Introduction
Calcific aortic valve disease (CAVD) is a slow
progressive pathologic process that exhibits mild thickening of the
aortic valve and calcification of valve leaflets (1). CAVD may be an actively regulated
process that includes chronic inflammation, lipoprotein deposition,
the renin-angiotensin system, extracellular matrix remodeling, the
activation of specific osteogenic signaling pathways and apoptosis.
However, degeneration of the aortic valve is the key etiology of
CAVD (occurring in 81.9% of CAVD patients) (2), and specific osteogenic signaling
pathways determine the activation and differentiation of the
resident fibroblasts or quiescent valve interstitial cells (VICs)
into myofibroblasts (activated VICs) and osteoblast-like cells
(osteoblastic VICs) with subsequent micro- and macro-calcification
(3–5). Therefore, inflammatory responses
participate in all stages of CAVD and promote its
pathophysiological progression (6,7).
High mobility group box 1 (HMGB1) is a non-histone
DNA binding protein (8). HMGB1 is
involved in the stabilization of DNA and promotion of gene
transcription (9). However,
previous studies have demonstrated that HMGB1 participates in
inflammation through effects on macrophages (10,11).
The expression level of HMGB1 was demonstrated to be increased in
acute and chronic inflammatory diseases, such as type 2 diabetes
(12), chronic asthma (13) and chronic rhinosinusitis (14). HMGB1 signals through receptor of
advanced glycation end products, toll-like receptor (TLR) 2 and
TLR4, thereby stimulating an inflammatory response in cells
(15). However, the role of HMGB1
in CAVD remains to be elucidated.
A previous study demonstrated that TLR4 proteins are
highly expressed in human aortic valve leaflets and aortic valve
interstitial cells (AVICs) (16).
Stimulation with LPS may result in the activation of the nuclear
factor-κB (NF-κB) signaling pathway (17). The TLR4 signaling pathway has been
demonstrated to enhance the pro-osteogenic response in AVICs and
may promote the progression of CAVD (18).
Chronic inflammation is crucial for the development
of CAVD, as it was observed to promote the osteoblastic
differentiation of AVICs (19).
The present study used immunohistochemistry to demonstrate that
HMGB1 and TLR4 were expressed in the aortic valve of CAVD samples.
In addition, it was revealed that HMGB1 is important for
osteoblastic differentiation in AVICs, which is mediated by TLR4.
TLR4 was demonstrated to be essential for HMGB1-induced
runt-related transcription factor 2 (Runx2), msh homeobox 2 (Msx2),
bone morphogenetic protein 2 (BMP2) and osteopontin (OPN)
expression in AVICs. These findings provide a novel mechanism for
CAVD progression and improve the understanding of aortic valve
disease.
Materials and methods
Reagents and antibodies
HMGB1 (R&D Systems, Inc., Minneapolis, MN, USA)
was used to stimulate AVICs. The antibodies used to detect HMGB1
(catalog no. ab18256) and TLR4 (catalog no. ab13556) by
immunohistochemical analysis were purchased from Abcam (Cambridge,
UK). The 3,3′-Diaminobenzidine Liquid Substrate system, Alkaline
Phosphatase Diethanolamine Activity kit and alizarin red S stain
were purchased from Sigma-Aldrich; Merck Millipore (Darmstadt,
Germany). Dulbecco's modified Eagle's medium (DMEM): F12,
supplemented with 20% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 µg/l streptomycin, was obtained from Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). The primary antibodies used
were specific to Runx2 (catalog no. ab23981), Msx2 (catalog no.
ab69058), BMP2 (catalog no. ab14933), OPN (catalog no. ab91655;
Abcam), β-actin (catalog no. 4970S; Cell Signaling Technology,
Inc., Danvers, MA, USA), p38 mitogen-activated protein kinase
(MAPK; catalog no. 8690S; Cell Signaling Technology, Inc.),
phosphorylated (p)-p38-MAPK (p-p38-MAPK; catalog no. 9215S; Cell
Signaling Tecnhology, Inc.), NF-κB (catalog no. 8242S; Cell
Signaling Technology, Inc.) and p-NF-κB (catalog no. 3033S; Cell
Signaling Technology, Inc.). SignalStain® Boost
Detection reagent (catalog no. 8114; Cell Signaling Technology,
Inc.) was used to detect primary antibodies. Activation of TLR4 was
inhibited using an anti-mouse TLR4/MD-2 antibody (cat. no. 16-9924;
eBioscience, Inc., San Diego, CA, USA), and a mouse IgG isotype
control antibody (catalog no. 16-4714; eBioscience, Inc.) was used
as a negative control.
