Introduction
The pathogenesis of cardiovascular disease (CVD) and
cardiovascular complications, such as atherosclerosis, is
associated with endothelial dysfunction (ED) (1,2). It
is well established that ED contributes to the high rates of
cardiovascular mortality (3).
Excessive oxidative stress, particularly reactive oxygen species
(ROS), exerts a diverse atherogenic effect that induces vascular
and tissue injuries (4), which are
considered to serve an important role in the development of ED.
The renin-angiotensin system (RAS) has long been
known to have an important role in the maintenance of
cardiovascular function, and electrolyte and fluid balance.
Emerging evidence has suggested that angiotensin II (AngII) is a
marker of the RAS system, and higher AngII levels correlate with a
higher incidence of CVD (5).
Furthermore, AngII has been shown to directly regulate vascular
resistance and induce cardiac fibrosis-associated gene expression
in a mouse model of cardiac dysfunction (6). It has also been hypothesized that
overproduction of AngII participates in the progression of CVD via
ROS (7). Furthermore, in model
rats with hypertensive disease, AngII levels were much higher in
endothelial cells compared with in plasma (8). Beyond vasoconstriction, previous
studies have demonstrated that AngII exerts cytotoxic effects via
the induction of apoptotic endothelial cell death, these effects
were associated with phosphorylated-endothelial nitric oxide
synthase (p-eNOS) and inducible nitric oxide synthase (iNOS) in a
concentration-dependent manner (9–12).
Notably, p-eNOS and iNOS have been indicated as biomarkers and
novel therapeutic targets in oxidative stress-associated ED
(12–15).
The endoplasmic reticulum (ER) is a subcellular
organelle in which protein translation, folding and trafficking
occur. In addition, inflammation generated by ER stress is
cytotoxic to several cell types, and may facilitate the progression
of ED. Previous studies have demonstrated that increased ER stress
levels are a marker for cardiovascular risk, and are associated
with higher levels of oxidative stress and hypoxia (16,17).
In addition, numerous studies have reported that several
conditions, such as diabetes mellitus and cardiovascular disorders,
are correlated with increased ER stress levels (18,19).
Glucose-regulated protein 78 (GRP78) and C/EBP-homologous protein
(CHOP) are biological markers for ER stress (20,21).
Furthermore, ER stress is associated with decreased p-eNOS
expression (14,22).
A previous study demonstrated that AngII induces ED
via ER stress (23). In addition,
increased levels of GRP78 and CHOP have been reported to correlate
with vascular ED, and decrease the activity of antioxidant enzymes,
such as eNOS, in AngII-treated mice, thus suggesting that
AngII-induced ED is associated with ER stress (23,24).
Hydrogen sulfide (H2S) can attenuate ER stress by
increasing superoxide dismutase expression and reducing ROS levels
(25). Furthermore, H2S
may attenuate RAS activation via reduced ROS overproduction
(26). The present study
hypothesized that H2S exerts cytoprotective effects
against AngII-induced ED in human umbilical vein endothelial cells
(HUVECs) via the inhibition of ER stress.
It has previously been demonstrated that
H2S exerts cytoprotective effects against oxidative
stress-induced endothelial cell injury via antioxidant defense
mechanisms (27). However, to the
best of our knowledge, no previous studies have verified whether
H2S has any effect on AngII-induced ER stress and ED in
HUVECs.
In the present study, HUVECs were treated with AngII
to establish a cellular model of ED, and the effects of AngII on ER
stress, which is considered a main cause of ED development, were
detected. Furthermore, the protective effects of H2S on
AngII-induced ER stress were investigated. The effects of
H2S on AngII-induced apoptosis and ROS accumulation were
also determined, which are important processes in the development
of ED.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were obtained from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Sodium hydrosulfide (NaHS),
AngII, N-acetyl-L-cysteine (NAC), Hoechst 33258, deoxynucleotidyl
transferase-mediated dUTP-biotin nick end labeling (TUNEL) and
2′,7-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased
from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany).
Antibodies targeting iNOS (cat. no. ab15323), p-eNOS (cat. no.
ab183046), GRP78 (cat. no. ab21685) and CHOP (cat. no. ab11419),
p-eNOS (cat. no. ab183046), GAPDH (cat. no. ab8245) and caspase-12
(cat. no. ab62484) were provided by Abcam (Cambridge, UK).
Cystathionine-c-lyase (CSE) antibody was purchased from by
Proteintech Group Inc. (Rosemont, IL, USA; cat. no. 12032-1-AP).
Cell Counting kit-8 (CCK-8) was obtained from Dojindo China Co.,
Ltd. (Shanghai, China). Horseradish peroxidase-conjugated IgG (H+L)
secondary antibodies were purchased from Beyotime Institute of
Biotechnology (Shanghai, China; cat. nos. A0192 and A0208). All
other reagents, unless specified, were purchased from Beyotime
Institute of Biotechnology.
Cell culture
HUVECs (Cell Bank of the Type Culture Collection of
the Chinese Acadmy of Sciences, Shanghai, China) were cultured in
DMEM supplemented with 10% FBS at 37°C in a humidified atmosphere
containing 5% CO2. Prior to each experiment, the medium
was replaced with fresh serum-containing medium unless otherwise
indicated. Cells were divided into the following treatment groups:
i) Untreated; ii) AngII, consisting of 1×106 HUVEC cells
treated with 10−6 M AngII for 0–24 h; iii) NaHS + AngII,
consisting of 1×106 HUVEC cells exposed to 200 µM NaHS
for 60 min prior to treatment with 10−6 M AngII for 24
h; iv) NAC + AngII, consisting of 1×106 HUVEC cells
exposed to 100 µM NAC for 60 min prior to treatment with
10−6 M AngII for 24 h; v) NaHS, consisting of
1×106 HUVEC cells exposed to 200 µM NaHS for 24 h; and
vi) NAC, consisting of 1×106 HUVEC cells exposed to 100
µM NAC for 24 h.
Cell viability assay
After the HUVECs were cultured in 96-well plates and
had received various treatments, 100 µl CCK-8 was added to each
well at a 1/10 dilution, and the eight plates were incubated for a
further 2 h at 37°C. Absorbance was then measured at 450 nm using a
microplate reader. The mean optical density (OD) of four wells in
the indicated groups was used to calculate the percentage of cell
viability, according to the following formula: Cell viability (%) =
OD treatment group / OD control group × 100%.
Detection of intracellular ROS
generation
After the HUVECs were cultured in 96-well plates and
had received various treatments, 10 µmol/l DCFH-DA in serum-free
DMEM was added to each well and incubated for a further 30 min at
37°C. The cells were then washed three times with serum-free DMEM
prior to the detection of dichlorofluorescein fluorescence. A
fluorescent microscope connected to an imaging system was used to
observe the entire field of vision. Mean fluorescence intensity
(MFI) from three random fields was analyzed using ImageJ 1.41o
software (National Institutes of Health, Bethesda, MD, USA); MFI
was used to represent the amount of ROS.
