Introduction
Previous studies have demonstrated the importance of
mitochondria and oxidative stress in the development of neuronal
diseases, such as Parkinson's disease (PD) and Alzheimer's disease
(1,2). In human eyes, the retina is extremely
vulnerable to oxidative damage. Oxidative stress has been
demonstrated to be the predominant mechanism associated with the
pathogenesis of age-related macular degeneration (AMD), diabetic
retinopathy (DR), glaucoma and retinitis pigmentosa (RP) (3–6).
Novel strategies targeting mitochondria and reactive oxygen species
(ROS) generation may provide promising prospects towards
antioxidant therapy in these retinal diseases.
A novel mitochondria-targeted peptide, MTP-131 (also
known as SS-31 or Bendavia), originally designed by Hazel H. Szeto
and Peter W. Schiller (7), is a
water-soluble peptide with an alternating aromatic cationic motif
(D-Arg-dimethylTyr-Lys-Phe-NH2) and a 3+ net charge at
physiological pH (8). The fraction
of the peptide partitioned to mitochondria has been estimated to be
1,000 to 5,000-fold compared with the extra-mitochondrial
concentration, and the peptide is predominately localized to the
inner mitochondrial membrane (7).
MTP-131 reduces ROS production in a dose-dependent manner and
prevents oxidative damage in neuronal cell lines (9). In addition, treatment of mice with
MTP-131 prevents loss of neurons and increases survival in PD and
amyotrophic lateral sclerosis animal models (10).
Since MTP-131 has a unique mitochondria-targeted
delivery, and exhibits potent antioxidant and neuroprotective
effect both in vitro and in vivo, the present study
aimed to evaluate the protective effect of MTP-131 against hydrogen
peroxide (H2O2)-induced oxidative damage in
RGC-5 cells.
Materials and methods
Reagents and chemicals
MTP-131
(D-Arg-(2′6′-dimethylTyr)-Lys-Phe-NH2) was kindly
provided by Stealth BioTherapeutics, Inc. (Newton, MA, USA). Gibco
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum
(FBS) were obtained from Thermo Fisher Scientific (Waltham, MA,
USA). Lactate dehydrogenase (LDH) assay kit was obtained from Roche
Diagnostics Gmbh (Mannheim, Germany). Tetramethylrhodamine methyl
ester perchlorate (TMRM), 2′,7′-dichlorodihydrofluorescein
diacetate (H2DCFDA) and MitoTracker Deep Red FM were
obtained from Thermo Fisher Scientific, Inc. and prepared as 50 µM,
1 and 1 mM stock solutions in dimethyl sulfoxide, respectively.
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
apoptosis assay kit was from Multisciences Lianke Biotech Co., Ltd.
(Hangzhou, China). Unless specified, all other reagents were
obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Cell culture
The RGC-5 cell line was kindly provided by Dr Zhiqun
Tan (Department of Neurology, UC Irvine School of Medicine, Irvine,
CA, USA). Undifferentiated RGC-5 cells were used in the present
study. Cells were cultured in DMEM containing 10% FBS, and 1%
penicillin-streptomycin (100 U/ml penicillin and 100 µg/ml
streptomycin) at 37°C and 5% CO2. Cells were passaged by
trypsinization every 3 days. All cell culture dishes and plates
were obtained from BD Biosciences (Franklin Lakes, NJ, USA).
MTP-131 pretreatment and induction of
oxidative stress
RGC-5 cells were seeded at a density of 1×104
cells/well in 6-well plates and incubated in 5% CO2 at
37°C for 24 h. Cells at approximately 70% confluence were
pretreated with 0.01, 0.1 or 1 µM MTP-131 in serum-free DMEM at
37°C for 1 h and then rinsed twice with PBS. Then, RGC-5 cells were
exposed to 500 µM H2O2 in serum-free DMEM for
24 h to induce a sustained oxidative stress in vitro.
Untreated cells and cells treated with H2O2
alone were used as normal and H2O2 control,
respectively.
Measurement of cell viability
Cell viability was assessed by LDH assay using the
Cytotoxicity Detection kitPLUS (Roche Diagnostics Gmbh) according
to the manufacturer's instructions and as described previously
(11). Briefly, RGC-5 cells were
seeded in 96-well plates at a density of 5×103/well, and treated as
described above. Culture supernatant (50 µl) from each well was
transferred to a new 96-well plate and 100 µl reaction mixture
solution was added to each well. Following incubation at room
temperature in the dark for 30 min, 50 µl of stop solution was
added to each well to terminate the reaction. Absorbance was
measured using a microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), at 490 nm test wavelength and at 630 nm
reference wavelength. Cell viability was estimated as follows:
cytotoxicity (%) = (experimental value-low control) / (high
control-low control) ×100%.
Measurement of mitochondrial membrane
potential changes (ΔΨm)
TMRM, a cationic fluorescent probe which is taken up
by mitochondria in a potential-dependent manner, was used to
evaluate ΔΨm, as described previously (9). To quantify ΔΨm by flow cytometry,
RGC-5 cells (1×104 cells/well in a 6-well plate) were pretreated
with 0.01, 0.1 or 1 µM MTP-131 for 1 h and then incubated with 500
µM H2O2 for 2 h. Cells were harvested and
suspended in freshly prepared TMRM (200 nM) in phenol red and
serum-free DMEM for 30 min at 37°C in the dark. All samples were
rinsed twice with PBS and analyzed immediately by flow cytometry
using a FACSAria II (BD Biosciences, Franklin Lakes, NJ, USA) and
excitation/emission (ex/em) wavelengths 548/573 nm. A total of
1×104 cells were routinely collected, and results were expressed in
arbitrary units as the mean fluorescence intensity (MFI) from the
average of at least three separate experiments.
