gp compared with K562 cells, in a manner closely
associated with their drug-resistance (11,21).
The results of the present study suggested that the quick ATP
supply in K562/ADM cells may activate P-gp and maintain the drug
efflux via glycolysis and depletion of ATP by glycolysis inhibition
blocked pump function of P-gp. Furthermore, increased aerobic
glycolysis caused an increase in lactate production, resulting in
acidification of the intracellular microenvironment, which may
additionally decrease drug absorption and efficiency by
upregulating H+-linked ATPases and transporters
(7,22). In addition, enhanced glycolysis has
been demonstrated to produce numerous intermediate metabolites,
including nucleotides, lipids and proteins, which support the
synthesis of macromolecules required for cell proliferation
(23). Therefore, metabolic
alterations may confer adaptive, proliferative, survival and
drug-resistant advantages to K562/ADM cells.
It is not established why MDR cells favour
glycolysis in normoxic conditions, or how glycolysis regulates MDR
in resistant leukaemia cells. Previous studies have indicated that
the AKT/mTOR signalling pathway is associated with a wide array of
cellular processes, including cell proliferation, metabolism, cell
cycle regulation and drug resistance (5,13,15,24).
Recent findings have demonstrated that AKT may increase
transcription of c-Myc and reduce degradation of c-Myc indirectly
(25). Notably, mTOR regulates
c-Myc-driven carcinogenesis (26,27),
and c-Myc, a versatile transcription factor, directly triggers
transcription of genes encoding glycolytic enzymes, including
HK-II, LDHA, and GLUTs (28). The
present study demonstrated significant increases in AKT, mTOR and
c-Myc expression levels in K562/ADM cells compared with
treatment-sensitive K562 cells, accompanied by enhancement of p-AKT
(Thr308) and p-mTOR (Ser2448). Thr308 and Ser473 are the two
critical phosphorylation sites of AKT; increased phosphorylation of
Thr308 may acutely increase the enzymatic activity of AKT. These
results suggested that the AKT/mTOR/c-Myc signalling pathway is
involved in maintenance of the glycolysis-mediated MDR phenotype of
drug-resistant leukaemia cells. It is possible that increased
expression levels and enhanced phosphorylation of AKT led to
over-activation of mTOR, which increased the transcription of
c-Myc, sequentially facilitating the expression levels and activity
of LDHA and HK-II to accelerate aerobic glycolysis activity in
K562/ADM cells. In addition, significantly increased GLUT4
expression levels further confirmed the over-activation of the
AKT/mTOR signalling pathway in K562/ADM cells, which may reinforce
the ability of MDR cells to rapidly transport and consume glucose
by glycolysis to generate ATP.
Numerous previous studies have suggested that
hypoxia is an important factor contributing to the Warburg effect
in cancer cells. Hypoxia activates HIF-1α, which regulates the
transcription of a variety of glycolysis-associated target genes
against hypoxia-induced injury. In hypoxic conditions, HIF-1α
primarily regulates genes involved in glycolysis, whereas c-Myc
regulates the same genes in normoxic conditions (29). In the present study, no significant
differences were observed in HIF-1α expression levels between K562
and K562/ADM cells in normoxia; however, K562/ADM cells expressed
significantly increased levels of c-Myc compared with K562 cells.
Therefore, in normoxic conditions, adaptive glucose metabolic
alterations in drug-resistant cells were potentially mediated by
c-Myc rather than HIF-1α, and increased glycolysis in MDR leukaemia
may not be due to the intracellular hypoxia. This requires
clarification in future studies. According to these findings, it is
assumed that during the process of acquiring drug resistance,
inherently increased glycolytic leukaemia cell populations are
selected, or induced, due to proliferation or survival advantages,
and may ultimately be responsible for MDR.
In conclusion, the results of the present study
indicated that leukaemia MDR cells exhibit enhanced aerobic
glycolytic activity in normoxic conditions, and the inhibition of
glycolysis is more damaging to resistant leukaemia cells and
potently reverses the resistance of MDR cells to anticancer agents.
Increased glycolysis in MDR cells is potentially mediated by
activation of the AKT-mTOR/c-Myc pathway. Glycolytic inhibition
leading to depletion of ATP and acidification of the
microenvironment, causing blockade of the ATP-dependent drug-efflux
functions of P-gp, may be a potential strategy to reverse MDR. The
present study indicated that a combination of glycolysis inhibitors
represents a potential chemotherapeutic strategy to overcome MDR in
relapsed/refractory leukaemia or cancer.
Acknowledgements
The present study was supported by the Fundamental
Research Funds for the Central Universities (grant no.
lzujbky-2014-136) and the National Natural Science Foundation of
China (grant nos. 81541025 and 81141053).
References
|
1
|
Szakàcs G, Paterson JK, Ludwig JA,
Booth-Genthe C and Gottesman MM: Targeting multidrug resistance in
cancer. Nat Rev Drug Discov. 5:219–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holleman A, Cheok MH, den Boer ML, Yang W,
Veerman AJ, Kazemier KM, Pei D, Cheng C, Pui CH, Relling MV, et al:
Gene-expression patterns in drug-resistant acute lymphoblastic
leukemia cells and response to treatment. N Engl J Med.