Measurement of CSE activity
After receiving the various treatments, HUVECs were
collected and homogenized in 50 mM ice-cold potassium phosphate
buffer (pH 6.8). The reaction mixture contained 100 mmol/l
potassium phosphate buffer (pH 7.4), 10 mmol/l L-cysteine, 2 mmol/l
pyridoxal 5′-phosphate and 10% (w/v) homogenate. Cryovial test
tubes (2 ml) were used as the center wells, into which 0.5 ml 1%
zinc acetate was added as the trapping solution and 2×2.5 cm filter
paper was used to increase air:liquid contact surface. The
reactions were performed in Erlenmeyer flasks. The flasks
containing the reaction mixture and the center wells were flushed
with N2, before being sealed with a double layer of
parafilm. The reactions were initiated by transferring the flasks
from ice to a 37°C shaking water bath. Following a 90 min
incubation at 37°C, 0.5 ml of 50% trichloroacetic acid was added to
terminate the reaction. The flasks were then resealed and incubated
at 37°C for a further 60 min to ensure the complete trapping of
H2S released from the mixture. The contents of the
center wells were transferred to test tubes, each containing 3.5 ml
water. Subsequently, 0.5 ml 20 mM N,N-dimethyl-p-phenylenediamine
sulphate in 7.2 M HCl was added, immediately followed by the
addition of 0.5 ml 30 mM FeCl3 in 1.2 M HCl. After 20
min, the absorbance of the resultant solution was measured at 670
nm using a spectrophotometer.
Western blot analysis
After the indicated treatments, the cells were
washed three times with PBS and were collected according to the
cell suspension method. The cells were then lysed in lysis buffer
on ice for 30 min. The resulting cell lysates were clarified by
centrifugation at 10,943 × g for 15 min at 4°C. Proteins
were quantified using the bicinchoninic acid method.
Total proteins (300 ng) were separated by 10%
SDS-PAGE and were electroblotted onto polyvinylidene fluoride
membranes. The membranes were blocked with 5% (w/v) non-fat dry
milk containing 0.1% (v/v) Tween 20 for 1 h, and were then
incubated with GRP78 (1:800), CHOP (1:2,000), p-eNOS (Ser615;
1:1,000), iNOS (1:1,000), CSE (1:1,000) and caspase-12 (1:2,000)
antibodies overnight at 4°C. Subsequently, secondary antibodies
(1:1,000) were incubated for 1 h at room temperature. Membranes
were visualized using an enhanced chemiluminescence kit according
to the manufacturer's protocols (Beyotime Institute of
Biotechnology) and relative protein expression levels were
semi-quantitively analyzed by densitometry using QuantityOne
software (version 4.62; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Determination of HUVEC apoptosis
The apoptosis of HUVECs was detected using TUNEL and
Hoechst 33258 staining. TUNEL staining was performed using an In
Situ Cell Death Detection kit, according to the manufacturer's
protocol. Briefly, after the indicated treatments, cells were fixed
with 4% paraformaldehyde in PBS for 10 min. After three washes in
PBS, the cells were stained with 50 µl TUNEL dye for 1 h, after
which the cells were rinsed briefly with PBS and were air-dried.
Subsequently, the cells were visualized under a florescence
microscope. Apoptotic cells with condensed nuclei exhibited green
fluorescence, whereas viable cells displayed normal nuclear size
and uniform florescence.
Hoechst 33258 staining was also
conducted
Briefly, after the indicated treatments, cells were
fixed with 4% paraformaldehyde in PBS for 10 min. After three
washes in PBS, cells were stained with 5 mg/l Hoechst 33258 dye for
10 min before being rinsed briefly with PBS and air-dried. The
cells were then visualized under a florescence microscope.
Apoptotic cells with condensed nuclei exhibited blue fluorescence,
whereas viable cells displayed normal nuclear size and uniform
florescence. The ratios of different fluorescent densities from 4
random fields was analyzed using ImageJ (version 1.41o; imagej.nih.gov/ij/). The experiment was repeated 3
times.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Differences between groups were analyzed by one-way
analysis of variance followed by Bonferroni's correction. Data were
analyzed using SPSS 15.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
AngII induces cytotoxicity and ER
stress by inhibiting the expression and activity of CSE in
HUVECs
iNOS and phosphorylation of eNOS (ser615) (an
essential step in eNOS activation) have been reported to be
involved in AngII cytotoxicity (28,29)
and ED (30). Therefore, the
present study investigated whether AngII could induce ED through
alterations in the expression levels of p-eNOS (ser615) and iNOS in
HUVECs. As presented in Fig. 1,
untreated HUVECs failed to produce detectable levels of iNOS,
whereas AngII-treated cells produced high levels of iNOS.
Conversely, the other ED marker, p-eNOS (ser615), was reduced in
AngII-treated cells. ER stress has been reported to be associated
with AngII in mice (31,32); therefore, the present study further
observed the effects of AngII on the expression of ER
stress-associated molecules, such as GRP78 and CHOP. Similar to
iNOS, AngII was able to induce GRP78 and CHOP expression.
Conversely, CSE expression and activity was reduced in a
time-dependent manner following AngII treatment.
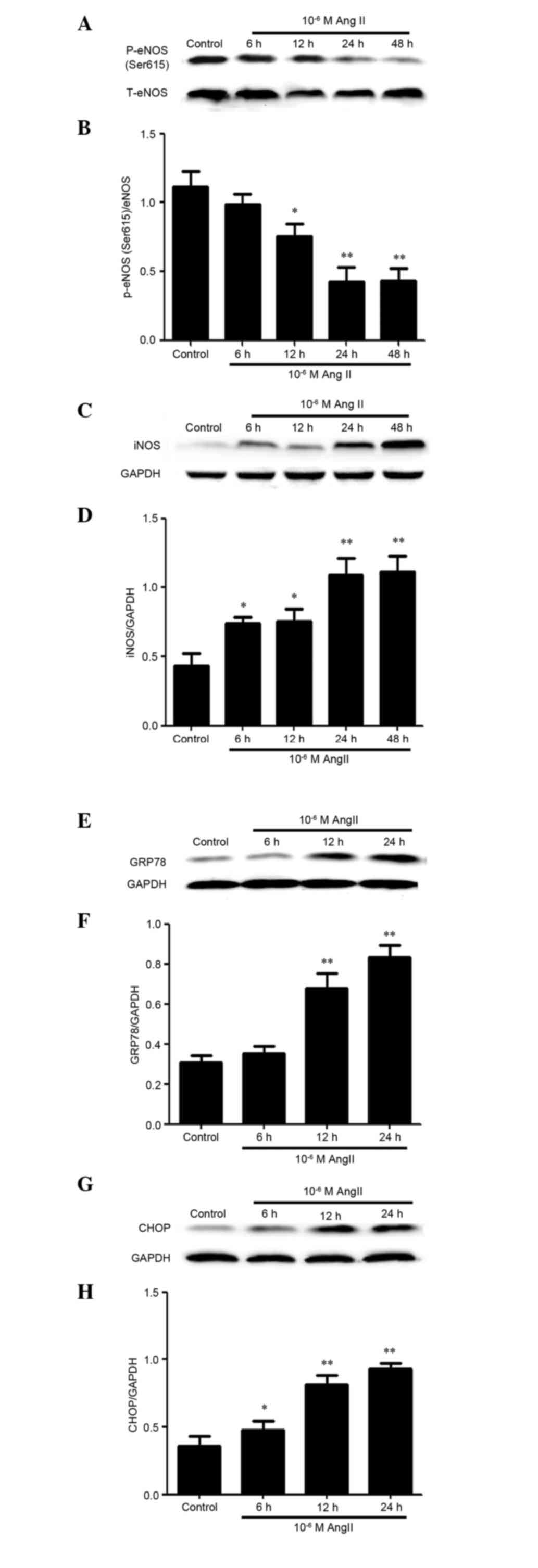 | Figure 1.AngII induced cytotoxicity and
endoplasmic reticulum stress by inhibiting the expression and
activity of cystathionine-c-lyase in human umbilical vein
endothelial cells. Cells were treated with 10−6M AngII
for the indicated times. The protein expression levels of (A and B)
p-eNOS (ser615) and (C and D) iNOS, (E and F) GRP78, (G and H) CHOP
and (I and J) CSE were determined by western blotting. were
detected by western blotting. (B, D, F, H and J) Blots from A, C,
E, G and I were semi-quantified by densitometric analysis. (K)
Activity levels of CSE were determined by methylene blue
spectrophotometric assay. Data are presented as the mean ± standard
error of the mean (n=3). **P<0.01, *P<0.05 vs. the control
group. AngII, angiotensin II; p-eNOS, phosphorylated-endothelial
nitric oxide synthase; T total; iNOS, inducible nitric oxide
synthase; GRP78, glucose-regulated protein 78; CHOP,
C/EBP-homologous protein; CSE, cystathionine-c-lyase. |
Stimulation of ROS production inhibits
the expression and activity of CSE in AngII-treated HUVECs
Oxidative stress, which is generated by AngII, can
induce endothelial cell injury (33). Since AngII can induce ROS
production, the present study aimed to determine the mechanism
underlying ROS production in AngII-treated HUVECs. As presented in
Fig. 2, HUVECs pretreated with NAC
before AngII exhibited increased CSE expression and activity
compared with cells treated with AngII. In addition, a well-known
exogenous ROS, hydrogen peroxide (H2O2), was
used to determine whether ROS induced the downregulation of CSE
expression and activity. H2O2-treated HUVECs
exhibited almost undetectable CSE expression and activity compared
with untreated control cells. These data indicate that
AngII-induced ROS may contribute to the downregulatory effects of
AngII on the expression and activity of CSE in HUVECs.