For confocal microscopy, RGC-5 cells
were plated in Petri dishes
Following MTP-131 pretreatment and
H2O2 incubation, cells were rinsed twice and
loaded with freshly prepared 500 nM TMRM for 30 min at 37°C in the
dark. Following two rinses with PBS, cells were visualized by
confocal microscopy using a LSM 510 microscope (Carl Zeiss AG,
Oberkochen, Germany) and ex/m wavelengths 548/573 nm.
Measurement of intracellular ROS
Intracellular ROS in RGC-5 cells was measured with
H2DCFDA, as described previously (12). This nonfluorescent probe
accumulates within cells and transforms into
2′,7′-dichlorodihydrofluorescein (H2DCF) by
deacetylation, which then reacts with ROS to form the fluorescent
dichlorofluorescein (DCF). To quantify ROS by flow cytometry, RGC-5
cells (1×104 cells/well in a 6-well plate) were pretreated with
0.01, 0.1 or 1 µM MTP-131 for 1 h and then incubated with 500 µM
H2O2 for 24 h. Cells were harvested and
suspended in freshly prepared H2DCFDA (5 µM) in phenol
red and serum-free DMEM for 30 min at 37°C in the dark. All samples
were rinsed twice with PBS and analyzed immediately by flow
cytometry (ex/em: 488/530 nm). Ten thousand cells were routinely
collected, and results were expressed as the MFI in arbitrary units
from the average of at least three separate experiments.
For visualization by confocal microscopy, cells were
plated in Petri dishes. Following pretreatment with MTP-131 and
incubation with H2O2, 5 µM freshly prepared
H2DCFDA was added into cells and incubated at 37°C for
30 min in the dark. Following two rinses with PBS, cells were
immediately imaged by confocal microscopy (ex/em: 495/525 nm).
Measurement of apoptosis
Apoptosis was measured using an Annexin V-FITC/PI
apoptosis assay kit, according to the manufacturer's instructions.
Briefly, cells were pretreated with 0.01, 0.1 or 1 µM MTP-131 for 1
h and then incubated with 500 µM H2O2 for 4,
8, 12 and 24 h. To quantify apoptosis by flow cytometry, cells were
collected by trypsinization and centrifugation (100 × g for 5 min,
at room temperature) at the appropriate time point. Following two
rinses with PBS, cells were resuspended in 500 µl loading buffer
and incubated with 5 µl Annexin V and 10 µl PI at room temperature
for 5 min in the dark. Flow cytometry analysis was immediately
performed (ex/em: 488/530 nm), using a FITC signal detector (FL1
channel) and PI emission signal detector (FL2 channel) to analyze
Annexin V-FITC binding and PI staining, respectively. A total of
1×104 cells were collected and divided into four groups: viable
cells (Annexin V-/PI-, Q3), early apoptotic cells (Annexin V+/PI-,
Q4), late apoptotic cells (Annexin V+/PI+, Q2) or dead cells
(Annexin V-/PI+, Q1). The apoptotic rate was calculated as the sum
of the percentages of early apoptotic cells (Q4) and late apoptotic
cells (Q2).
Measurement of cytochrome c
release
The release of cytochrome c from mitochondria to the
cytoplasm was measured by confocal microscopy as previously
described (13). Briefly, cells
were seeded onto Fisherbrand cover glass (Thermo Fisher Scientific,
Inc.) at a density of 2,000 cells/chamber. Following pretreatment
with 1 µM MTP-131 for 1 h, cells were incubated in the presence or
absence of 500 µM H2O2 for 6 h at 37°C in 5%
CO2. Freshly prepared MitoTracker (500 nm) was added to
the cells and incubated for 30 min, just before the end of the 6 h
incubation period. Cells were rinsed twice with PBS and fixed in 4%
paraformaldehyde for 15 min, followed by 5 min permeabilization
with methanol on ice. Following blocking with 5% bovine serum
albumin (BSA, Jackson ImmunoResearch Europe Ltd. Suffolk, UK) for
30 min, cells were incubated with mouse monoclonal anti-cytochrome
c antibody (sc-4280; 1:100, diluted with 1% BSA; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at 4°C overnight. Following
three rinses with PBS, cells were incubated with DyLight
488-conjugated goat anti-mouse immunoglobulin G antibody
(85–11-4011-85; 1:500, diluted with 1% BSA; Multisciences Lianke
Biotech Co., Ltd.) for 30 min at 37°C in the dark. Finally, cells
were stained with Hoechst 33342 (1:1,000, Thermo Fisher Scientific,
Inc.) for 5 min, washed and mounted with anti-fade fluorescence
mounting medium (Applygen Technologies, Inc., Beijing, China).
Images were visualized by confocal microscopy. Translocation of
cytochrome c from mitochondria to cytoplasm was analyzed by
overlapping staining of cytochrome c and MitoTracker. In each
group, six slides were selected and five fields of view from each
were assessed.
Analysis of cell morphology
RGC-5 cells were seeded at a density of 1×104
cells/well on a 6-well plate. Following pretreatment with 1 µM
MTP-131 for 1 h and subsequent treatment with 500 µM
H2O2 for 24 h, micrographs of all cultures
were captured using a Zeiss LSM 510 Cell Observer system (Carl
Zeiss, AG) to examine changes in cell morphology.
