Introduction
The heart is the first organ to form during
embryogenesis and a sufficient supply of oxygen and nutrients
through the circulatory system is an essential prerequisite for
embryonic growth and survival (1).
The formation of a fully functional four-chambered heart from the
early embryo involves a precisely coordinated process of cellular
differentiation and integrated multi-cellular morphogenesis, and
even slight perturbations of this biological process may lead to
cardiac developmental abnormalities, which is highlighted by the
fact that congenital heart disease (CHD) is the most common form of
birth defect in humans, with an estimated prevalence of 1% in live
births, and up to 10% of stillbirths (1–4).
Congenital heart defects can be classified into two major
categories of morphological malformations and functional anomalies,
including cardiac arrhythmias and cardiomyopathies. Cardiac
structural deformities are a leading contributor to infant
morbidity and mortality rates, whereas defects in the development
of cardiac conduction system confer a significantly increased risk
of mortality throughout life (5).
Although the genetic basis underpinning a number of these defects
remains to be elucidated, the core cardiac transcription factors,
which are expressed predominantly in the heart and mediate the
expression of genes encoding cardiac structural proteins or
regulatory proteins, are increasingly recognized as being important
in the normal development of the heart (6). Mutations in certain genes encoding
core cardiac transcription factors, including
homeodomain-containing NK2 homeobox 5 (NKX2.5) and NKX2.6 (7–16),
GATA-binding protein (GATA) 4, GATA5 and GATA6 (17–24),
T-box protein (TBX) 5 and TBX20 (25–29),
and a paired-like homeobox transcription factor, PITX2 (30–33),
have emerged as major contributors to several types of congenital
heart defects (2,5,6,34).
During the last decade, increasing evidence has
demonstrated the essential role of the NKX2.5 transcription factor
in embryonic cardiogenesis and postnatal cardiac adaptation,
particularly in the development of the cardiac conduction system
(1). In mice, NKX2.5 is expressed
at high levels in the early heart progenitor cells of primary and
secondary heart fields during embryonic morphogenesis, and
continues to be expressed at a high level in the heart through
adulthood (1). Of note, the
expression of NKX2.5 is transiently elevated in specialized
myocardial conduction cells during formation of the conduction
system, indicating the importance of NKX2.5 in the development of
the conduction system (1). In
murine embryos completely null for Nkx2.5, embryonic death
occurred at ~E9-10 due to arrested looping morphogenesis of the
heart tube and growth retardation (35,36).
In addition, in Nkx2.5-knockoutmice, the number of cells in
the cardiac conduction system were found to be directly associated
with Nkx2.5 gene dosage, with homozygous mutant embryos
lacking the primordium of the atrioventricular node (37). By contrast, mice homozygous for a
ventricle-restricted knockout of Nkx2.5 showed no
cardiovascular structural defects, but exhibited marked overgrowth
of trabecular muscle and progressive complete heart block owing to
hypoplastic atrioventricular node at birth (38). In humans, mutations in
NKX2.5 have been associated with a wide spectrum of
cardiovascular diseases, including congenital heart defects, such
as atrial septal defect (ASD), ventricular septal defect, tetralogy
of Fallot, bicuspid aortic valve, mitral valve deformations,
subvalvular aortic stenosis, abnormal systemic venous return,
hypoplasia of left heart and visceral situs inversus (2,5–9,39),
dilated cardiomyopathy (10),
arrhythmias, such as atrioventricular block (AVB), atrial
fibrillation and ventricular tachycardia (10–12,34,40–42),
and sudden cardiac death (12,43,44),
of which ASD and AVB are the two most frequent phenotypes in
patients carrying NKX2.5 mutations. Of note, in a murine knock-in
model generated by knocking in a comparable NKX2.5 missense
mutation (p.R52G) previously identified in patients, pleiotropic
cardiac anomalies, including ASD, ventricular septal defect,
atrioventricular septal defect, Ebstein malformation of the
tricuspid valve and ventricular noncompaction were observed, in
addition to progressive AVB, and this was similar or more marked,
compared with the cardiac anomalies found in humans harboring a
heterozygous mutation of p.R52 G in NKX2.5 (45,46).
These observational results suggest that NKX2.5 mutations
are responsible for ASD and AVB, and evaluation of the prevalence
and spectrum of NKX2.5 mutations in patients with congenital
ASD and AVB is warranted. This study was sought to evaluate the
prevalence and spectrum of NKX2.5 mutations in patients with
congenital ASD and AVB.
Materials and methods
Study population
In the present study, 62 unrelated patients with
congenital ASD and AVB were recruited from the Chinese Han
population, who were admitted to Shanghai Chest Hospital from
January 2011 to December 2013. The available family members of the
index patient carrying an identified NKX2.5 mutation were also
included. In addition, 300 unrelated healthy individuals, who were
matched to the patients with ASD and AVB in ethnicity, gender and
age, were recruited as controls. All study subjects underwent
detailed clinical investigation, including familial, personal and
medical histories, physical examination, routine laboratory tests
in hospitals, transthoracic echocardiography, and standard 12-lead
electrocardiogram. The clinical types of CHDs were determined using
two-dimensional continuous wave Doppler and color Doppler
techniques on transthoracic echocardiography. When there was a
strong indication, transesophageal echocardiography, cardiac
catheterization and angiography were performed to further clarify
the cardiovascular anatomic malformations. The definition and
classification of AVB, which are widely used clinically and are
based on the extent (first, second or third degree) of a block and
the site within the conduction system of a block, were described
previously (47). In brief,
first-degree AVB is referred to as prolongation of the PR interval,
which is the time interval between onset of P-wave and onset of QRS
complex, on the surface electrocardiogram. In second-degree AVB,
certain atrial impulses are conducted to the ventricles;
electrocardiographic manifestation is the association of certain
QRS complexes with P waves. Second-degree AVB is further classified
as type 1 or type 2. In type 1 block, progressive prolongation of
the PR interval is observed prior to the occurrence of AVB
(Wenckebach periodicity). In type 2, the block occurs abruptly
without PR interval prolongation. In third-degree or complete AVB,
no atrial impulses conduct to the ventricles; on the
electrocardiogram, QRS complexes are independent of P waves. In
certain cases, AVB is described as progressive if the
electrocardiographic characterization of atrioventricular
conduction worsens over time, for example when the block progresses
from second to third degree (47).