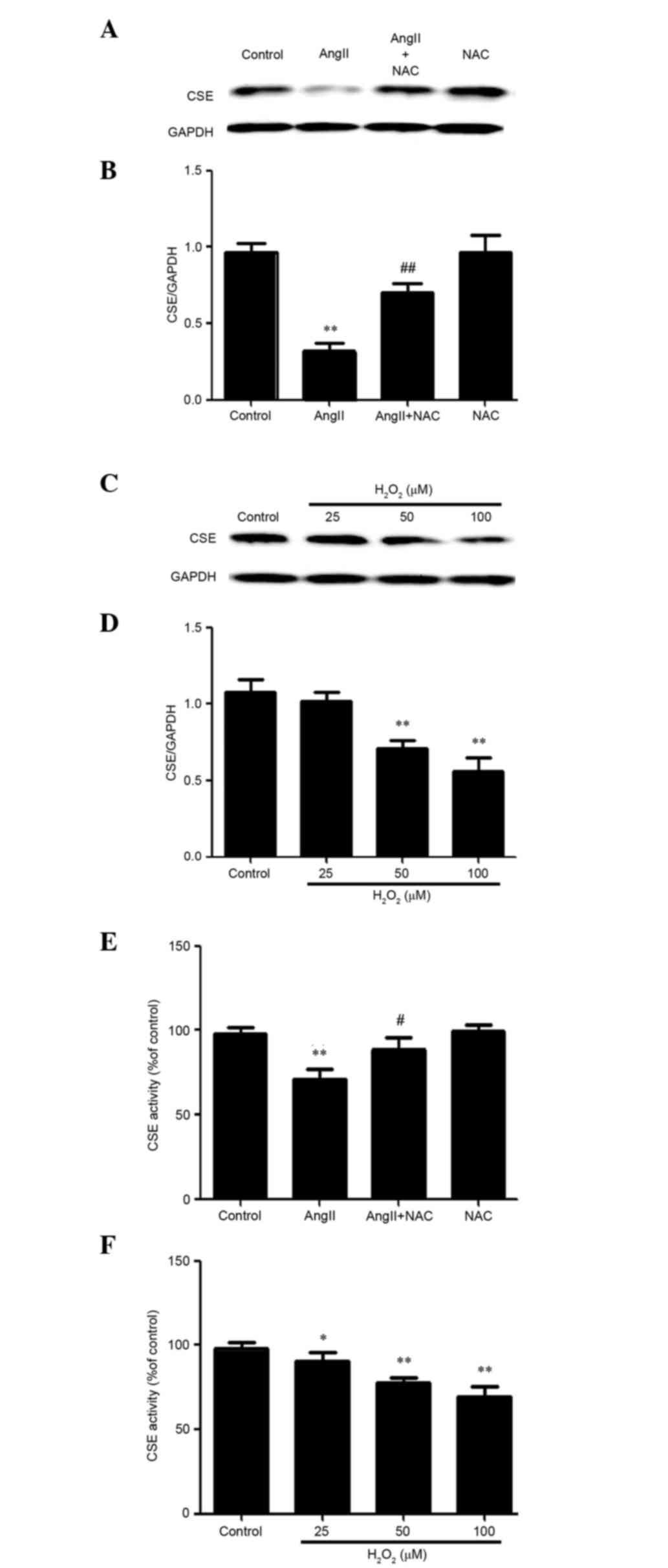 | Figure 2.Stimulation of reactive oxygen
species production suppressed the expression and activity of CSE in
AngII-treated human umbilical vein endothelial cells. (A, B and E)
Cells were treated with 10−6M AngII for 24 h, or were
pretreated with 1 mM NAC for 1 h prior to AngII treatment for 24 h.
(A and B) Protein expression levels of CSE were determined by
western blotting, and (E) the activity levels of CSE were
determined by methylene blue spectrophotometric assay. (C, D and F)
Cells were treated with the indicated concentrations of
H2O2 for 24 h. (C and D) Protein levels of
CSE were determined by western blotting, and (F) the activity
levels of CSE were determined by methylene blue spectrophotometric
assay. (B and D) Blots in A and C were determined by densitometric
analysis. Data are presented as the means ± standard error of the
mean (n=3). **P<0.01, *P<0.05 vs. the control group,
##P<0.01, #P<0.05 vs. the AngII
treatment group. AngII, angiotensin II; NAC, N-acetyl-L-cysteine;
CSE, cystathionine-c-lyase; H2O2, hydrogen
peroxide. |
Exogenous H2S ameliorates
AngII-induced ED mediated by ROS in HUVECs
The present study demonstrated that AngII can
inhibit activation of the CSE/H2S pathway in
AngII-treated HUVECs; therefore, the present study used a
well-known endothelial protective agent, NaHS, to determine whether
NaHS could reduce AngII-induced ED. As presented in Fig. 3, cells treated with NaHS and AngII
had lower levels of iNOS, compared with NaHS-unstimulated
AngII-treated cells. Conversely, NaHS markedly ameliorated
AngII-reduced p-eNOS (ser615) expression. The results suggest that
treatment with 200 or 400 µM NaHS may inhibit AngII-induced ED.
Similarly, treatment with NAC significantly ameliorated the
expression levels of AngII-suppressed p-eNOS (ser615) (Fig. 3E and F) and AngII-enhanced iNOS
(Fig. 3G and H). Notably, cells
treated with either NaHS or NAC alongside AngII exhibited improved
cell viability compared with AngII-treated cells, whereas treatment
with either NaHS or NAC alone did not alter cell viability in
HUVECs (Fig. 3I). These data
indicate that antioxidants may mediate the effects of
H2S on AngII-induced ED.