Statistical analysis
All assays were performed in at least three
independent experiments and data are presented as the mean +
standard error of the mean. Statistical analysis was performed by
one-way analysis of variance and Newman-Keuls multiple comparison
test, using GraphPad Prism 5.0 software (GraphPad Software Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
MTP-131 treatment reduces
H2O2-induced LDH release in RGC-5 cells
LDH assay was performed to assess cell viability in
RGC-5 cells following H2O2-induced sustained
oxidative stress. Incubation with 500 µM H2O2
for 24 h resulted in loss of cell viability compared with untreated
cells, and the percentage of LDH release was 28.62±2.94% relative
to the untreated cells (Fig. 1).
Pretreatment with 1, 0.1 and 0.01 µM MTP-131 reduced LDH release in
a dose-dependent manner compared to cells treated with
H2O2 alone, with LDH release measured at
6.05±0.44, 13.08±0.53 and 22.39±1.73% relative to the untreated
cells, respectively (P<0.01, P<0.01 and P<0.05,
respectively, compared with H2O2 alone;
Fig. 1).
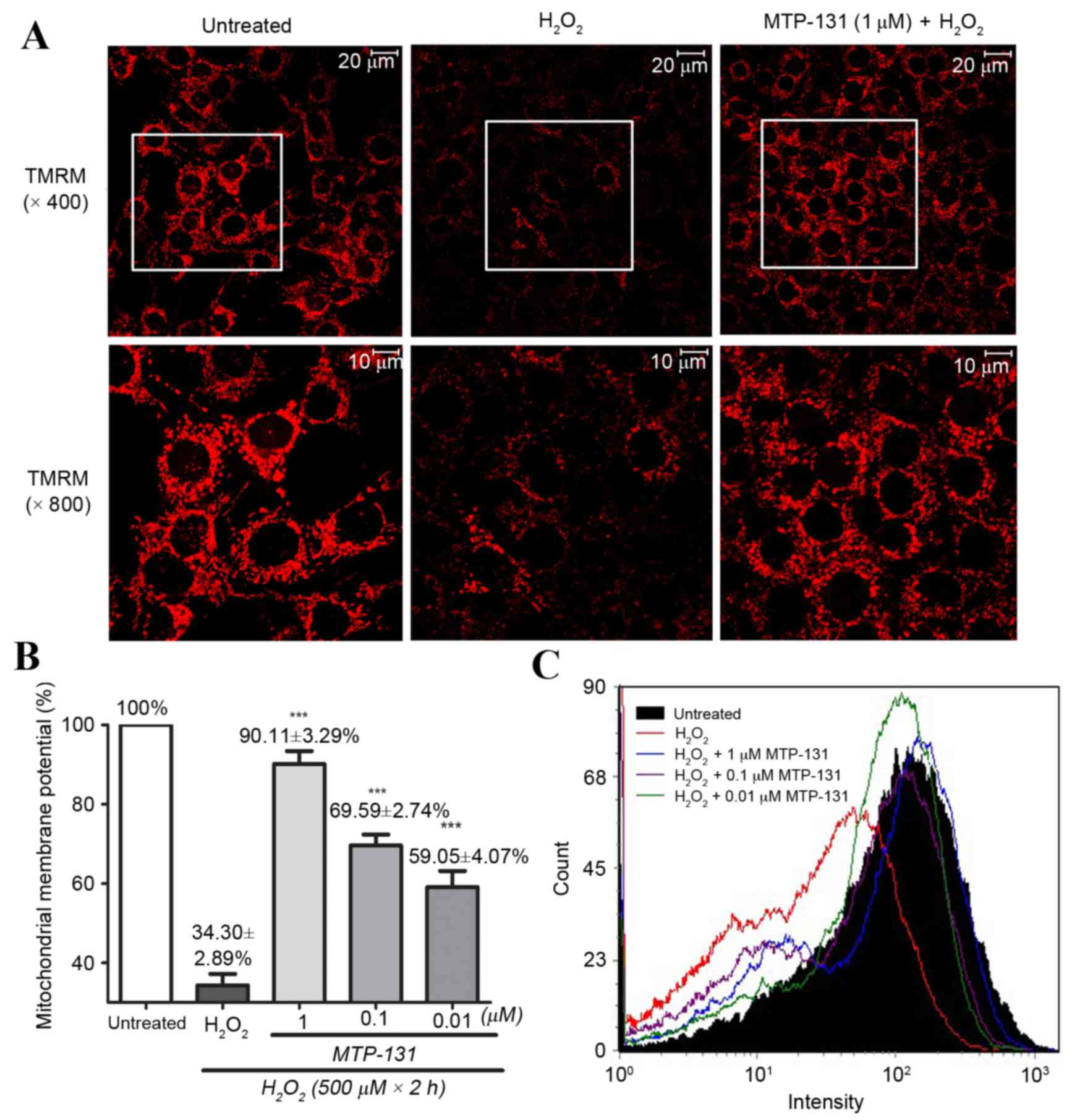
MTP-131 treatment prevents
H2O2-induced mitochondrial
depolarization
RGC-5 cells were pretreated with 1 µM MTP-131 for 1
h, and then incubated with 500 µM H2O2 for 2
h. TMRM, a lipophilic cation which accumulates in mitochondria in a
manner dependent on the membrane potential, was used to demonstrate
ΔΨm in RGC-5 cells. As expected, untreated RGC-5 cells with healthy
mitochondrial membrane potential exhibited high fluorescence
intensity for TMRM (Fig. 2A, left
panel), while incubation with 500 µM H2O2
alone for 2 h led to mitochondrial depolarization, resulting in
loss of the TMRM dye from the mitochondria and a decrease in
fluorescence intensity (Fig. 2A,
middle panel). Pretreatment of the cells with 1 µM MTP-131 for 1 h,
however, inhibited H2O2-induced mitochondria
depolarization, as evidenced by the higher fluorescence intensity
of TMRM in the MTP-131-treated cells (Fig. 2A, right panel) compared with the
cells treated with H2O2 alone. Flow cytometry
analysis confirmed that pretreatment with MTP-131 significantly
prevented H2O2-induced mitochondria
depolarization in a dose-dependent manner, compared with cells
treated with H2O2 alone (Fig. 2B and C).