Clinical samples
Human aortic valves with calcific regions were
obtained from 3 patients who underwent a valve replacement surgery.
Normal aortic valve leaflets were collected from the explanted
hearts of 3 patients undergoing heart transplantation surgery. All
patients were admitted to The Affiliated Tongji Hospital (Shanghai,
China) between August 2015 and December 2016. All patients were
male (weight, 65±5 kg; age, 70±3 years). The protocols of the
present study were approved by the Ethics Committee of Tongji
Hospital, Shanghai Tongji University School of Medicine (Shanghai,
China) and written informed consent was obtained from all
patients.
Primary AVIC culture
Male, 6-week-old (weight, 10–15 g) wild-type
(C57BL/6; n=20) and TLR4 knockout mice (TLR4−/−; n=20)
were obtained from the Model Animal Research Center of Nanjing
University (Nanjing, China). To avoid the stress response, the
animals had free access to water and a normal diet for 2 weeks and
housed at 5 mice per cage, under a 12-h light/dark cycle at room
temperature. The mice were anesthesized with 100 mg/kg sodium
pentobarbital and their aortic valves were obtained for the culture
of primary AVICs using the methods described previously (20). Valve leaflets were subjected to
collagenase digestion and gently scraped to expose the endothelial
layer. The leaflets were then divided into 1–2 mm2
pieces and cultured in DMEM: F12 (1:1) supplemented with 20% FBS, 2
mmol/l L-glutamine (Gibco; Thermo Fisher Scientific, Inc.), 100
U/ml penicillin and 100 µg/l streptomycin. The cells were cultured
in 5% CO2 at 37°C. When 80% confluence was reached,
AVICs were passaged using 0.25% trypsin-EDTA. AVICs at passages 3
and 8 were used for further experiments. The animal experimental
procedures employed in the current study complied with the Animal
Management Rules of the Chinese Ministry of Health (Document no.
55, 2001), and were approved by the Animal Care Committee of Tongji
University (Shanghai, China).
Immunohistochemistry
Human calcific (n=3) and non-calcific aortic valves
(n=3) were used for histological and immunochemical analysis. The
samples were fixed in 4% paraformaldehyde overnight and divided
into serial 5-µm cryosections. The sections were stained with 5%
hematoxylin and 0.5% eosin for 15 min at room temperature.
Immunohistochemical analysis was performed using anti-HMGB1
(dilution, 1:50) and anti-TLR4 (dilution, 1:50) antibodies at room
temperature for 2 h. Following incubation with HRP-conjugated
secondary antibodies (dilution, 1:100) at room temperature for 1 h,
the sections were incubated with 3,3′-diaminobenzidine (catalog no.
D8001; Sigma-Aldrich; Merck Millipore). The expression levels of
HMGB1 and TLR4 detected by immunohistochemistry were determined
using Image Pro-Plus software version 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA). The results were determined as integrated
optical density/area.