Patients with ASD who suffered from AVB following cardiac surgery
or intervention for CHD, or had chromosomal abnormalities or
syndromic cardiovascular anomalies, including Holt-Oram syndrome,
Di George syndrome, Alagille syndrome and Axenfeld-Rieger syndrome,
were excluded from the present study. The present study was
performed in accordance with the principles outlined in the
Declaration of Helsinki. The study protocol was reviewed and
approved by the Ethics Committee of Shanghai Chest Hospital,
Shanghai Jiao Tong University (Shanghai, China). Written informed
consent was obtained from all adult participants and from parents
of minors prior to the investigation.
Mutational analysis of NKX2.5
Peripheral venous blood samples (3 ml) were
collected from the patients and control individuals. Genomic DNA
was extracted from blood leukocytes (for most steps in the DNA
purification procedure, centrifugation at a speed of 2,000 × g for
10 min at −4°C) using a Wizard Genomic DNA Purification kit
(Promega, Madison, WI, USA), according to the manufacturer's
protocol. With the assistance of online Primer 3 software
(http://bioinfo.ut.ee/primer3-0.4.0/), the primers used
to amplify the coding exons and flanking introns of NKX2.5
by polymerase chain reaction (PCR) were designed as shown in
Table I. The referential genomic
DNA sequence of NKX2.5 was derived from GenBank (https://www.ncbi.nlm.nih.gov/nuccore/NG_013340.1;
accession no. NG_01,3340.1). PCR was performed with HotStar
Taq DNA polymerase (Qiagen GmbH) on a Veriti thermal cycle
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). The total volume of a PCR mixture was 25 µl, encompassing 2.5
µl 10X buffer, 11.25 µl deionized water, 5 µl 5X Q solution, 1 µl
of each primer at 20 µM, 2 µl of dNTPs (2.5 mM each), 0.25 µl of
HotStar Taq DNA Polymerase at 5 U/µl (Qiagen GmbH) and 2 µl of
genomic DNA at 200 ng/µl. The PCR cycling parameters were as
follows: Pre-denaturation of template and activation of the DNA
polymerase at 95°C for 15 min, followed by 35 cycles of
denaturation at 94°C for 30 sec, annealing at 62°C for 30 sec and
extension at 72°C for 1 min, with a final extension at 72°C for 6
min. The PCR-generated amplicons were purified and sequenced with
NKX2.5-specific primers using a BigDye®
Terminator v3.1 Cycle Sequencing kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) under an ABI PRISM 3130 XL DNA analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). For an
identified sequence variation in NKX2.5, its numbering began
from nucleotide A of the initial translation codon ATG (accession
no. NM_004387.3). To verify the novelty of an identified
NKX2.5 sequence variation, the single nucleotide
polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) database, the 1000
Genomes Project (1000 G; http://www.1000genomes.org) database, the exome
variant server (EVS; http://evs.gs.washington.edu/EVS) database and the
human genome mutation (HGM; http://www.hgmd.org/) database were queried.
 | Table I.Primers for amplification of coding
exons and flanking introns of NK2 homeobox 5. |
Table I.
Primers for amplification of coding
exons and flanking introns of NK2 homeobox 5.
| Exon | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Amplicon size
(bp) |
|---|
| 1 |
CTTGTGCTCAGCGCTACCTG |
TCTTGGGGACGAAAGCGACC | 543 |
| 2-a |
CCGTAGGTCAAGCCGCTCTT |
ACGAAGTTGTTGTTGGCGGC | 599 |
| 2-b |
GCGTGCTGAAACTCACGTCC |
GGTCATGTTGGGAGCCCCTT | 545 |
Alignment of multiple NKX2.5 proteins
across species
The NKX2.5 protein sequences of humans were aligned
with those of chimpanzee, monkey, dog, cattle, mouse, rat, fowl,
zebrafish and frog using the online MUSCLE program (version, 3.6;
http://www.ncbi.nlm.nih.gov/homologene?cmd=Retrieve&dopt=MultipleAlignment&list_uids=3230).
Expression plasmids and site-directed
mutagenesis
The recombinant expression plasmidsNKX2.5-pEFSA and
GATA4-pSSRa, and the atrial natriuretic factor (ANF)-luciferase;
ANF-luc) reporter plasmid, which contained 2,600 base pairs
upstream of the transcriptional start site of the ANF gene
and expresses Firefly luciferase, were provided by Dr Ichiro
Shiojima at the Department of Cardiovascular Science and Medicine,
Chiba University Graduate School of Medicine (Chiba, Japan). The
identified mutation was introduced into the wild-type NKX2.5-pEFSA
construct by site-directed mutagenesis using a complementary pair
of primers (forward, 5′-AAACTCACGTCCACGTAGGTCAAGATCTGGT-3′;
reverse, 5′-ACCAGATCTTGACCTACGTGGACGTGAGTTT-3′) and the Quick
Change II XL Site-Directed Mutagenesis kit (Stratagene; Agilent
Technologies, Santa Clara, CA, USA), and confirmed by direct
sequencing.