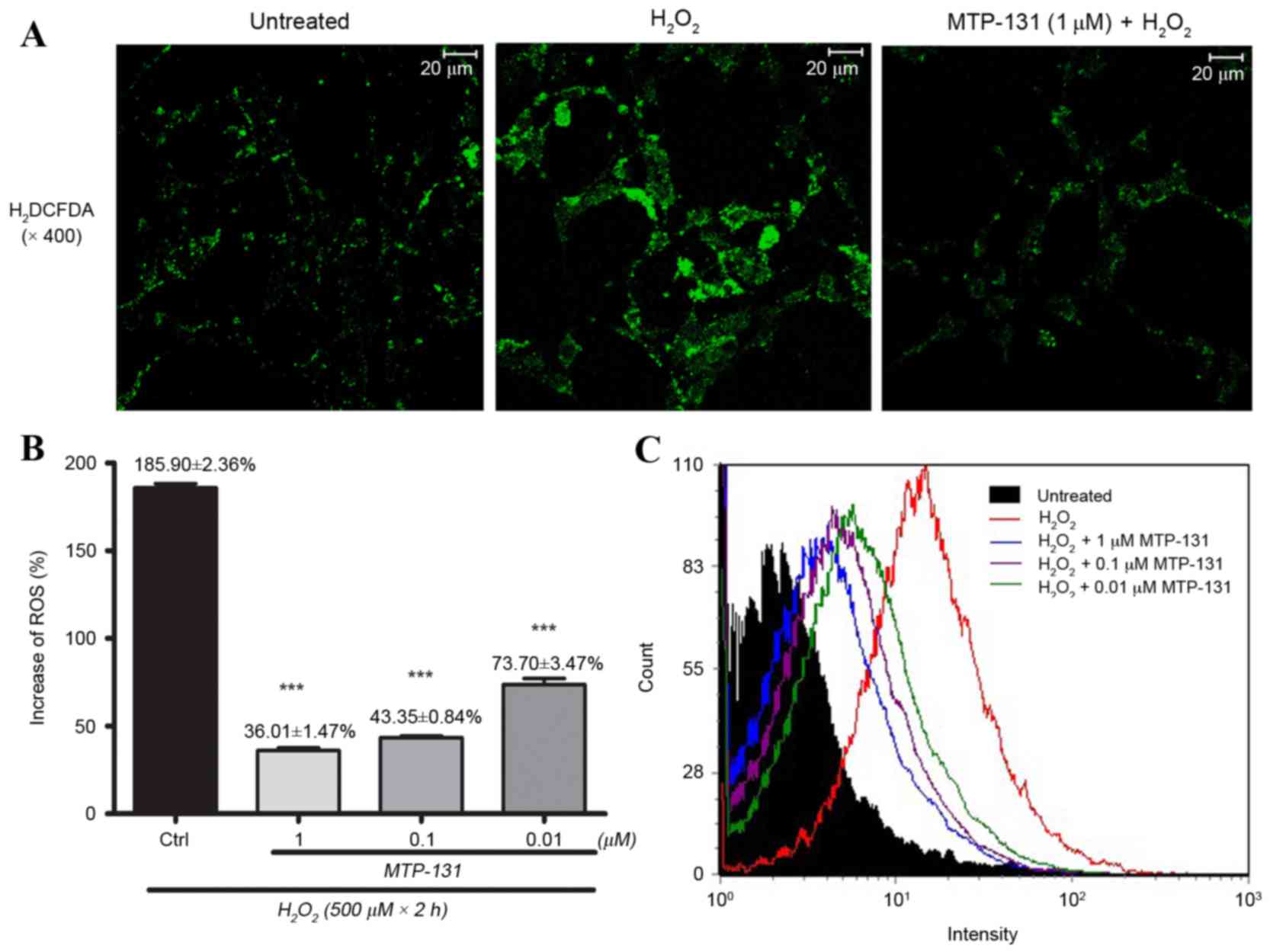
MTP-131 treatment inhibits
H2O2-induced intracellular ROS
production
RGC-5 cells were pretreated with 1 µM MTP-131 for 1
h, then incubated with 500 µM H2O2 for 24 h.
Cells were then loaded with H2DCFDA for 30 min and DCF
fluorescence was measured by flow cytometry and confocal
microscopy. As demonstrated in Fig.
3A, incubation with H2O2 increased ROS
production, as evidenced by the elevated DCF fluorescence intensity
in the H2O2-treated cells (middle panel)
compared with the untreated cells (left panel). Pretreatment with
MTP-131, however, inhibited H2O2-induced ROS
production, as evidenced by the lower DCF fluorescence intensity in
MTP-131-treated cells (right panel; Fig. 3A) compared with the
H2O2-treated cells (middle panel; Fig. 3A). Flow cytometry analysis further
confirmed that pretreatment with MTP-131 reduced ROS generation in
RGC-5 cells in a dose-dependent manner compared with cells treated
with H2O2 alone (Fig. 3B and C).