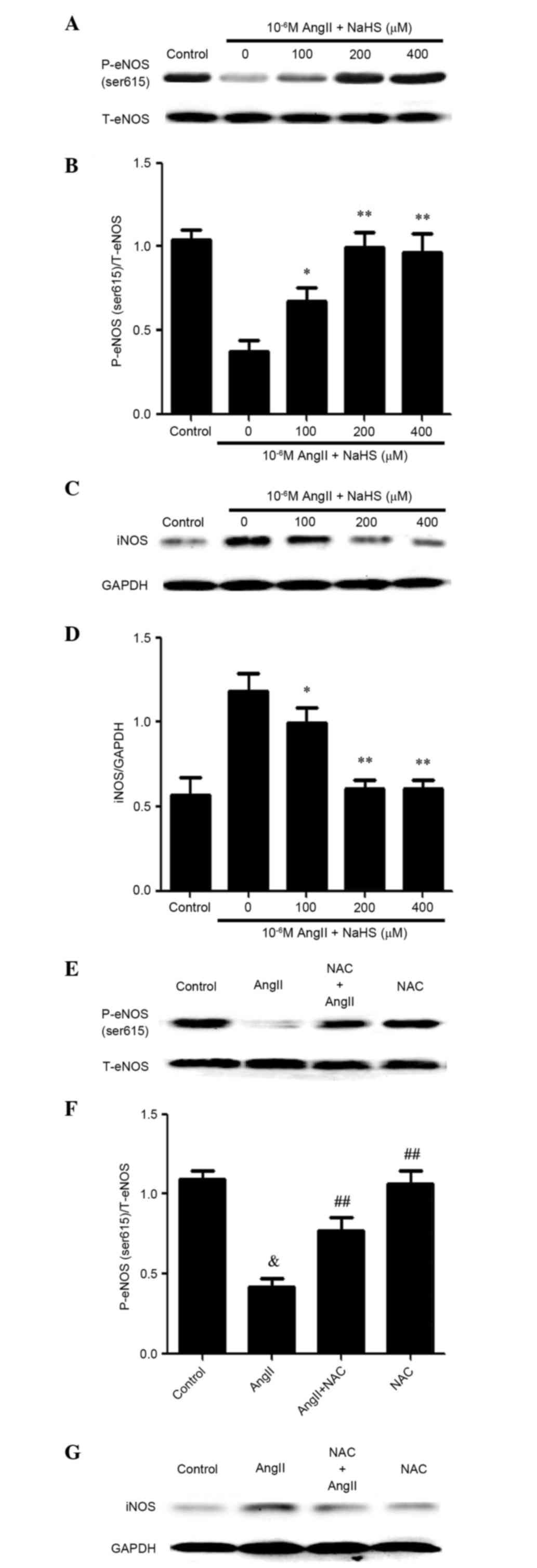 | Figure 3.Exogenous H2S ameliorates
AngII-induced endothelial dysfunction mediated by reactive oxygen
species in human umbilical vein endothelial cells. Cells were
pretreated with the indicated concentrations of NaHS for 1 h and
were then treated with 10−6 M AngII for 24 h. The
protein expression levels of (A and B) p-eNOS (ser615) and (C and
D) iNOS were determined by western blotting. (B and D) Blots from A
and C were semi-quantified by densitometric analysis. Data are
presented as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01 vs. the 0 µM group. Cells were pretreated
with 1 mM NAC for 1 h and were then treated with treated with AngII
for 24 h. The protein expression levels of (E and F) p-eNOS
(ser615) and (G and H) iNOS were determined by western blotting. (F
and H) Blots from E and G were semi-quantified by densitometric
analysis. (I) Cells were incubated with 1 mM NAC or 200 µM NaHS
only for 24 h, or were pre-incubated with 1 mM NAC or 200 µM NaHS
for 1 h before being incubated with 10−6M AngII for 24
h. Cell viability was measured using the Cell Counting kit-8 assay.
Data are presented as the mean ± standard error of the mean (n=3).
##P<0.01, #P<0.05 vs. the AngII
treatment group, &P<0.01 vs. the control group.
AngII, angiotensin II; NaHS, sodium hydrosulfide; NAC,
N-acetyl-L-cysteine; p-eNOS, phosphorylated-endothelial nitric
oxide synthase; T, total; iNOS, inducible nitric oxide
synthase. |
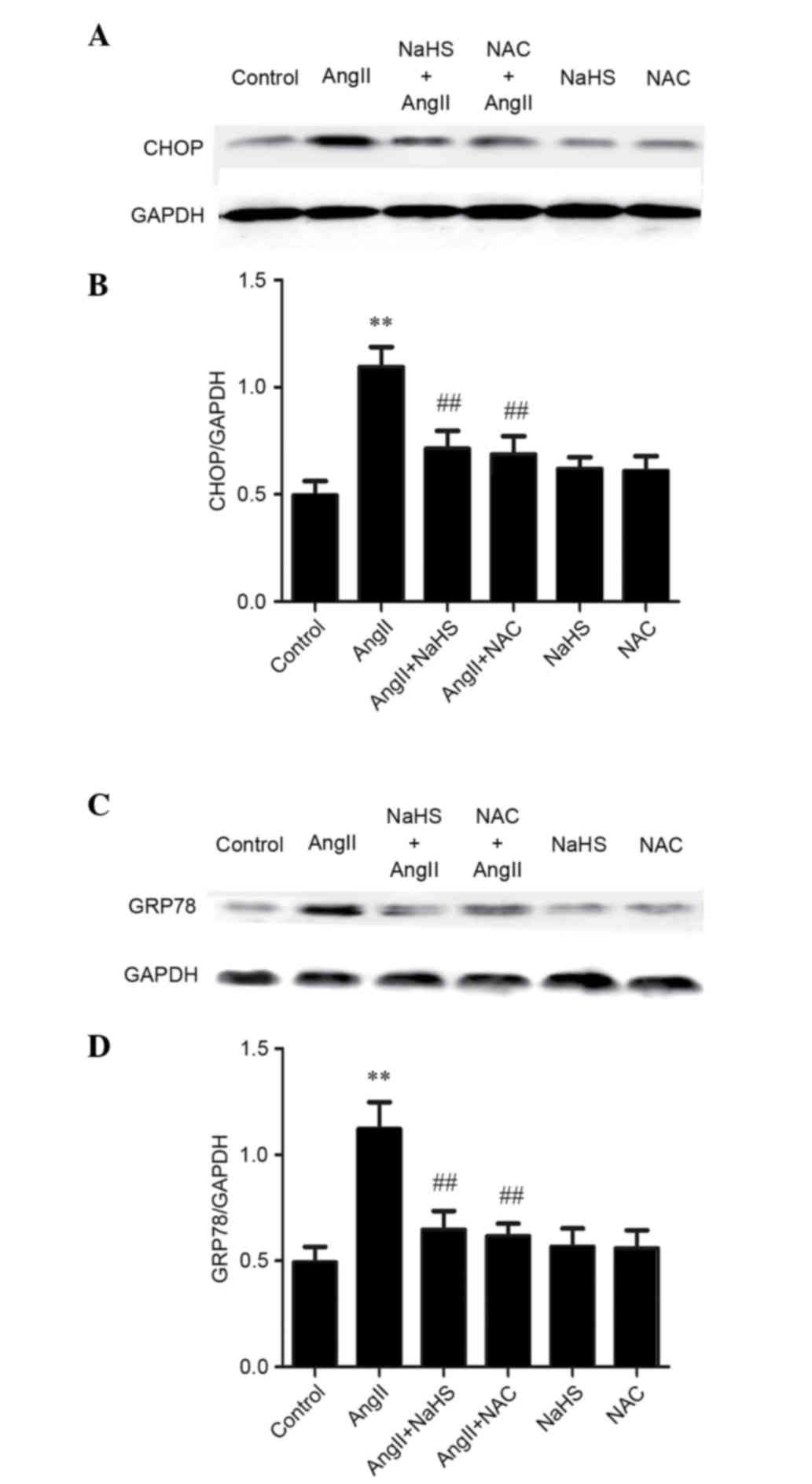
H2S supplementation or NAC
ameliorates AngII-stimulated GRP78 and CHOP overexpression in
HUVECs
The present study detected the potential mechanism
underlying the protective effects of H2S against
AngII-induced ED in HUVECs. It has previously been demonstrated
that ROS may induce ER stress. The present results indicated that
treatment with the antioxidants NaHS or NAC could suppress
AngII-induced GRP78 and CHOP expression, whereas either NaHS or NAC
alone did not alter the expression levels of GRP78 and CHOP in
HUVECs (Fig. 4). These data
suggest that the inhibitory effects of H2S may protect
against AngII-induced dysfunction in HUVECs via suppressing ER
stress.