351:533–542. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hulleman E, Kazemier KM, Holleman A,
VanderWeele DJ, Rudin CM, Broekhuis MJ, Evans WE, Pieters R and Den
Boer ML: Inhibition of glycolysis modulates prednisolone resistance
in acute lymphoblastic leukemia cells. Blood. 113:2014–2021. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP,
Wilson GL, Voellmy R, Lin Y, Lin W, Nahta R, et al: Overcoming
trastuzumab resistance in breast cancer by targeting dysregulated
glucose metabolism. Cancer Res. 71:4585–4597. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heiden MG Vander, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aghaee F, Pirayesh Islamian J and
Baradaran B: Enhanced radiosensitivity and chemosensitivity of
breast cancer cells by 2-deoxy-d-glucose in combination therapy. J
Breast Cancer. 15:141–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Y, Butler EB and Tan M: Targeting
cellular metabolism to improve cancer therapeutics. Cell Death Dis.
4:e5322013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hua G, Liu Y, Li X, Xu P and Luo Y:
Targeting glucose metabolism in chondrosarcoma cells enhances the
sensitivity to doxorubicin through the inhibition of lactate
dehydrogenase-A. Oncol Rep. 31:2727–2734. 2014.PubMed/NCBI
|
|
9
|
Hamanaka RB and Chandel NS: Targeting
glucose metabolism for cancer therapy. J Exp Med. 209:211–215.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei HL, Yao XJ, Li YN, Wang P, Zhao HS,
Bai DC, Peng X and Ma LF: Arsenic trioxide inhibits P-glycoprotein
expression in multidrug-resistant human leukemia K562/ADM cell line
that overexpresses mdr-1 gene and enhances their chemotherapeutic
sensitivity. Zhonghua Xue Ye Xue Za Zhi. 24:28–31. 2003.(In
Chinese). PubMed/NCBI
|
|
12
|
Cao X, Fang L, Gibbs S, Huang Y, Dai Z,
Wen P, Zheng X, Sadee W and Sun D: Glucose uptake inhibitor
sensitizes cancer cells to daunorubicin and overcomes drug
resistance in hypoxia. Cancer Chemother Pharmacol. 59:495–505.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Elstrom RL, Bauer DE, Buzzai M, Karnauskas
R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and
Thompson CB: Akt stimulates aerobic glycolysis in cancer cells.
Cancer Res. 64:3892–3899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yeung SJ, Pan J and Lee MH: Roles of p53,
MYC and HIF-1 in regulating glycolysis-the seventh hallmark of
cancer. Cell Mol Life Sci. 65:3981–3999. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mungamuri SK, Yang X, Thor AD and
Somasundaram K: Survival signaling by Notch1: Mammalian target of
rapamycin (mTOR)-dependent inhibition of p53. Cancer Res.
66:4715–4724. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang J and Mills GB: AMPK: A contextual
oncogene or tumor suppressor? Cancer Res. 73:2929–2935. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cerella C, Gaigneaux A, Dicato M and
Diederich M: Antagonistic role of natural compounds in
mTOR-mediated metabolic reprogramming. Cancer Lett. 356:251–262.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miller DM, Thomas SD, Islam A, Muench D
and Sedoris K: c-Myc and cancer metabolism. Clin Cancer Res.
18:5546–5553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan B, Piwnica-Worms D and Ratner L:
Multidrug resistance transporters and modulation. Curr Opin Oncol.
12:450–458. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen J, Wei HL, Xie B, Wang B and Cheng J
and Cheng J: Endoplasmic reticulum stress contributes to arsenic
trioxide-induced apoptosis in drug-sensitive and -resistant
leukemia cells. Leuk Res. 36:1526–1535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gatenby RA and Gillies RJ: Why do cancers
have high aerobic glycolysis? Nat Rev Cancer. 4:891–899. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu J, Blenis J and Yuan J: Activation of
PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by
phosphorylating and promoting the degradation of Mad1. Proc Natl
Acad Sci USA. 105:6584–6589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pourdehnad M, Truitt ML, Siddiqi IN,
Ducker GS, Shokat KM and Ruggero D: Myc and mTOR converge on a
common node in protein synthesis control that confers synthetic
lethality in Myc-driven cancers. Proc Natl Acad Sci USA.
110:11988–11993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Masui K, Tanaka K, Akhavan D, Babic I,
Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, et al: mTOR
complex 2 controls glycolytic metabolism in glioblastoma through
FoxO acetylation and upregulation of c-Myc. Cell Metab. 18:726–739.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dang CV: MYC, metabolism, cell growth, and
tumorigenesis. Cold Spring Harb Perspect Med. 3:pii.a0142172013.
View Article : Google Scholar
|
|
29
|
Dang CV: The interplay between MYC and HIF
in the Warburg effect. Ernst Schering Found Symp Proc. 35–53.
2007.PubMed/NCBI
|