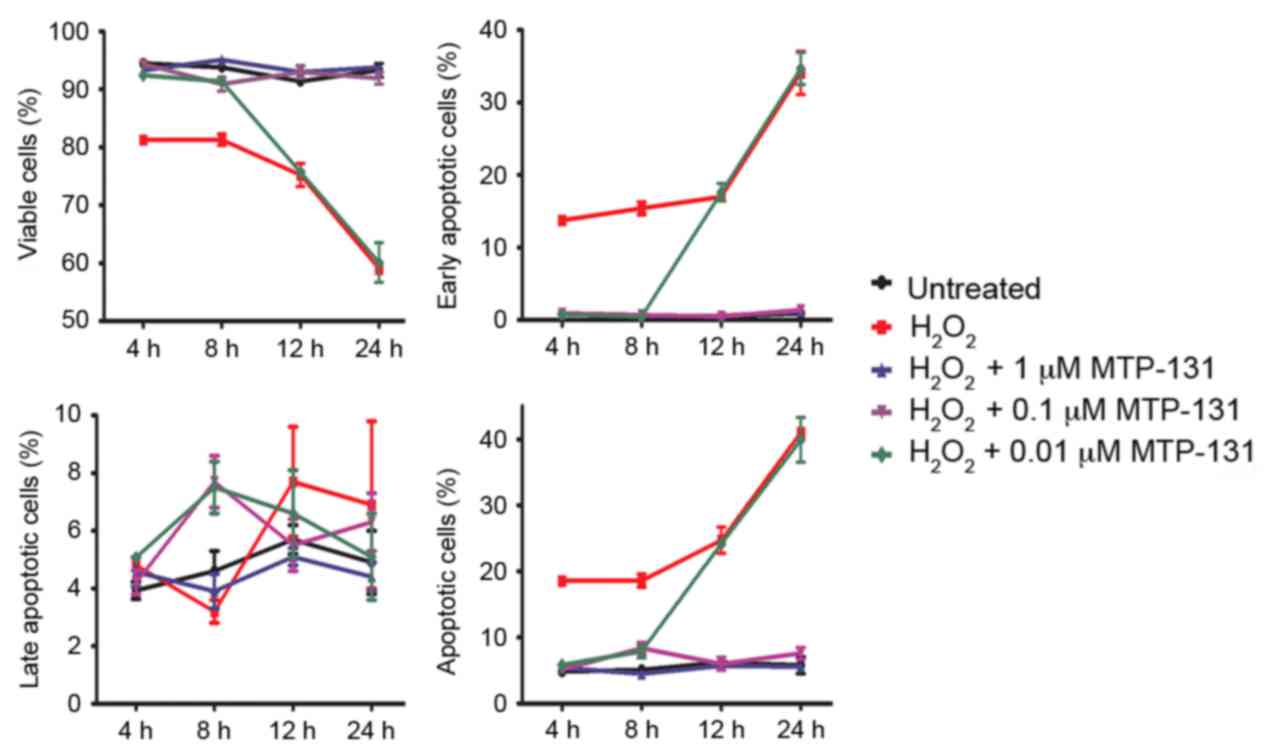
MTP-131 treatment protects against
H2O2-induced apoptosis
Apoptosis was measured by Annexin V-FITC/PI staining
and flow cytometry analysis. RGC-5 cells were pretreated with
MTP-131 for 1 h and then incubated with 500 µM
H2O2 for 4, 8, 12 and 24 h. Following
incubation with H2O2, the survival rate of
RGC-5 cells decreased in a time-dependent manner compared with
untreated cells (Fig. 4 and
Table I). Pretreatment with 1 and
0.1 µM MTP-131 inhibited H2O2-induced
apoptosis at 4, 8, 12 and 24 h compared with cells treated with
H2O2 alone at the same time points
(P<0.001 compared with H2O2 alone;
Fig. 4 and Table I). Pretreatment with 0.01 µM
MTP-131 also alleviated apoptosis at 4 and 8 h following
H2O2 incubation, but no effect was observed
at 12 and 24 h compared with cells treated with
H2O2 alone (Fig.
4 and Table I).
 | Table I.Summary of flow cytometry data
measuring apoptosis. |
Table I.
Summary of flow cytometry data
measuring apoptosis.