H2S supplementation or NAC
ameliorates AngII-induced ROS generation in HUVECs
To determine whether the cytoprotective effects of
H2S against AngII-induced ED were due to its
antioxidative activity, NaHS and NAC were used to treat HUVECs.
AngII exposure markedly enhanced intracellular ROS generation in
HUVECs (Fig. 5B). However,
pretreatment with either NaHS or NAC for 60 min markedly attenuated
AngII-induced ROS accumulation in HUVECs (Fig. 5C and D). Pretreatment with NAC had
a similar antioxidant effect to NaHS against AngII-induced ROS
accumulation, whereas NaHS or NAC treatment alone did not alter
base intracellular ROS levels (Fig. 5E
and F).
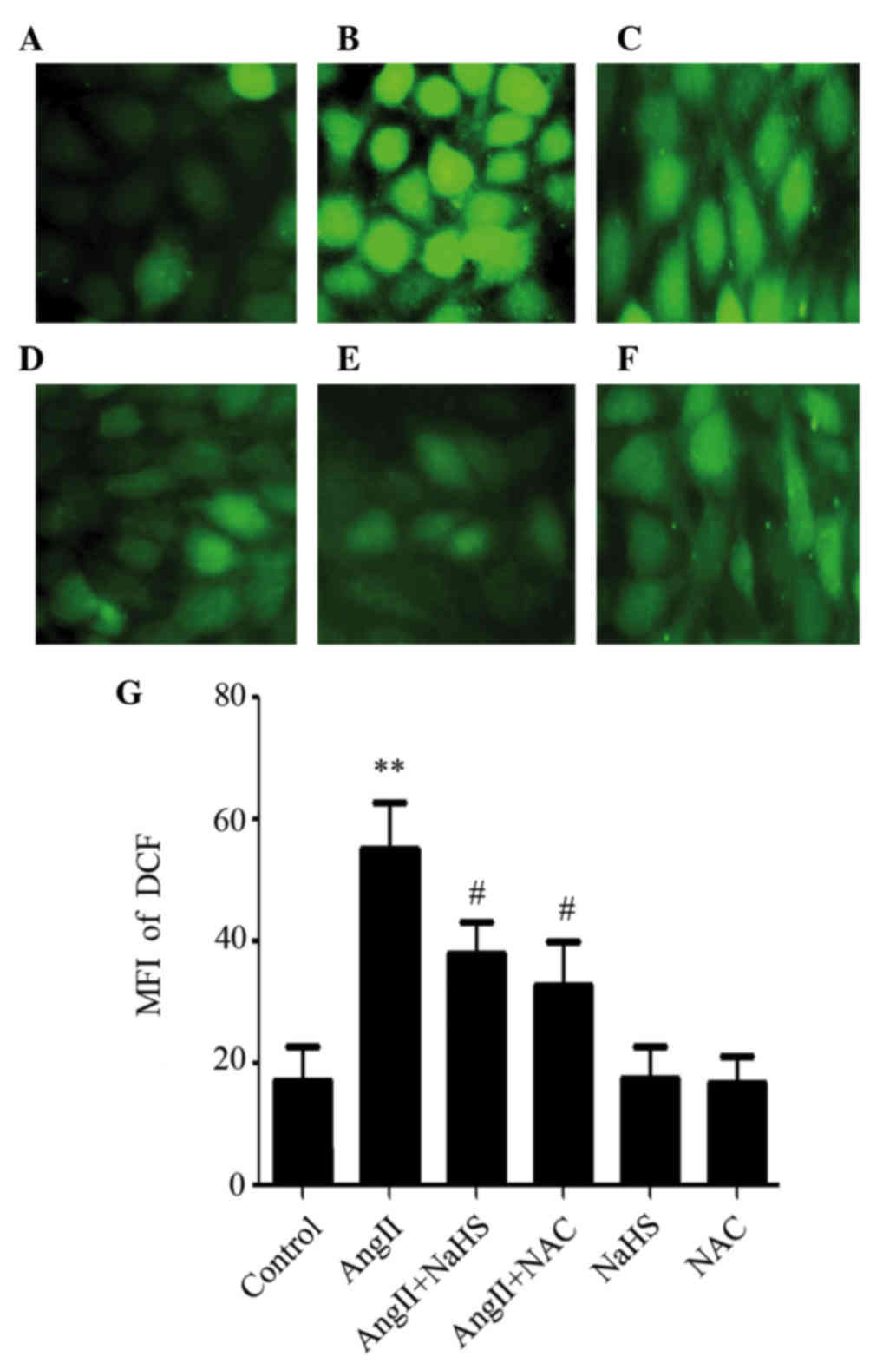 | Figure 5.Hydrogen sulfide supplementation or
NAC ameliorates AngII-induced reactive oxygen species generation in
human umbilical vein endothelial cells. Cells were incubated with
10−6M AngII, 200 µM NaHS or 1 mM NAC for 24 h, or were
pretreated with 200 µM NaHS or 1 mM NAC for 1 h before being
treated with 10−6M AngII for 24 h. Cellular ROS
accumulation was observed by microscopy following
2′,7-dichlorodihydrofluorescein diacetate staining. (A) Control;
(B) AngII; (C) NaHS + AngII; (D) NAC + AngII; (E) NaHS and (F) NAC
groups. Magnification, ×400.(G) MFI was determined using ImageJ
1.41o software with quantitative analysis. Data are presented as
the mean ± standard error of the mean (n=5). **P<0.01 vs. the
control group, #P<0.05 vs. the AngII treatment group.