Cell culture, transfection and
reporter gene assays
COS-7 cells, which were obtained from the cell bank
at the Cardiovascular Research Laboratory of Shanghai Chest
Hospital (Shanghai, China). The cells were cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 µg/ml penicillin and 100 µg/ml streptomycin
under an atmosphere of 5% CO2 at 37°C. Transient
transfections of cells at ~90% confluence were performed with
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) 24 h post-seeded at a cell density of about
1×105 per well in 12-well plates. The internal control
pGL4.75 plasmid (Promega Corp.), which expresses Renilla
luciferase, was used in transfection assays to normalize
transfection efficiency. The COS-7 cells were transfected with 0.4
µg wild-type or Q181X-mutant NKX2.5-pEFSA, in combination with 1.0
µg ANF-luc and 0.04 µg pGL4.75. For co-transfection experiments,
0.2 µg wild-type NKX2.5-pEFSA, 0.2 µg of Q181X-mutant NKX2.5-pEFSA,
1.0 µg of ANF-luc and 0.04 µg of pGL4.75 were used. For analysis of
the synergistic transcriptional activation between NKX2.5 and
GATA4, the COS-7 cells were transfected with the same quantity (0.2
µg) of each plasmid DNA (wild-type NKX2.5-pEFSA, GATA4-pSSRa, or
Q181X-mutant NKX2.5-pEFSA) was used alone or in combination, in the
presence of 1.0 µg ANF-luc and 0.04 µg pGL4.75. At 6 h
post-transfection, the media was replaced with growth media and
cells were maintained for 48 h. The transfected cells were washed
twice with PBS and lysed using PLB buffer (Promega Corp.) for
incubation at room temperature for 15 min. The Firefly and
Renilla luciferase activities were measured using a
Dual-Luciferase Reporter Assay system (Promega Corp.) according to
the manufacturer's protocol. The activity of the ANF
promoter was determined as the fold activation of Firefly
luciferase relative to Renilla luciferase. Three independent
experiments were performed in triplicate for each cell
transfection, and each value presented as the average of triplicate
samples.
Statistical analysis
Statistical analyses were performed using SPSS
version 18.0 (SPSS, Inc., Chicago, IL, USA). Continuous variables
with normal distribution are expressed as mean and standard
deviations. Categorical variables are expressed as numbers and
percentages. Differences between two groups were compared using
Student's unpaired t-test for continuous variables, and with the
χ2 test or Fisher's exact test for categorical
variables. P<0.05 (two-tailed) was considered to indicate a
statistically significant difference.
Results
Clinical characteristics of the study
population
In the present study, 62 unrelated patients with ASD
and AVB were clinically evaluated for comparison with 300 unrelated
healthy individuals. All patients had congenital ASD, confirmed by
echocardiogram, and AVB, confirmed by electrocardiogram. There were
six patients who also had a positive family history of ASD and AVB.
Based on their medical histories, echocardiographic records and
electrocardiographic results, the control individuals had neither
CHD nor AVB. None of the control individuals had a family history
of ASD or AVB. No statistical differences were found in ethnicity,
gender or age between the patient and control groups. The baseline
clinical characteristics of the study population are shown in
Table II.
| Table II.Baseline clinical characteristics of
the study population. |
Table II.
Baseline clinical characteristics of
the study population.
| Variable | Patients (n=62) n
(%) | Controls (n=300) n
(%) | P-value |
|---|
| Age (years) | 31±15 | 32±7 |
0.4200 |
| Male | 39 (63) | 190 (63) |
0.9490 |
| Family history of
ASD and AVB | 6 (10) | 0 (0) | <0.0001 |
| History of
syncope | 15 (24) | 0 (0) | <0.0001 |
| Implanted cardiac
pacemaker | 18 (29) | 0 (0) | <0.0001 |
| Surgical or
catheter-based ASD closure | 41 (66) | 0 (0) | <0.0001 |
| Distribution of
types of ASD |
| Ostium secundum
ASD | 51 (82) | 0 (0) | <0.0001 |
| Ostium primum
ASD | 9 (15) | 0 (0) | <0.0001 |
| Sinus venosus
ASD | 2 (3) | 0 (0) |
0.0289 |
| Distribution of
types of AVB |
| First-degree
AVB | 34 (55) | 0 (0) | <0.0001 |
| Second-degree
AVB | 12 (19) | 0 (0) | <0.0001 |
| Third-degree
AVB | 16 (26) | 0 (0) | <0.0001 |
Identification of a novel NKX2.5
mutation
Sequence analysis of the NKX2.5 gene in 62
unrelated patients with ASD and AVB revealed a heterozygous
substitution of thymine for cytosine in the first nucleotide of
codon 181 (c.541C>T) in the index patient; this mutation was
predicted to introduce a stop codon at amino acid 181, resulting in
the production of a truncated protein with only the
NH2-terminal 180 amino acids remaining (p.Q181X). The
DNA sequencing electropherograms showing the NKX2.5 mutation
of c.541C>T and its control sequence are shown in Fig. 1A. A schematic diagram of the NKX2.5
protein showing the functionally important structural domains and
the location of the mutation identified in the present study is
shown in Fig. 1B. The nonsense
mutation was not detected in the 300 control individuals or in the
SNP, 1000 G, EVS and HGM databases. Genotyping NKX2.5 in the
proband's available family members showed that the heterozygous
mutation was present in all three affected family members and
absent in all four unaffected family members. Genetic analysis of
the proband's pedigree unveiled that the mutation co-segregated
with ostium secundum ASD and progressive AVB, which were
transmitted in an autosomal dominant pattern with complete
penetrance. In addition, the proband's father had ventricular
septal defect and atrial fibrillation, and succumbed to mortality
at the age of 60 years. The proband's younger sister also had an
incomplete right bundle branch block. All the living affected
family members underwent catheter-based closure of ASD. The
pedigree structure of the family is illustrated in Fig. 1C. The clinical features of the
affected family members are shown in Table III.