|
|
H2O2 (500
µM)+MTP-131 (µM) |
|---|
|
|
|
|---|
| Time point | Quadrant | RGC-5 |
H2O2 (500 µM) | 1 | 0.1 | 0.01 |
|---|
| 4 h | Q3 | 94.50±0.10 | 81.27±0.50 |
93.27±0.59a |
94.33±0.45a |
92.40±0.09a |
|
| Q4 |
0.90±0.06 | 13.77±0.38 |
0.83±0.03a |
0.93±0.06a |
0.77±0.09a |
|
| Q2 |
3.93±0.30 |
4.80±0.15 |
4.56±0.50 |
4.20±0.42 |
5.07±0.13 |
|
| Q2+Q4 |
4.83±0.24 | 18.57±0.45 |
5.40±0.47a |
5.13±0.48a |
5.83±0.22a |
| 8 h | Q3 | 93.80±0.74 | 81.30±1.01 |
95.07±0.58a |
90.93±1.17a |
91.37±0.75a |
|
| Q4 |
0.53±0.03 | 15.40±0.90 |
0.57±0.09a |
0.67±0.12a |
0.47±0.09a |
|
| Q2 |
4.60±0.70 |
3.20±0.40 |
3.93±0.57 |
7.70±0.85b |
7.47±0.92b |
|
| Q2+Q4 |
5.13±0.73 | 18.60±1.01 |
4.50±0.57a |
8.37±0.93a |
7.93±0.96a |
| 12 h | Q3 | 91.40±0.30 | 75.20±1.99 |
93.00±0.31a |
93.00±1.11a | 75.70±0.55 |
|
| Q4 |
0.53±0.17 | 17.03±0.22 |
0.63±0.07a |
0.53±0.07a | 17.63±1.17 |
|
| Q2 |
5.67±0.52 |
7.67±1.88 |
5.10±0.25 |
5.50±0.92 |
6.57±1.45 |
|
| Q2+Q4 |
6.20±0.68 | 24.70±1.99 |
5.73±0.28a |
6.03±0.98a | 24.20±0.60 |
| 24 h | Q3 | 93.30±1.20 | 58.97±0.56 |
93.77±0.46a |
91.87±0.96a | 60.10±3.38 |
|
| Q4 |
0.90±0.02 | 34.10±2.96 |
1.20±0.17 |
1.37±0.17 | 34.73±2.19 |
|
| Q2 |
4.87±1.11 |
6.93±2.85 |
4.43±0.52 |
6.27±0.95 |
5.13±1.53 |
|
| Q2+Q4 |
5.76±1.23 | 41.03±0.56 |
5.63±0.54a |
7.63±0.90a | 39.87±3.38 |
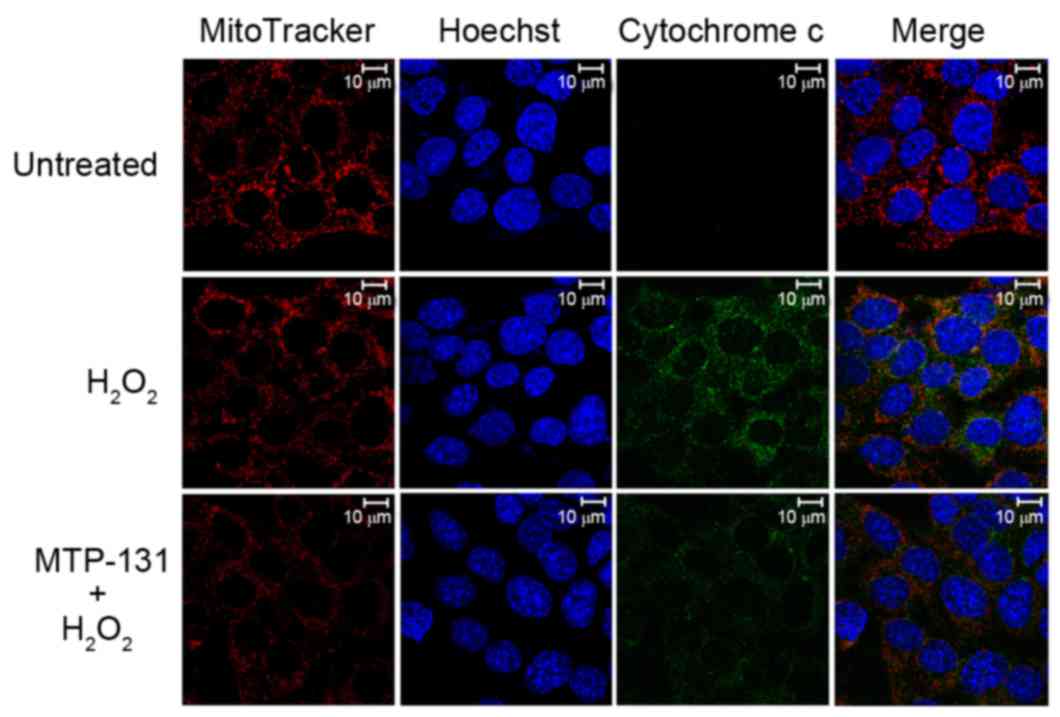
MTP-131 treatment inhibits
H2O2-induced release of cytochrome c from
mitochondria to cytoplasm
The release of cytochrome c from mitochondria into
the cytoplasm was examined by confocal microscopy. As demonstrated
in Fig. 5, no release of
cytochrome c (green fluorescence signal) from mitochondria (red
fluorescence signal) was evident in untreated RGC-5 control cells
(upper panels). Incubation with 500 µM H2O2
for 6 h, however, resulted in enhanced cytochrome c staining in the
cytoplasm of RGC-5 cells (middle panels) compared with the
untreated RGC-5 cells (Fig. 5),
indicating that H2O2 induced the release of
cytochrome c from mitochondria to cytoplasm. Pretreatment with 1 µM
MTP-131 for 1 h inhibited the H2O2-induced
release of cytochrome c, as evidenced by the decreased cytochrome c
(green) fluorescent staining in MTP-131-treated cells (lower
panels) compared with cells treated with H2O2
alone (Fig. 5).
MTP-131 treatment alleviates
H2O2-induced morphological changes in RGC-5
cells
Morphological changes in RGC-5 cells were analyzed
by phase contrast microscopy. As demonstrated in Fig. 6, normal untreated RGC-5 cells
exhibit a neuron-like appearance, with thin axons extending from
the cell bodies (left panel). However, following incubation with
500 µM H2O2 for 24 h, most cells exhibited a
degenerative appearance, evidenced by the presence of vacuolar cell
bodies and shrinkage of the axons (middle panel, Fig. 6) compared with the untreated cells.
Pretreatment with 1 µM MTP-131 prevented the
H2O2-induced morphological changes, with
MTP-131-treated cells maintaining a relatively healthy appearance,
with flat cell bodies and regular axon extensions (right panel,
Fig. 6).