In vitro calcification of AVICs
Primary AVICs were isolated from the aortic valves
of C57BL/6 and TLR4−/− knockout mice. AVIC calcification
was induced in an osteogenic medium containing DMEM, supplemented
with 15% FBS, 50 mg/ml ascorbate-2-phosphate, 10 nM dexamethasone
and 10 mM β-glycerol phosphate (all purchased from Gibco; Thermo
Fisher Scientific) as described previously (21). The culture medium was refreshed
every 48–72 h, and the cells were harvested after 3 weeks. The
extent of calcification of AVICs was determined by alizarin red S
staining. The cells were washed in distilled water and then exposed
to freshly prepared 2% alizarin red S (pH 4.1–4.3) for 5 min, where
a red/orange color indicated positive staining. For quantitative
analysis of alizarin red S staining, the dye was released from the
cell matrix by incubating the cells with cetylpyridinium chloride
for 15 min at room temperature. The quantity of released dye was
determined using a spectrophotometer at 540 nm. Alkaline
phosphatase (ALP) activity was determined using spectrophotometry
to quantify the level of the p-nitrophenol in AVICs, according to
the methods described previously (22). The quantity of alizarin red S
staining and ALP activity were normalized to the total quantity of
cellular protein using Image Pro-Plus software version 6.0. The
quantity of cellular protein was measured by Bichinchoninic Acid
assay.
Inhibition of TLR4 activation
Primary AVICs from C57BL/6 mice were seeded onto
6-well plates at a density of 5×106 cells/well, before
they were pretreated with 5 µg/ml anti-TLR4 antibody (1:20) or 5
µg/ml mouse IgG (1:20) as a negative control for 1 h, and
subsequently stimulated with HMGB1 at concentrations of 0.5, 1 or 2
µg/ml for 60 min or 3 weeks.
Western blotting
AVICs (5×106 cells) were lysed with the
ProteoJET Mammalian Cell Lysis reagent (Fermentas; Thermo Fisher
Scientific, Inc.) to extract cytoplasmic proteins. Equal quantities
of protein (50 µg) were subjected to 10% SDS-PAGE and blotted onto
polyvinylidene difluoride membranes. The membrane was blocked with