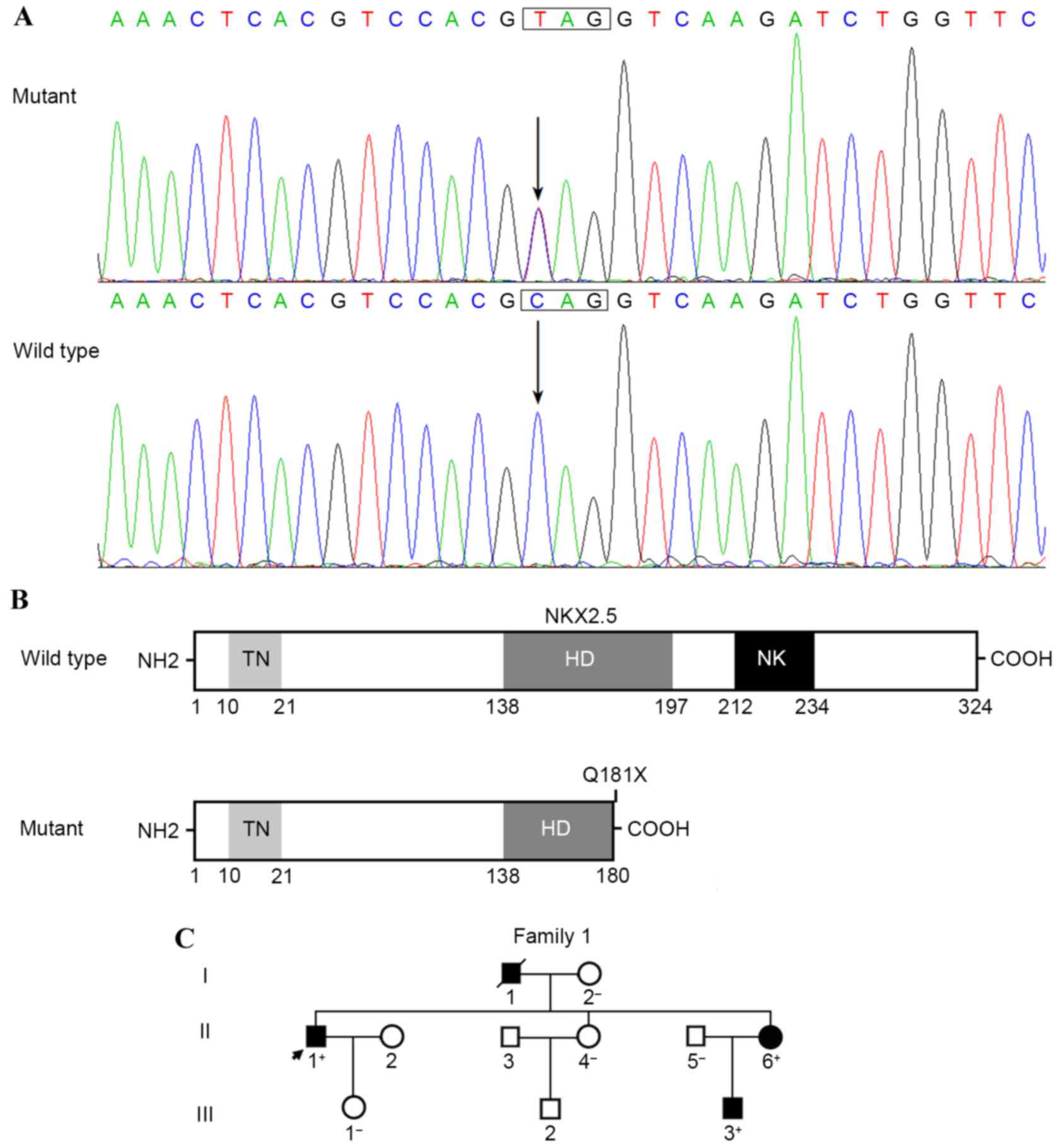 | Figure 1.A novel NKX2.5 mutation associated
with familial ASD and AVB. (A) Sequence electropherograms show the
heterozygous NKX2.5 mutation and its wild-type control. The
arrow indicates the heterozygous nucleotides of C/T in the index
patient with congenial ASD and AVB (mutant) or the homozygous
nucleotides of C/C in the corresponding control subject
(wild-type). The rectangle marks the nucleotides that comprise a
codon of NKX2.5. (B) Schematic diagram illustrating the
structural domains of NKX2.5. The mutation identified in patients
with congenial ASD and AVB is shown above the truncated protein
(mutant). (C) Pedigree structure of a family with congenial ASD and
AVB, designated as family 1. Family members are identified as
generations and numbers. Squares, male family members; circles,
female family members; closed symbols, affected; open symbols,
unaffected; symbol with a slash, deceased member; arrow, proband;
+, carriers of the heterozygous mutation; -, non-carriers; NKX2.5,
NK2 homeobox 5; NH2, amino-terminus; TN, tinman domain; HD,
homeodomain; NK, nucleotide kinase domain; COOH, carboxyl-terminus;
ASD, atrial septal defect; AVB, atrioventricular block. |
| Table III.Clinical features and mutational
status of NKX2.5 in affected family members. |
Table III.