Discussion
In the present study, the antioxidant effect of
MTP-131 against H2O2-induced sustained
oxidative stress was examined in undifferentiated RGC-5 cells. The
present findings demonstrated that MTP-131 treatment prevented
mitochondrial depolarization, decreased intracellular ROS
production, and inhibited apoptosis induced by sustained oxidative
stress in RGC-5 cells.
H2O2 is widely used to induce
oxidative stress in experimental studies, leading to an increase in
intracellular accumulation of ROS and RGC apoptosis (14–16).
In the present study, H2O2 (500 µM for 24 h)
was used to induce a sustained oxidative stress, and was
demonstrated to cause significant cell damage in RGC-5 cells, which
is consistent with previous studies (14). MTP-131 pretreatment protected RGC-5
cells from mitochondrial membrane depolarization, reduced ROS
production and inhibited cytochrome c release from mitochondria to
cytoplasm. These effects were demonstrated to alleviate apoptosis
in RGC-5 cells, thus suggesting that MTP-131 exhibited a potential
protective effect against oxidative stress in RGC-5 cells through
direct inhibition of mitochondria-mediated pathways.
RGC-5 cells have been widely used in in vitro
experimental studies (17–19). However, the validity of this cell
line is now questioned. It has been demonstrated that the RGC-5
cell line is of mouse origin, not rat as it was first described,
and that it express photoreceptor markers instead of retinal
ganglion markers (20,21). However, RGC-5 cells still have
several properties in common with retinal progenitor cells and is
therefore appropriate for neuronal cell studies (20). A series of retinal disorders have
been related to oxidative damage, such as AMD, DR and RP (3,4,6). The
retina is constantly exposed to visible light, which makes it
extremely vulnerable to oxidative damage, due to its great energy
demands and high oxygen consumption (5). A previous study have demonstrated
that oxidative stress is extremely important in the retinal pigment
epithelium (RPE) cells. ROS induces mitochondrial dysfunction in
RPE, leading to RPE cell apoptosis and death of photoreceptor cells
(22).
Compared with traditional antioxidants, MTP-131
represents the only known class of cell-permeable compounds that
specifically concentrates in the inner mitochondrial membrane
(7). With a 3+ net charge, MTP-131
selectively binds to cardiolipin and modulates its interaction with
cytochrome c. By inhibiting the cytochrome c/cardiolipin complex
peroxidase activity, MTP-131 optimizes mitochondrial electron
transport and ATP synthesis (23,24).
Numerous in vitro and animal studies have demonstrated the
remarkable efficacy of MTP-131 in age-associated diseases. For
example, daily treatment with MTP-131 reverses mitochondrial
dysfunction and inhibits neuronal apoptosis and inflammation in
mice with sepsis-associated encephalopathy (25). In addition, MTP-131 exhibits
protective effects against cardiac ischemia-reperfusion injury
(26). The effectiveness of
cardiolipin combined with MTP-131 is currently being evaluated in a
multinational clinical trial for reperfusion injury in patients
with acute coronary events (ClinicalTrials.gov Identifier: NCT01572909), and a
Phase 2 trial is underway assessing the effectiveness of MTP-131 on
improving renal function following angioplasty for severe renal
artery stenosis (ClinicalTrials.gov Identifier: NCT01755858) (27). Since MTP-131 has a unique
mitochondria-targeted delivery, and exhibits potent antioxidant and
neuroprotective effects both in vitro and in vivo,
the present study aimed to evaluate the protective effect of
MTP-131 against H2O2-induced oxidative damage
in RGC-5 cells. The present findings demonstrated that MTP-131
inhibited H2O2-induced mitochondria
depolarization, reduced intracellular ROS and prevented apoptosis
in RGC-5 cells.
In conclusion, this novel class of targeted peptide
therapeutics, including MTP-131, has the potential to restore
mitochondrial bioenergetics. Further studies using animal models
will be needed in order to fully explore this novel antioxidant
approach for the treatment of age-related retinal diseases.
Acknowledgements
The authors would like to thank Stealth Peptides
International Inc. for providing us with MTP-131 and a research
grant. The present study was supported by the Zhejiang Provincial
Natural Science Foundation of China (grant nos. LQ15H120001 and
LY12H12008) and the National Natural Science Foundation of China
(grant no. 81372930).
References
|
1
|
Bhat AH, Dar KB, Anees S, Zargar MA,
Masood A, Sofi MA and Ganie SA: Oxidative stress, mitochondrial
dysfunction and neurodegenerative diseases; a mechanistic insight.
Biomed Pharmacother. 74:101–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thanan R, Oikawa S, Hiraku Y, Ohnishi S,
Ma N, Pinlaor S, Yongvanit P, Kawanishi S and Murata M: Oxidative
stress and its significant roles in neurodegenerative diseases and
cancer. Int J Mol Sci. 16:193–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonilha VL, Rayborn ME, Yang X, Xie C and
Cai H: Oxidative stress regulation by DJ-1 in the retinal pigment
epithelium. Adv Exp Med Biol. 801:649–654. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu Y, Tang L and Chen B: Oxidative stress:
Implications for the development of diabetic retinopathy and
antioxidant therapeutic perspectives. Oxid Med Cell Longev.