5% bovine serum albumin (catalog no. V900933; Sigma-Aldrich; Merck
Millipore) and probed with antibodies against TLR4 (dilution,
1:500), Runx2 (dilution, 1:1000), Msx2 (dilution, 1:1,000), BMP2
(dilution, 1:1,000), OPN (dilution, 1:1,000), β-actin (dilution,
1:2,000), p38-MAPK (dilution, 1:1,000), p-p38-MAPK (dilution,
1:1,000), NF-κB (dilution, 1:1,000) and p-NF-κB (dilution, 1:1,000)
overnight at 4°C, followed by incubation with HRP-conjugated
secondary antibodies (dilution, 1:5,000) for 1 h at room
temperature. The blots were developed using an enhanced
chemiluminescence detection system (Merck Millipore). Images were
captured, and the intensity of each band was analyzed with Quantity
One software version 4.62 (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Student's paired t-test was performed to compare paired samples and
analysis of variance was used for multiple group comparisons,
followed by Friedman's post-hoc test. P<0.05 was considered to
indicate statistically significant difference. Statistical analyses
were performed using SPSS software version 20 (IBM SPSS, Armonk,
NY, USA).
Results
Calcific aortic valves exhibit HMGB1
and TLR4 expression
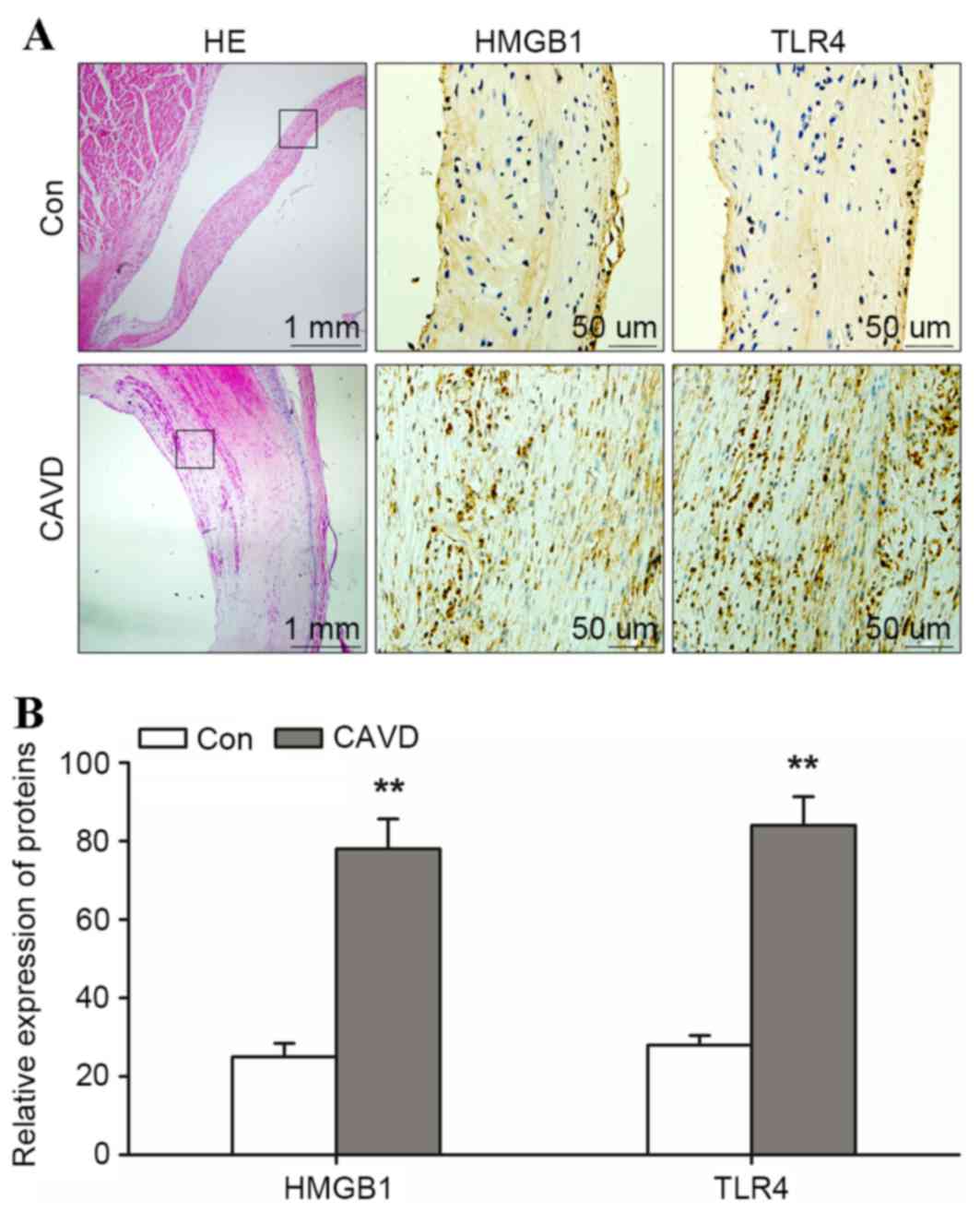
To investigate the pathology of calcific aortic
valves, calcific (n=3) and non-calcific aortic valves (n=3) were
stained with HE. Routine pathological examination of calcific
aortic valves revealed calcification, macrophage rupture and AVIC
thickening close to the region of calcification (Fig. 1A). In calcific aortic valves, the
positive expression of HMGB1 and TLR4 was detected across the
region of calcification and the integrated optical density/area
level of HMGB1 and TLR4 was significantly higher when compared with
those in the control aortic valve group (HMGB1, 78.1±7.6 and TLR4,
84.2±7.3 in calcific aortic valves vs. HMGB1, 25.1±3.4 and TLR4,
28.0±2.4 in non-calcific aortic valves; P<0.01; Fig. 1B). These results indicated that
HMGB1 and TLR4 may contribute to the calcification of the aortic
valves.