Clinical features and mutational
status of NKX2.5 in affected family members.
| Individual | Gender | Age (years) | Cardiac structural
defects | Cardiac
arrhythmias | Q181X mutation |
|---|
| I-1 | M | 60a | ASD, VSD | III0
AVB, AF | NA |
| II-1 | M | 47 | ASD | II0
AVB | +/− |
| II-6 | F | 41 | ASD | II0 AVB,
IRBBB | +/− |
| III-3 | M | 16 | ASD | I0
AVB | +/− |
Alignment of the NKX2.5 proteins from
various species
Alignment of the human NKX2.5 protein with those of
chimpanzee, monkey, dog, cattle, mouse, rat, fowl, zebrafish and
frog showed that the altered amino acid, p.Q181, was completely
conserved evolutionarily (Fig.
2).
Functional failure of NKX2.5 protein
resulted from mutation
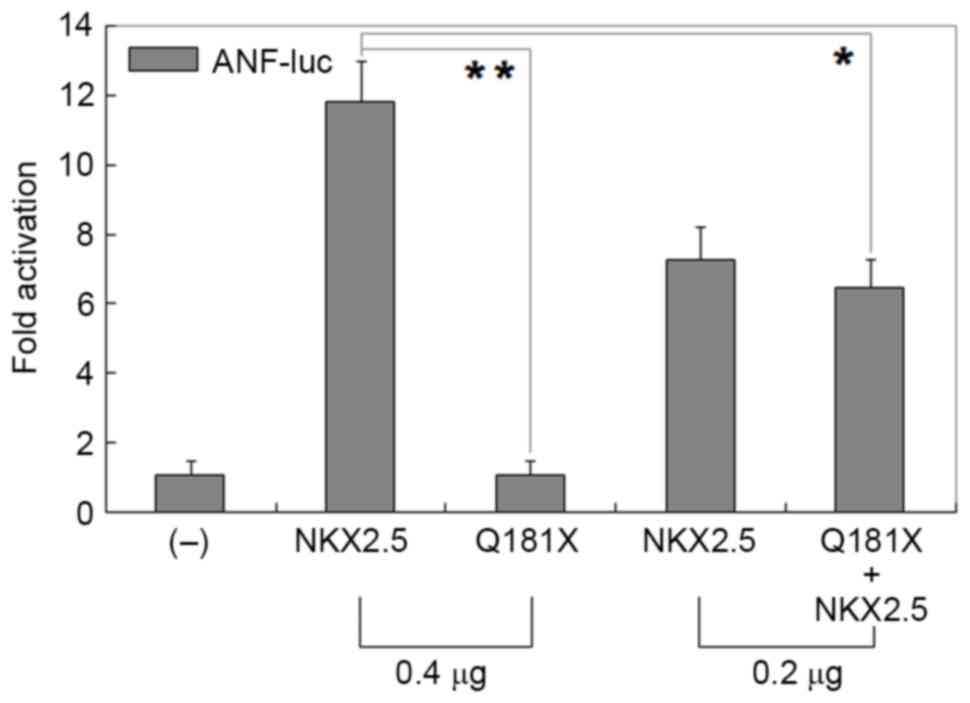
As shown in Fig. 3,
the same quantity (0.4 µg) of wild-type and Q181X-mutant NKX2.5
transcriptionally activated the C_ItalicsCleanCSS_12>ANF
promoter by ~12-fold and ~1-fold, respectively (wild-type, vs.
mutant; t=15.0303, P=0.0001); whereas 0.2 µg of wild-type NKX2.5
with 0.2 µg Q181X-mutant NKX2.5 activated the ANF promoter
by ~6-fold (0.4 µg of wild-type, vs. 0.2 µg mutant + 0.2 µg
wild-type; t=6.5294, P=0.0028).
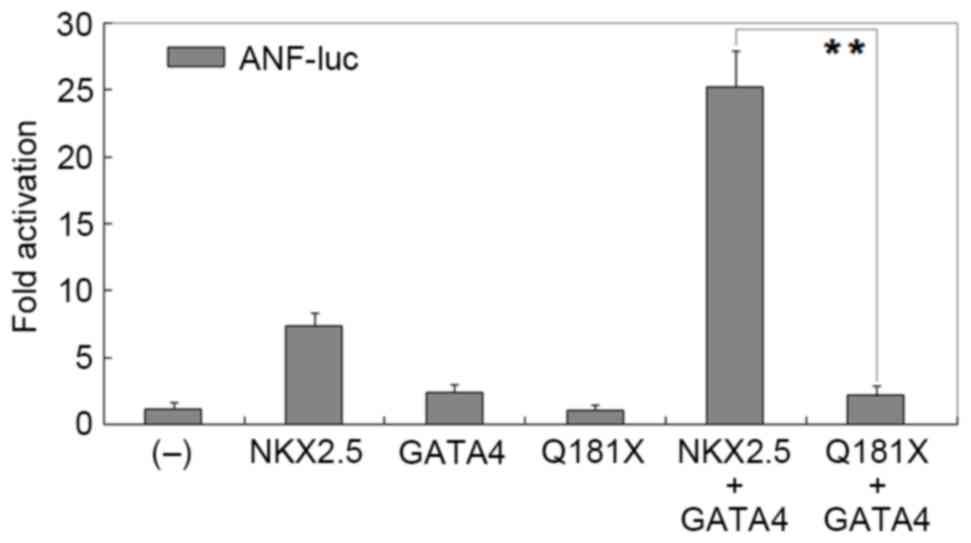
Disrupted synergistic activation
between NKX2.5 and GATA4 by the mutation
As shown in Fig. 4,
the same quantity (0.2 µg) of wild-type NKX2.5, GATA4 and
Q181X-mutant NKX2.5 activated the ANF promoter by ~7-fold,
~2-fold and ~1-fold, respectively. In the presence of 0.2 µg
wild-type GATA4, the same quantity (0.2 µg) of wild-type and
Q181X-mutant NKX2.5 activated the ANF promoter by ~25-fold
and ~2-fold, respectively (wild-type, vs. mutant; t=14.1488,
P=0.0001).