AngII, angiotensin II; NaHS, sodium hydrosulfide; NAC,
N-acetyl-L-cysteine; MFI, mean fluorescent intensity; DCF,
dichlorofluorescein. |
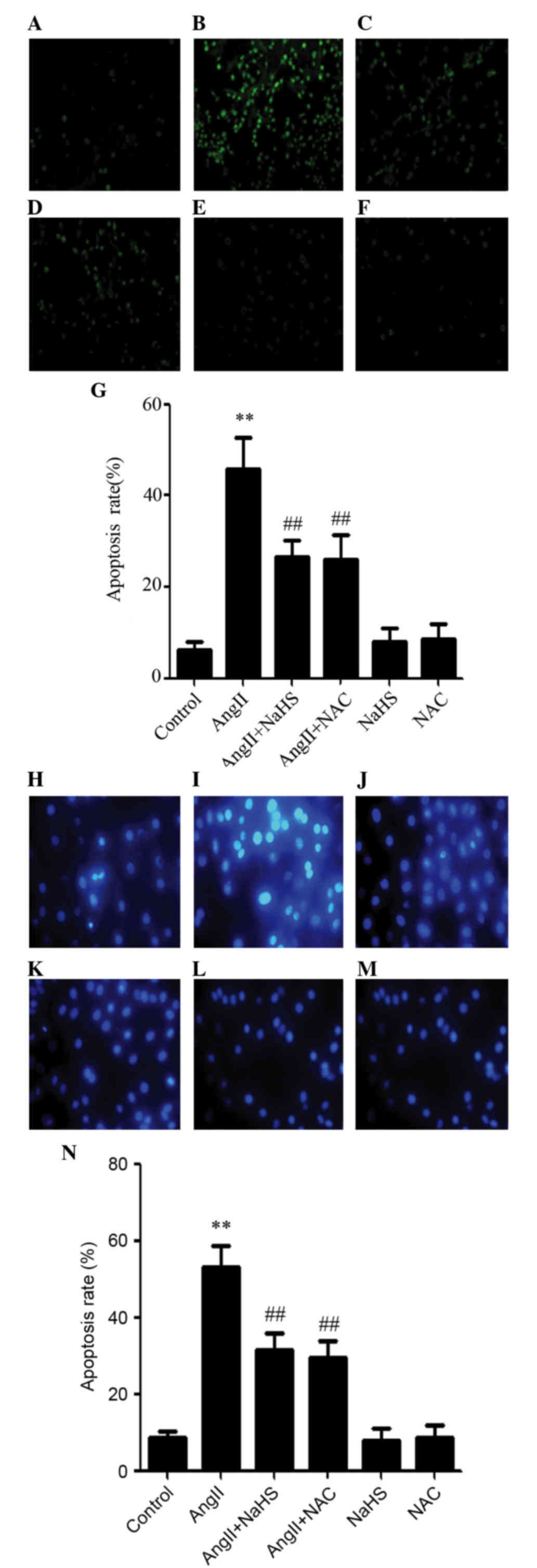
Exogenous H2S and NAC
reduce AngII-induced apoptosis in HUVECs
The present study also determined the effects of
NaHS and NAC on AngII-induced apoptosis. As shown in Fig. 6, AngII-stimulated HUVECs exhibited
typical characteristics of apoptosis, including chromatin
condensation, nuclear shrinkage, and the presence of apoptotic
bodies. However, treatment with either NaHS or NAC, alongside
AngII, resulted in amelioration of the typical characteristics of
apoptosis, including chromatin condensation, nuclear shrinkage, and
the presence of apoptotic bodies compared with in cells treated
with AngII only. Treatment with NaHS or NAC alone did not markedly
alter cell morphology or the percentage of apoptotic HUVECs.
Consistent with these results, the protein expression levels of
cleaved caspase-12 were examined by western blot analysis, and were
reduced in AngII-treated cells following NaHS or NAC treatment.
However, treatment with either NaHS or NAC alone did not alter the
expression of cleaved caspase-12. These findings indicate that
exogenous H2S protects HUVECs against AngII-induced ER
stress.
Discussion
RAS activity and oxidative stress are two major
interacting pathogenic factors associated with ED, which is the
initiating factor and pathological basis of CVD (34,35).
As an important mediator of endothelial function maintenance,
reduced levels of nitric oxide (NO) could accelerate the ED process
(36). Cellular and animal studies
have demonstrated that NO, via constitutive NOS isoforms, modulates
endothelial cell dilation and blood flow distribution, and low
levels of p-eNOS activity are associated with injured endothelial
function (37,38). However, high levels of iNOS also
exert a similar effect (39).
Nakao et al (40)
demonstrated that suppressed p-eNOS and enhanced iNOS expression
are associated with AngII cytotoxicity; together with the present
findings, these results suggested that AngII may result in HUVECs
dysfunction.
Emerging evidence from experimental and clinical
research has indicated that ER stress is involved in CVD with ED
(41) and RAS activity (42). ER stress modulates various factors,
including GRP78 and CHOP. Oxidative stress (43) or AngII stimulation (44) have been suggested to trigger GRP78
and CHOP overexpression. Furthermore, AngII can upregulate the
expression of inflammatory mediators, including GRP78 and CHOP,
leading to cytotoxicity in HUVECs. The present findings are in
agreement with those of a previous study, which demonstrated that
high levels of AngII led to cardiac damage and vascular ED via ER
stress (23). CSE serves
atherogenic and endothelial cytoprotective roles in CVD (27,45).
Conversely, AngII exerts cytotoxic effects, and in the present
study AngII was shown to downregulate the expression and activity
of CSE in HUVECs, which was accompanied by decreased cell
viability. In addition, insufficient production of endogenous
H2S or inhibition of its synthesis have been reported to
enhance the overproduction of ROS in HUVECs (46). Taken together, it may be
hypothesized that AngII-induced ER stress leads to ED in HUVECs via
the inhibition of endogenous H2S production.
Insufficient production of endogenous H2S
has been reported to be associated with AngII-induced cytotoxicity
via oxidative stress (47). The
present study, alongside a previous study, confirmed that ROS, such
as H2O2, serve a primary role in ER stress,
are regulated by AngII and are responsible for the consequent
generation of ED (48).
Furthermore, the present study demonstrated that treatment with the
exogenous ROS, H2O2, downregulated the
expression and activity of CSE; the inhibitory effects of
H2O2 were similar to those of AngII in
HUVECs. Subsequently, HUVECs were treated with NAC prior to AngII
exposure; the results demonstrated that NAC suppressed
overproduction of ROS and insufficient productions of endogenous
H2S. These findings suggested that ROS serves a key role
in the inhibition of CSE induced by AngII in HUVECs.
The present study also demonstrated that the effects
of exogenous H2S on AngII-induced ED were due to
antioxidative mechanisms. The findings indicated that
H2S supplementation prior to AngII treatment in HUVECs
resulted in a reduction in the expression levels of iNOS, an
increase in p-eNOS (Ser615) expression and increased cell
viability. Furthermore, treatment with NAC before AngII exposure
not only exerted inhibitory effects on iNOS expression, but also
enhanced p-eNOS (Ser615) expression and cell viability, whereas
supplementation of H2S (NaHS) prior to AngII-treated
HUVECs had similar effects. The present study further explored the
potential mechanism underlying the protective effects of exogenous
H2S, by treating cells with either NaHS or NAC prior to
AngII exposure. Since NaHS or NAC exert similar antioxidative
effects, NAC, similar to NaHS, was able to reduce ER stress,
including GRP78 and CHOP expression. In addition, DCFH-DA was used
to detect cellular ROS accumulation. The results indicated that the
antioxidative effects of H2S may exert protection
against AngII-induce ED by suppressing ER stress. ROS are
associated with accelerated ER malfunction (49); the present study demonstrated that
in HUVECs pretreated with NaHS or NAC prior to AngII exposure, ROS
overproduction was inhibited.