HMGB1 induces AVIC calcification
The present study investigated whether HMGB1 was
involved in the calcification of AVICs in mice. Primary AVICs were
stimulated with HMGB1 in order to assess its effects on the
calcification of AVICs and the expression levels of Runx2, Msx2,
BMP2 and OPN. The ALP activity and alizarin red S staining assays
demonstrated that HMGB1 treatment promoted calcification in a
dose-dependent manner (Fig. 2A and
B). The levels of ALP activity and alizarin red S staining
increased significantly at concentrations of 1 and 2 µg/ml HMGB1
when compared with the control group (ALP activity, 1 µg/ml HMGB1,
434.32±34.51 vs. 285.33±34.95, P<0.01 and 2 µg/ml HMGB1,
510.34±29.28 vs. 285.33±34.95, P<0.01; alizarin red S staining
assay, 1 µg/ml HMGB1, 148.21±7.43 vs. 114.33±17.50, P<0.05 and 2
µg/ml HMGB1, 174.67±9.03 vs. 114.33±17.50, P<0.01; Fig. 2A and B). Runx2, Msx2, BMP2 and OPN
have been previously demonstrated to be involved in and promote the
calcification of AVICs (23). The
present study used western blotting to determine the effects of
HMGB1 on their protein expression levels. Following exposure of
AVICs to HMGB1, the protein expression levels of Runx2, Msx2, BMP2
and OPN demonstrated a dose-dependent increase (Fig. 2C). High doses (1 and 2 µg/ml) of
HMGB1 significantly induced the expression of these genes
(P<0.01; Fig. 2D). These
results indicated that, HMGB1 upregulated calcification and the
expression of associated genes in primary AVICs in a dose-dependent
manner, which suggests that HMGB1 may contribute to the
calcification process in AVICs.
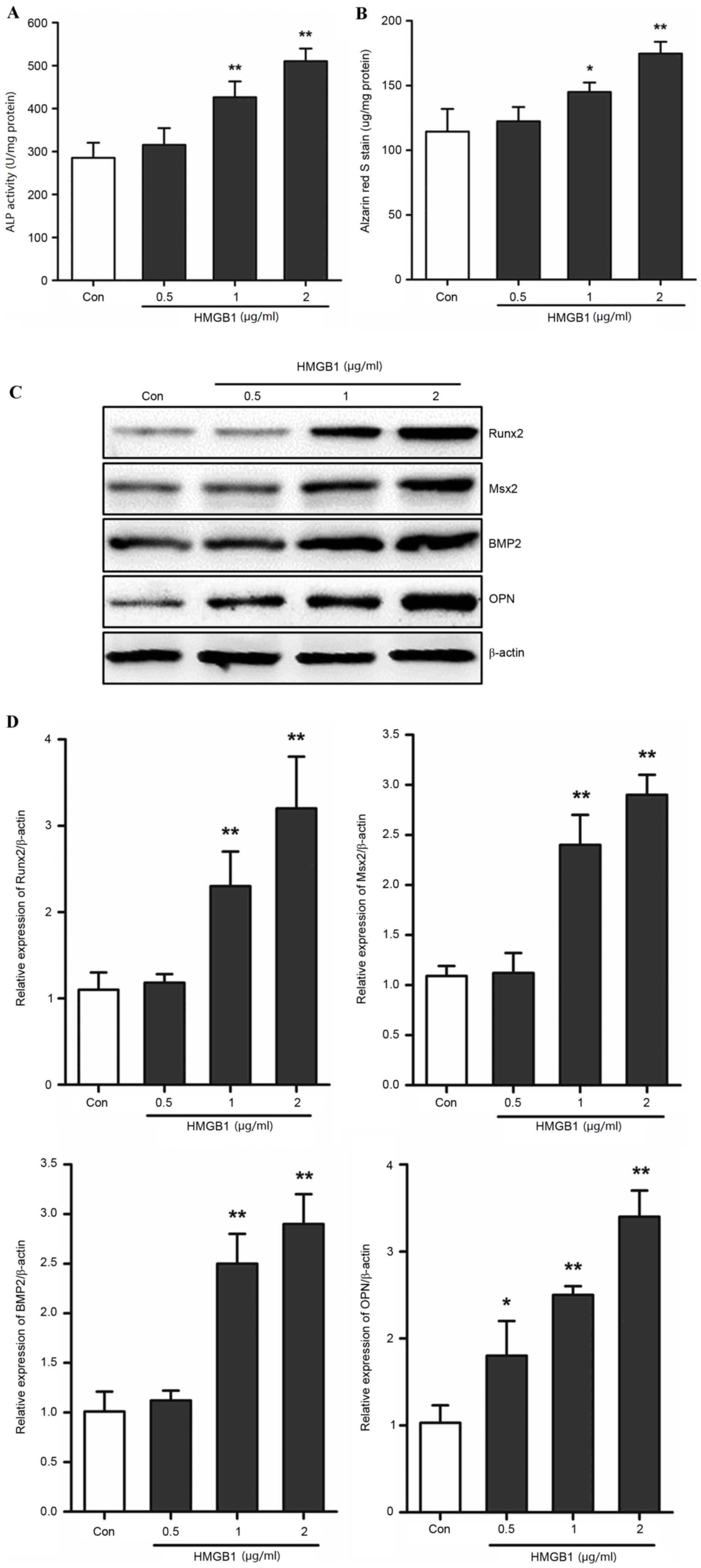 | Figure 2.HMGB1 induced calcification of AVICs.