Discussion
In the present study, a novel heterozygous NKX2.5
mutation of p.Q181X was identified in a family with congenital ASD
with AVB. The nonsense mutation was absent in 600 control
chromosomes from a control population matched for ethnicity, gender
and age. The mutation was predicted to yield a truncated protein
lacking multiple functionally important domains. Reporter gene
assays demonstrated that the Q181X-mutant NKX2.5 protein showed
loss of transcriptional activity. Furthermore, the Q181X mutation
abrogated the synergistic activation between NKX2.5 and GATA4.
Therefore, it is possible that genetically compromised
NKX2.5contributed to ASD and AVB in these mutation carriers.
In humans, the NKX2.5 gene is mapped to
chromosome 5q34, coding for a protein of 324 amino acids, which is
expressed at a high level in the human heart (11). The NKX2.5 protein contains an
evolutionarily conserved homeodomain (HD), which is centrally
located at amino acid positions 138–197, and functions to
specifically recognize and bind to a consensus DNA motif, AAGTG,
which is key in the transcriptional regulation of target genes,
including ANF, brain natriuretic peptide and α-actin, either
alone or in synergy with other transcription factors, including
GATA4, TBX5, TBX20, heart and neural crest derivatives expressed 2
and paired-like homeodomain 2 (11,19,26,29,31,48,49).
The NKX2.5 mutation of p.Q181X identified in the present study was
located in the HD, and functional analyses revealed that the mutant
protein showed loss of the ability to transcriptionally activate
ANF, alone or together with GATA4. These findings supported
NKX2.5 haploinsufficiency as an alternative pathogenic
mechanism of ASD and AVB.
ASD is the second most common form of CHD with an
estimated incidence of 1/1,500 live births worldwide, accounting
for ~10% of all CHDs (43,44). Although small ASDs may close
spontaneously during infancy or childhood, large ASDs or those
remaining open into adulthood may cause a significant volume of
blood being shunted from the left side of the heart to the right,
leading to pneumonia, embolism, pulmonary vascular disease,
congestive heart failure, atrial arrhythmias and sudden cardiac
death (43). In addition, familial
AVB has been observed as congenital or adult-onset type to occur
together with ASD (43).
Congenital complete AVB is associated with mortality rates ranging
between 33 and 80% if heart rate is <50 beats per minute or it
co-occurs with structural heart diseases; whereas the adult-onset
type of familial AVB is of a progressive nature, and there are
several reports of patients with normal electrocardiogram or a
harmless first-degree AVB followed by sudden onset of second- and
third-degree AVB or sudden cardiac death later in life (12,43,44).
Therefore, identification of a novel NKX2.5 mutation
underpinning ASD and AVB is of important clinical significance,
particularly for genetic counseling in patients carrying the
NKX2.5 mutation.
To date, >60 mutations in NKX2-5 have been
reported to be associated with a wide range of cardiac phenotypes,
of which ASD and AVB are the most commonly reported phenotypes, in
~68 and 66% of patients, respectively (43,44).
In addition, >97% of familial cases present with clinically
documented ASD and AVB (44). Of
note, of all reported non-synonymous mutations, ~1/3 occur within
the HD region (44). Functional
deciphers of these NKX2.5 mutations have shown reduced
transcriptional activity, alone or in synergy with its cooperative
partners (44), similar to the
present study. Additionally, the results of the present and
previous studies (43,44) suggest that there appears to be a
genotype-phenotype correlation of NKX2.5 mutations. All
mutations causing truncations and all missense mutations in the HD
region result in ASD with AVB.
In conclusion, the present study expands the
mutational spectrum of NKX2.5 linked to ASD and AVB, providing
additional evidence that the NKX2.5 loss-of-function mutation is an
uncommon cause of ASD and AVB.
Acknowledgements
This study was supported in part by grants from the
Key Program for Basic Research of Shanghai, China (grant no.
14JC1405500), the National Natural Science Fund of China (grant
nos. 81270161, 81470372 and 81400244), the Natural Science Fund of
Shanghai, China (grant nos. 13ZR1438400, 14ZR1438000 and
15ZR1438100) and the Major Development Program for Science and
Technology of Shanghai Chest Hospital, Shanghai, China (grant nos.
2014YZDH10102 and 2014YZDH20500).