ER stress is particularly associated with apoptosis.
Song et al (50)
demonstrated that GRP78 and CHOP are markedly activated in
apoptosis. The present study pretreated HUVECs with NaHS or NAC,
prior to AngII exposure, and TUNEL and Hoechst 33258 staining were
used to detect apoptosis. The results indicated that pretreatment
with either NaHS or NAC significantly decreased AngII-induced
apoptosis and cleaved caspase-12 expression. In addition, Qabazard
et al (51) reported that
H2S significantly suppressed
H2O2-induced ER stress in bovine aortic
endothelial cells, and ameliorated cell apoptosis.
In conclusion, using AngII-treated HUVECs, the
present study demonstrated that ROS participates in AngII-induced
ER stress-mediated ED, by inhibiting the expression and activity of
CSE. In addition, H2S supplementation may be considered
a beneficial antioxidative therapy in AngII-induced ED via the
suppression of ER stress. The results of the present study provide
novel evidence to suggest that exogenous H2S may be
considered a novel therapeutic strategy for the treatment of
CVD.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81100106), Aid
Programme for Science and Technology Innovative Research Team.
References
|
1
|
Ding Z, Liu S, Wang X, Dai Y, Khaidakov M,
Romeo F and Mehta JL: LOX-1, oxidant stress, mtDNA damage,
autophagy, and immune response in atherosclerosis. Can J Physiol
Pharmacol. 92:524–530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Messner B and Bernhard D: Smoking and
cardiovascular disease: Mechanisms of endothelial dysfunction and
early atherogenesis. Arterioscler Thromb Vasc Biol. 34:509–515.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stam F, van Guldener C, Becker A, Dekker
JM, Heine RJ, Bouter LM and Stehouwer CD: Endothelial dysfunction
contributes to renal function-associated cardiovascular mortality
in a population with mild renal insufficiency: The Hoorn study. J
Am Soc Nephrol. 17:537–545. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geronikaki AA, Pitta EP and Liaras KS:
Thiazoles and thiazolidinones as antioxidants. Curr Med Chem.
20:4460–4480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang X, Yuan B, Dong W, Yang B, Yang Y,
Lin X and Gong G: Humid heat exposure induced oxidative stress and
apoptosis in cardiomyocytes through the angiotensin II signaling
pathway. Heart Vessels. 30:396–405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Li N, Pang W, Zhang X, Hammock BD,
Ai D and Zhu Y: Opposite effects of gene deficiency and
pharmacological inhibition of soluble epoxide hydrolase on cardiac
fibrosis. PLoS One. 9:e940922014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Johar S, Cave AC, Narayanapanicker A,
Grieve DJ and Shah AM: Aldosterone mediates angiotensin II-induced
interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase.
FASEB J. 20:1546–1548. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kittikulsuth W, Looney SW and Pollock DM:
Endothelin ET(B) receptors contribute to sex differences in blood
pressure elevation in angiotensin II hypertensive rats on a
high-salt diet. Clin Exp Pharmacol Physiol. 40:362–370. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marrachelli VG, Mastronardi ML, Sarr M,
Soleti R, Leonetti D, Martínez MC and Andriantsitohaina R: Sonic
hedgehog carried by microparticles corrects angiotensin II-induced
hypertension and endothelial dysfunction in mice. PLoS One.
8:e728612013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schäfer SC, Pellegrin M, Wyss C, Aubert
JF, Nussberger J, Hayoz D, Lehr HA and Mazzolai L: Intravital
microscopy reveals endothelial dysfunction in resistance arterioles
in Angiotensin II-induced hypertension. Hypertens Res. 35:855–861.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin LY, Lin CY, Su TC and Liau CS:
Angiotensin II-induced apoptosis in human endothelial cells is
inhibited by adiponectin through restoration of the association
between endothelial nitric oxide synthase and heat shock protein
90. FEBS Lett. 574:106–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matsumoto S, Shimabukuro M, Fukuda D,
Soeki T, Yamakawa K, Masuzaki H and Sata M: Azilsartan, an
angiotensin II type 1 receptor blocker, restores endothelial
function by reducing vascular inflammation and by increasing the
phosphorylation ratio Ser (1177)/Thr(497) of endothelial nitric
oxide synthase in diabetic mice. Cardiovasc Diabetol. 13:302014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
FT IV Billings, Pretorius M, Schildcrout
JS, Mercaldo ND, Byrne JG, Ikizler TA and Brown NJ: Obesity and
oxidative stress predict AKI after cardiac surgery. J Am Soc
Nephrol. 23:1221–1228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsumoto T, Kobayashi T and Kamata K:
Relationships among ET-1, PPARgamma, oxidative stress and
endothelial dysfunction in diabetic animals. J Smooth Muscle Res.
44:41–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xing S, Yang X, Li W, Bian F, Wu D, Chi J,
Xu G, Zhang Y and Jin S: Salidroside stimulates mitochondrial
biogenesis and protects against H2O2-induced endothelial
dysfunction. Oxid Med Cell Longev. 2014:9048342014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kei A, Tellis C, Liberopoulos E, Tselepis
A and Elisaf M: Effect of switch to the highest dose of
rosuvastatin versus add-on-statin fenofibrate versus add-on-statin
nicotinic acid/laropiprant on oxidative stress markers in patients
with mixed dyslipidemia. Cardiovasc Ther. 32:139–146. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bozaykut P, Sozen E, Yazgan B, Karademir B
and Kartal-Ozer N: The role of hypercholesterolemic diet and
vitamin E on Nrf2 pathway, endoplasmic reticulum stress and
proteasome activity. Free Radic Biol Med. 75 Suppl 1:S242014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hasnain SZ, Prins JB and McGuckin MA:
Oxidative and endoplasmic reticulum stress in β-cell dysfunction in
diabetes. J Mol Endocrinol. 56:R33–R54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cimellaro A, Perticone M, Fiorentino TV,
Sciacqua A and Hribal ML: Role of endoplasmic reticulum stress in
endothelial dysfunction. Nutr Metab Cardiovasc Dis. 26:863–871.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Simon-Szabó L, Kokas M, Mandl J, Kéri G
and Csala M: Metformin attenuates palmitate-induced endoplasmic
reticulum stress, serine phosphorylation of IRS-1 and apoptosis in
rat insulinoma cells. PLoS One. 9:e978682014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang XY, Yang CT, Zheng DD, Mo LQ, Lan AP,
Yang ZL, Hu F, Chen PX, Liao XX and Feng JQ: Hydrogen sulfide
protects H9c2 cells against doxorubicin-induced cardiotoxicity
through inhibition of endoplasmic reticulum stress. Mol Cell
Biochem. 363:419–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li PC, Yang CC, Hsu SP and Chien CT:
Repetitive progressive thermal preconditioning hinders thrombosis
by reinforcing phosphatidylinositol 3-kinase/Akt-dependent
heat-shock protein/endothelial nitric oxide synthase signaling. J
Vasc Surg. 56:159–170. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kassan M, Galán M, Partyka M, Saifudeen Z,
Henrion D, Trebak M and Matrougui K: Endoplasmic reticulum stress
is involved in cardiac damage and vascular endothelial dysfunction
in hypertensive mice. Arterioscler Thromb Vasc Biol. 32:1652–1661.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Bai YP, Hong D, Gao HC, Li LF, Li
CC, Zhu LP, Sun Q and Zhang GG: Ang II induces capillary formation
from endothelial cells via the AT1R-dependent inositol requiring
enzyme 1 pathway. Biochem Biophys Res Commun. 434:552–558. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen ZF, Zhao B, Tang XY, Li W, Zhu LL,
Tang CS, DU JB and Jin HF: Hydrogen sulfide regulates vascular
endoplasmic reticulum stress in apolipoprotein E knockout mice.