Primary AVICs were incubated with 0.5, 1 and 2 µg/ml HMGB1 for 3
weeks. Quantification of (A) ALP activity and (B) alizarin red S
staining in AVICs. (C) Western blotting was used to determine the
protein expression levels of Runx2, Msx2, BMP2 and OPN. (D)
Quantification of band densities by densitometry in 3 independent
experiments relative to β-actin (n=3). *P<0.05 and **P<0.01
vs. Con. HMGB1, high mobility group box 1; AVICs, aortic valve
interstitial cells; ALP, alkaline phosphatase; Runx2, runt-related
transcription factor 2; Msx2, msh homeobox 2; BMP2, bone
morphogenetic protein 2; OPN, osteopontin; Con, control. |
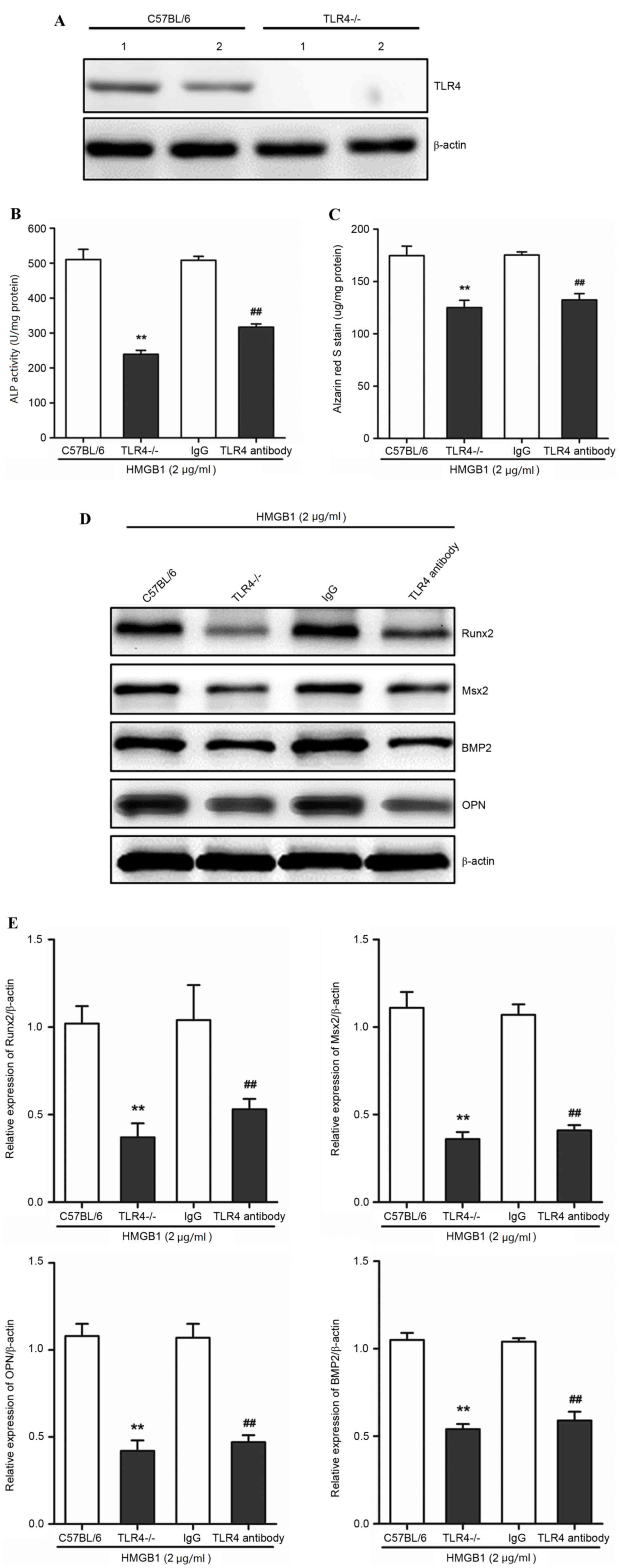
TLR4 mediates HMGB1-induced
calcification of AVICs
In order to determine whether TLR4 mediated the
HMGB1-induced calcification in AVICs, as well as the expression
levels of associated genes in AVICs, the calcification and protein
expression levels of Runx2, Msx2, BMP2 and OPN following HMGB1
treatment in primary AVICs derived from the valves of wild-type
(C57BL/6) or TLR4−/− mice, were compared. A TLR4 assay
was used in order to identify the phenotype of wild-type and
TLR4−/−. TLR4 protein expression was detected in the
wild-type, but not in TLR4−/−-derived AVICs (Fig. 3A). As 2 µg/ml HMGB1 induced the
highest calcification in AVICs (Fig.
2A and B), this concentration was used for further experiments.
Primary AVICs were incubated with 2 µg/ml HMGB1 for 3 weeks.
TLR4−/− AVICs exhibited a significant reduction in ALP
activity and alizarin red S staining when compared with wild-type
AVICs (P<0.01; Fig. 3B and C).
In addition, the protein expression levels of Runx2, Msx2, BMP2 and
OPN, were significantly reduced in TLR4−/−-derived AVICs
when compared with the wild-type AVICs (P<0.01; Fig. 3D and E), suggesting that TLR4 may