References
|
1
|
Akazawa H and Komuro I: Cardiac
transcription factor Csx/Nkx2-5: Its role in cardiac development
and diseases. Pharmacol Ther. 107:252–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fahed AC, Gelb BD, Seidman JG and Seidman
CE: Genetics of congenital heart disease: The glass half empty.
Circ Res. 112:707–720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marelli AJ, Ionescu-Ittu R, Mackie AS, Guo
L, Dendukuri N and Kaouache M: Lifetime prevalence of congenital
heart disease in the general population from 2000 to 2010.
Circulation. 130:749–756. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mozaffarian D, Benjamin EJ, Go AS, Arnett
DK, Blaha MJ, Cushman M, De Ferranti S, Després JP, Fullerton HJ,
Howard VJ, et al: Heart disease and stroke statistics-2015 update:
A report from the American Heart Association. Circulation.
131:e29–e322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Andersen TA, Troelsen K, de L and Larsen
LA: Of mice and men: Molecular genetics of congenital heart
disease. Cell Mol Life Sci. 71:1327–1352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM,
Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al: A novel NKX2.5
loss-of-function mutation associated with congenital bicuspid
aortic valve. Am J Cardiol. 114:1891–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Izumi K, Noon S, Wilkens A and Krantz ID:
NKX2.5 mutation identification on exome sequencing in a patient
with heterotaxy. Eur J Med Genet. 57:558–561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q,
Wang J and Sun K: Investigation of somatic NKX2-5 mutations in
Chinese children with congenital heart disease. Int J Med Sci.
12:538–543. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu
YJ, Liu X, Fang WY, Yang YQ and Liao DN: A novel NKX2-5
loss-of-function mutation predisposes to familial dilated
cardiomyopathy and arrhythmias. Int J Mol Med. 35:478–486.
2015.PubMed/NCBI
|
|
11
|
Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang
J, Li RG, Xu L, Jiang WF, Qiu XB, et al: Mutational spectrum of the
NKX2-5 gene in patients with lone atrial fibrillation. Int J Med
Sci. 11:554–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perera JL, Johnson NM, Judge DP and
Crosson JE: Novel and highly lethal NKX2.5 missense mutation in a
family with sudden death and ventricular arrhythmia. Pediatr
Cardiol. 35:1206–1212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ta-Shma A, El-lahham N, Edvardson S,
Stepensky P, Nir A, Perles Z, Gavri S, Golender J,
Yaakobi-Simhayoff N, Shaag A, et al: Conotruncal malformations and
absent thymus due to a deleterious NKX2-6 mutation. J Med Genet.
51:268–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Zhang DF, Sun YM, Li RG, Qiu XB,
Qu XK, Liu X, Fang WY and Yang YQ: NKX2-6 mutation predisposes to
familial atrial fibrillation. Int J Mol Med. 34:1581–1590.
2014.PubMed/NCBI
|
|
15
|
Zhao L, Ni SH, Liu XY, Wei D, Yuan F, Xu
L, Xin Li, Li RG, Qu XK, Xu YJ, et al: Prevalence and spectrum of
Nkx2.6 mutations in patients with congenital heart disease. Eur J
Med Genet. 57:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Mao JH, Ding KK, Xu WJ, Liu XY,
Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, et al: A novel NKX2.6
mutation associated with congenital ventricular septal defect.
Pediatr Cardiol. 36:646–656. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.PubMed/NCBI
|
|
19
|
Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG
and Yang YQ: Prevalence and spectrum of GATA4 mutations associated
with sporadic dilated cardiomyopathy. Gene. 548:174–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI
|
|
21
|
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu
L, Liu H, Li RG, Xu YJ, Wang Q, et al: GATA5 loss-of-function
mutations associated with congenital bicuspid aortic valve. Int J
Mol Med. 33:1219–1226. 2014.PubMed/NCBI
|
|
22
|
Zhang XL, Dai N, Tang K, Chen YQ, Chen W,
Wang J, Zhao CM, Yuan F, Qiu XB, Qu XK, et al: GATA5
loss-of-function mutation in familial dilated cardiomyopathy. Int J
Mol Med. 35:763–770. 2015.PubMed/NCBI
|
|
23
|
Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R,
Chen S and Sun K: Identification of two novel GATA6 mutations in
patients with nonsyndromic conotruncal heart defects. Mol Med Rep.
10:743–748. 2014.PubMed/NCBI
|
|
24
|
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li
RG, Xu YJ, Zhang M, Fang WY, Qu XK, et al: GATA6 loss-of-function
mutations contribute to familial dilated cardiomyopathy. Int J Mol
Med. 34:1315–1322. 2014.PubMed/NCBI
|
|
25
|
Baban A, Postma AV, Marini M, Trocchio G,
Santilli A, Pelegrini M, Sirleto P, Lerone M, Albanese SB, Barnett
P, et al: Identification of TBX5 mutations in a series of 94
patients with tetralogy of Fallot. Am J Med Genet A.
164A:3100–3107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM,
Li RG, Xu L, Xu YJ, Shi HY, Hou XM, et al: TBX5 loss-of-function
mutation contributes to familial dilated cardiomyopathy. Biochem
Biophys Res Commun. 459:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang
YQ and Qiu XB: A novel TBX5 loss-of-function mutation associated
with sporadic dilated cardiomyopathy. Int J Mol Med. 36:282–288.