Chin Med J (Engl). 124:3460–3467. 2011.PubMed/NCBI
|
|
26
|
Xue H, Yuan P, Ni J, Li C, Shao D, Liu J,
Shen Y, Wang Z, Zhou L, Zhang W, et al: H(2)S inhibits
hyperglycemia-induced intrarenal renin-angiotensin system
activation via attenuation of reactive oxygen species generation.
PLoS One. 8:e743662013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wen YD, Wang H, Kho SH, Rinkiko S, Sheng
X, Shen HM and Zhu YZ: Hydrogen sulfide protects HUVECs against
hydrogen peroxide induced mitochondrial dysfunction and oxidative
stress. PLoS One. 8:e531472013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Marzetti E, Calvani R, Cesari M, Buford
TW, Lorenzi M, Behnke BJ and Leeuwenburgh C: Mitochondrial
dysfunction and sarcopenia of aging: From signaling pathways to
clinical trials. Int J Biochem Cell Biol. 45:2288–2301. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tomada I, Negrão R, Almeida H and Neves D:
Long-term high-fat consumption leads to downregulation of Akt
phosphorylation of eNOS at Ser1177 and upregulation of Sirtuin-1
expression in rat cavernous tissue. Age (Dordr). 36:597–611. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ripa P, Ornello R, Pistoia F, Carolei A
and Sacco S: The renin-angiotensin system: A possible contributor
to migraine pathogenesis and prophylaxis. Expert Rev Neurother.
14:1043–1055. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang W, Qiu L, Howard A, Solis N, Li C,
Wang X, Kopp JB and Levi M: Protective effects of aliskiren and
valsartan in mice with diabetic nephropathy. J Renin Angiotensin
Aldosterone Syst. 15:384–395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y
and Cai L: Diabetes- and angiotensin II-induced cardiac endoplasmic
reticulum stress and cell death: Metallothionein protection. J Cell
Mol Med. 13:1499–1512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park MH, Heo SJ, Park PJ, Moon SH, Sung
SH, Jeon BT and Lee SH: 6,6′-bieckol isolated from ecklonia cava
protects oxidative stress through inhibiting expression of ROS and
proinflammatory enzymes in high-glucose-induced human umbilical
vein endothelial cells. Appl Biochem Biotechnol. 174:632–643. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu JJ, Li DL, Zhou J, Sun L, Zhao M, Kong
SS, Wang YH, Yu XJ, Zhou J and Zang WJ: Acetylcholine prevents
angiotensin II-induced oxidative stress and apoptosis in H9c2
cells. Apoptosis. 16:94–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wong WT, Tian XY and Huang Y: Endothelial
dysfunction in diabetes and hypertension: Cross talk in RAS, BMP4,
and ROS-dependent COX-2-derived prostanoids. J Cardiovasc
Pharmacol. 61:204–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dudzinski DM and Michel T: Life history of
eNOS: Partners and pathways. Cardiovasc Res. 75:247–260. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Heltianu C, Costache G, Gafencu A, Diaconu
M, Bodeanu M, Cristea C, Azibi K, Poenaru L and Simionescu M:
Relationship of eNOS gene variants to diseases that have in common
an endothelial cell dysfunction. J Cell Mol Med. 9:135–142. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
El Accaoui RN, Gould ST, Hajj GP, Chu Y,
Davis MK, Kraft DC, Lund DD, Brooks RM, Doshi H, Zimmerman KA, et
al: Aortic valve sclerosis in mice deficient in endothelial nitric
oxide synthase. Am J Physiol Heart Circ Physiol. 306:H1302–H1313.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Albrecht EW, Stegeman CA, Heeringa P,
Henning RH and van Goor H: Protective role of endothelial nitric
oxide synthase. J Pathol. 199:8–17. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakao T, Morita H, Maemura K, Amiya E,
Inajima T, Saito Y, Watanabe M, Manabe I, Kurabayashi M, Nagai R
and Komuro I: Melatonin ameliorates angiotensin II-induced vascular
endothelial damage via its antioxidative properties. J Pineal Res.
55:287–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou B, Li H, Liu J, Xu L, Zang W, Wu S
and Sun H: Intermittent injections of osteocalcin reverse
autophagic dysfunction and endoplasmic reticulum stress resulting
from diet-induced obesity in the vascular tissue via the
NFkB-p65-dependent mechanism. Cell Cycle. 12:1901–1913. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Finckenberg P, Eriksson O, Baumann M,
Merasto S, Lalowski MM, Levijoki J, Haasio K, Kytö V, Muller DN,
Luft FC, et al: Caloric restriction ameliorates angiotensin
II-induced mitochondrial remodeling and cardiac hypertrophy.
Hypertension. 59:76–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Watanabe Y, Suzuki O, Haruyama T and
Akaike T: Interferon-gamma induces reactive oxygen species and
endoplasmic reticulum stress at the hepatic apoptosis. J Cell
Biochem. 89:244–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wencker D, Chandra M, Nguyen K, Miao W,
Garantziotis S, Factor SM, Shirani J, Armstrong RC and Kitsis RN: A
mechanistic role for cardiac myocyte apoptosis in heart failure. J
Clin Invest. 111:1497–1504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Szczesny B, Módis K, Yanagi K, Coletta C,
Le Trionnaire S, Perry A, Wood ME, Whiteman M and Szabo C: AP39, a
novel mitochondria-targeted hydrogen sulfide donor, stimulates
cellular bioenergetics, exerts cytoprotective effects and protects
against the loss of mitochondrial DNA integrity in oxidatively
stressed endothelial cells in vitro. Nitric Oxide. 41:120–130.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pan LL, Liu XH, Gong QH, Wu D and Zhu YZ:
Hydrogen sulfide attenuated tumor necrosis factor-α-induced
inflammatory signaling and dysfunction in vascular endothelial
cells. PLoS One. 6:e197662011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Snijder PM, Frenay AS, De Boer RA, Pasch
A, Hillebrands JL, Leuvenink HG and van Goor H: Exogenous
administration of thiosulfate, a donor of hydrogen sulfide,
attenuates angiotensin II-induced hypertensive heart disease in
rats. Br J Pharmacol. 172:1494–1504. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chaudhari N, Talwar P, Parimisetty A,
d'Hellencourt C Lefebvre and Ravanan P: A molecular web:
Endoplasmic reticulum stress, inflammation, and oxidative stress.
Front Cell Neurosci. 8:2132014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Song XJ, Yang CY, Liu B, Wei Q, Korkor MT,
Liu JY and Yang P: Atorvastatin inhibits myocardial cell apoptosis
in a rat model with post-myocardial infarction heart failure by
downregulating ER stress response. Int J Med Sci. 8:564–572. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qabazard B, Ahmed S, Li L, Arlt VM, Moore
PK and Stürzenbaum SR: C. elegans aging is modulated by hydrogen
sulfide and the sulfhydrylase/cysteine synthase cysl-2. PLoS One.
8:e801352013. View Article : Google Scholar : PubMed/NCBI
|