2014:7523872014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pinazo-Durán MD, Gallego-Pinazo R,
García-Medina JJ, Zanón-Moreno V, Nucci C, Dolz-Marco R,
Martínez-Castillo S, Galbis-Estrada C, Marco-Ramírez C,
López-Gálvez MI, et al: Oxidative stress and its downstream
signaling in aging eyes. Clin Interv Aging. 9:637–652. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Campochiaro PA, Strauss RW, Lu L, Hafiz G,
Wolfson Y, Shah SM, Sophie R, Mir TA and Scholl HP: Is there excess
oxidative stress and damage in eyes of patients with retinitis
pigmentosa? Antioxid Redox Signal. 23:643–648. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao K, Zhao GM, Wu D, Soong Y, Birk AV,
Schiller PW and Szeto HH: Cell-permeable peptide antioxidants
targeted to inner mitochondrial membrane inhibit mitochondrial
swelling, oxidative cell death, and reperfusion injury. J Biol
Chem. 279:34682–34690. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao K, Luo G, Zhao GM, Schiller PW and
Szeto HH: Transcellular transport of a highly polar 3+ net charge
opioid tetrapeptide. J Pharmacol Exp Ther. 304:425–432. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao K, Luo G, Giannelli S and Szeto HH:
Mitochondria-targeted peptide prevents mitochondrial depolarization
and apoptosis induced by tert-butyl hydroperoxide in neuronal cell
lines. Biochem Pharmacol. 70:1796–1806. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Szeto HH: Mitochondria-targeted peptide
antioxidants: Novel neuroprotective agents. AAPS J. 8:E521–E531.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iizuka Y, Hong S, Kim CY, Kim SK and Seong
GJ: Agmatine pretreatment protects retinal ganglion cells (RGC-5
cell line) from oxidative stress in vitro. Biocell. 32:245–250.
2008.PubMed/NCBI
|
|
12
|
Shimazawa M, Nakajima Y, Mashima Y and
Hara H: Docosahexaenoic acid (DHA) has neuroprotective effects
against oxidative stress in retinal ganglion cells. Brain Res.
1251:269–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen M, Liu B, Gao Q, Zhuo Y and Ge J:
Mitochondria-targeted peptide MTP-131 alleviates mitochondrial
dysfunction and oxidative damage in human trabecular meshwork
cells. Invest Ophthalmol Vis Sci. 52:7027–7037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koriyama Y, Ohno M, Kimura T and Kato S:
Neuroprotective effects of 5-S-GAD against oxidative stress-induced
apoptosis in RGC-5 cells. Brain Res. 1296:187–195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou X, Su CF, Zhang Z, Wang CY, Luo JQ,
Zhou XW, Cai L, Yan L, Zhang W and Luo HM: Neuroprotective effects
of methyl 3,4-dihydroxybenzoate against H2O2-induced apoptosis in
RGC-5 cells. J Pharmacol Sci. 125:51–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jia WC, Liu G, Zhang CD and Zhang SP:
Formononetin attenuates hydrogen peroxide (H2O2)-induced apoptosis
and NF-κB activation in RGC-5 cells. Eur Rev Med Pharmacol Sci.
18:2191–2197. 2014.PubMed/NCBI
|
|
17
|
Wang R, Peng L, Zhao J, Zhang L, Guo C,
Zheng W and Chen H: Gardenamide A protects RGC-5 cells from
H2O2-induced oxidative stress insults by activating PI3K/Akt/eNOS
signaling pathway. Int J Mol Sci. 16:22350–22367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang P, Huang C, Wang W and Wang M: Early
changes in staurosporine-induced differentiated RGC-5 cells
indicate cellular injury response to nonlethal blue light exposure.
Photochem Photobiol Sci. 14:1093–1099. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ding W, Shang L, Huang JF, Li N, Chen D,
Xue LX and Xiong K: Receptor interacting protein 3-induced RGC-5
cell necroptosis following oxygen glucose deprivation. BMC
Neurosci. 16:492015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sippl C and Tamm ER: What is the nature of
the RGC-5 cell line? Adv Exp Med Biol. 801:145–154. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Al-Ubaidi MR: RGC-5: Are they really 661W?
The saga continues. Exp Eye Res. 119:1152014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mao H, Seo SJ, Biswal MR, Li H, Conners M,
Nandyala A, Jones K, Le YZ and Lewin AS: Mitochondrial oxidative
stress in the retinal pigment epithelium leads to localized retinal
degeneration. Invest Ophthalmol Vis Sci. 55:4613–4627. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Birk AV, Chao WM, Bracken C, Warren JD and
Szeto HH: Targeting mitochondrial cardiolipin and the cytochrome
c/cardiolipin complex to promote electron transport and optimize
mitochondrial ATP synthesis. Br J Pharmacol. 171:2017–2028. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szeto HH: First-in-class
cardiolipin-protective compound as a therapeutic agent to restore
mitochondrial bioenergetics. Br J Pharmacol. 171:2029–2050. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu J, Zhang M, Hao S, Jia M, Ji M, Qiu L,
Sun X, Yang J and Li K: Mitochondria-targeted peptide reverses
mitochondrial dysfunction and cognitive deficits in
sepsis-associated encephalopathy. Mol Neurobiol. 52:783–791. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ajith TA and Jayakumar TG:
Mitochondria-targeted agents: Future perspectives of mitochondrial
pharmaceutics in cardiovascular diseases. World J Cardiol.
6:1091–1099. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Birk AV, Liu S, Soong Y, Mills W, Singh P,
Warren JD, Seshan SV, Pardee JD and Szeto HH: The
mitochondrial-targeted compound SS-31 re-energizes ischemic
mitochondria by interacting with cardiolipin. J Am Soc Nephrol.
24:1250–1261. 2013. View Article : Google Scholar : PubMed/NCBI
|