2015.PubMed/NCBI
|
|
28
|
Pan Y, Geng R, Zhou N, Zheng GF, Zhao H,
Wang J, Zhao CM, Qiu XB, Yang YQ and Liu XY: TBX20 loss-of-function
mutation contributes to double outlet right ventricle. Int J Mol
Med. 35:1058–1066. 2015.PubMed/NCBI
|
|
29
|
Zhao CM, Bing Sun, Song HM, Wang J, Xu WJ,
Jiang JF, Qiu XB, Yuan F, Xu JH and Yang YQ: TBX20 loss-of-function
mutation associated with familial dilated cardiomyopathy. Clin Chem
Lab Med. 54:325–332. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wei D, Gong XH, Qiu G, Wang J and Yang YQ:
Novel PITX2c loss-of-function mutations associated with complex
congenital heart disease. Int J Mol Med. 33:1201–1208.
2014.PubMed/NCBI
|
|
31
|
Zhao CM, Peng LY, Li L, Liu XY, Wang J,
Zhang XL, Yuan F, Li RG, Qiu XB and Yang YQ: PITX2 loss-of-function
mutation contributes to congenital endocardial cushion defect and
Axenfeld-Rieger syndrome. PLoS One. 10:e01244092015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Zhang DF, Sun YM and Yang YQ: A
novel PITX2c loss-of-function mutation associated with familial
atrial fibrillation. Eur J Med Genet. 57:25–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiu XB, Xu YJ, Li RG, Xu L, Liu X, Fang
WY, Yang YQ and Qu XK: PITX2C loss-of-function mutations
responsiblefor idiopathic atrial fibrillation. Clinics (Sao Paulo).
69:15–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hong K and Xiong Q: Genetic basis of
atrial fibrillation. Curr Opin Cardiol. 29:220–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lyons I, Parsons LM, Hartley L, Li R,
Andrews JE, Robb L and Harvey RP: Myogenic and morphogenetic
defects in the heart tubes of murine embryos lacking the homeo box
gene Nkx2-5. Genes Dev. 9:1654–1666. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tanaka M, Chen Z, Bartunkova S, Yamasaki N
and Izumo S: The cardiac homeobox gene Csx/Nkx2.5 lies genetically
upstream of multiple genes essential for heart development.
Development. 126:1269–1280. 1999.PubMed/NCBI
|
|
37
|
Jay PY, Harris BS, Maguire CT, Buerger A,
Wakimoto H, Tanaka M, Kupershmidt S, Roden DM, Schultheiss TM,
O'Brien TX, et al: Nkx2-5 mutation causes anatomic hypoplasia of
the cardiac conduction system. J Clin Invest. 113:1130–1137. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pashmforoush M, Lu JT, Chen H, Amand TS,
Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, et
al: Nkx2-5 pathways and congenital heart disease; loss of
ventricular myocyte lineage specification leads to progressive
cardiomyopathy and complete heart block. Cell. 117:373–386. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lalani SR and Belmont JW: Genetic basis of
congenital cardiovascular malformations. Eur J Med Genet.
57:402–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gutierrez-Roelens I, De Roy L, Ovaert C,
Sluysmans T, Devriendt K, Brunner HG and Vikkula M: A novel
CSX/NKX2-5 mutation causes autosomal-dominant AV block: Are atrial
fibrillation and syncopes part of the phenotype? Eur J Hum Genet.
14:1313–1316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reamon-Buettner SM and Borlak J: NKX2-5:
An update on this hypermutable homeodomain protein and its role in
human congenital heart disease (CHD). Hum Mutat. 31:1185–1194.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Baruteau AE, Probst V and Abriel H:
Inherited progressive cardiac conduction disorders. Curr Opin
Cardiol. 30:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ellesøe SG, Johansen MM, Bjerre JV,
Hjortdal VE, Brunak S and Larsen LA: Familial atrial septal defect
and sudden cardiac death: Identification of a novel NKX2-5 mutation
and a review of the literature. Congenit Heart Dis. 11:283–290.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hassan OK Abou, Fahed AC, Batrawi M, Arabi
M, Refaat MM, DePalma SR, Seidman JG, Seidman CE, Bitar FF and
Nemer GM: NKX2-5 mutations in an inbred consanguineous population:
Genetic and phenotypic diversity. Sci Rep. 5:88482015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ashraf H, Pradhan L, Chang EI, Terada R,
Ryan NJ, Briggs LE, Chowdhury R, Zárate MA, Sugi Y, Nam HJ, et al:
A mouse model of human congenital heart disease: High incidence of
diverse cardiac anomalies and ventricular noncompaction produced by
heterozygous Nkx2-5 homeodomain missense mutation. Circ Cardiovasc
Genet. 7:423–433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chowdhury R, Ashraf H, Melanson M, Tanada
Y, Nguyen M, Silberbach M, Wakimoto H, Benson DW, Anderson RH and
Kasahara H: Mouse model of human congenital heart disease:
Progressive atrioventricular block induced by a heterozygous Nkx2-5
homeodomain missense mutation. Circ Arrhythm Electrophysiol.
8:1255–1264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Benson DW: Genetics of atrioventricular
conductiondisease in humans. Anat Rec A Discov Mol Cell Evol Biol.
280:934–939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu CX, Gong HR, Liu XY, Wang J, Zhao CM,
Huang RT, Xue S and Yang YQ: A novel HAND2 loss-of-function
mutation responsible for tetralogy of Fallot. Int J Mol Med.
37:445–451. 2016.PubMed/NCBI
|
|
49
|
Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li
RG, Qu XK, Huang RT, Xue S and Yang YQ: PITX2 loss-of-function
mutation contributes to tetralogy of Fallot. Gene. 577:258–264.
2016. View Article : Google Scholar : PubMed/NCBI
|