be important for HMGB1-mediated calcification of AVICs. The
activation of TLR4 was then inhibited using TLR4-specific
antibodies, as described previously (24). TLR4-specific antibodies
significantly inhibited the increase in HMGB1-induced calcification
in primary AVICs from C57BL/6 mice, as demonstrated by the
significant reduction in ALP activity and alizarin S staining when
compared with the mouse IgG control (P<0.01; Fig. 3B and C). The protein expression
levels of Runx2, Msx2, BMP2 and OPN in the TRL antibody group were
significantly reduced when compared with the mouse IgG control
(P<0.01; Fig. 3D and E). These
findings demonstrated that HMGB1-induced calcification was
significantly attenuated following TLR4 knockout and inhibition,
which suggests that AVIC calcification in response to
HMGB1stimulation may require TLR4.
HMGB1 activated p38 and NF-κB via TLR4
in AVICs
The authors hypothesized that TLR4 may interact with
MAPK in AVICs following exposure to HMGB1. The phosphorylation of
p38 was significantly increased following HMGB1 treatment at
concentrations of 0, 0.5, 1 and 2 µg/ml for 60 min (P<0.01;
Fig. 4A and B). Notably, the
phosphorylation of NF-κB, as the downstream effect of p38
phosphorylation, was significantly increased following 60 min
incubation with 0, 0.5, 1 and 2 µg/ml HMB1 (P<0.01; Fig. 4A and B). To determine whether the
HMGB1-induced activation of p38 MAPK was dependent on TLR4 in
AVICs, specific anti-TLR4 antibodies and TLR4−/−-derived
AVICs were used to reduce TLR4 expression levels and activity. TLR4
knockout and inhibition significantly decreased p38 and NF-κB
activation in AVICs when compared with the control treatments
(P<0.01; Fig. 4C and D). These
findings suggested that the molecular mechanism underlying HMGB1
treatment may involve TLR4 and p38 MAPK to mediate inflammatory
responses.
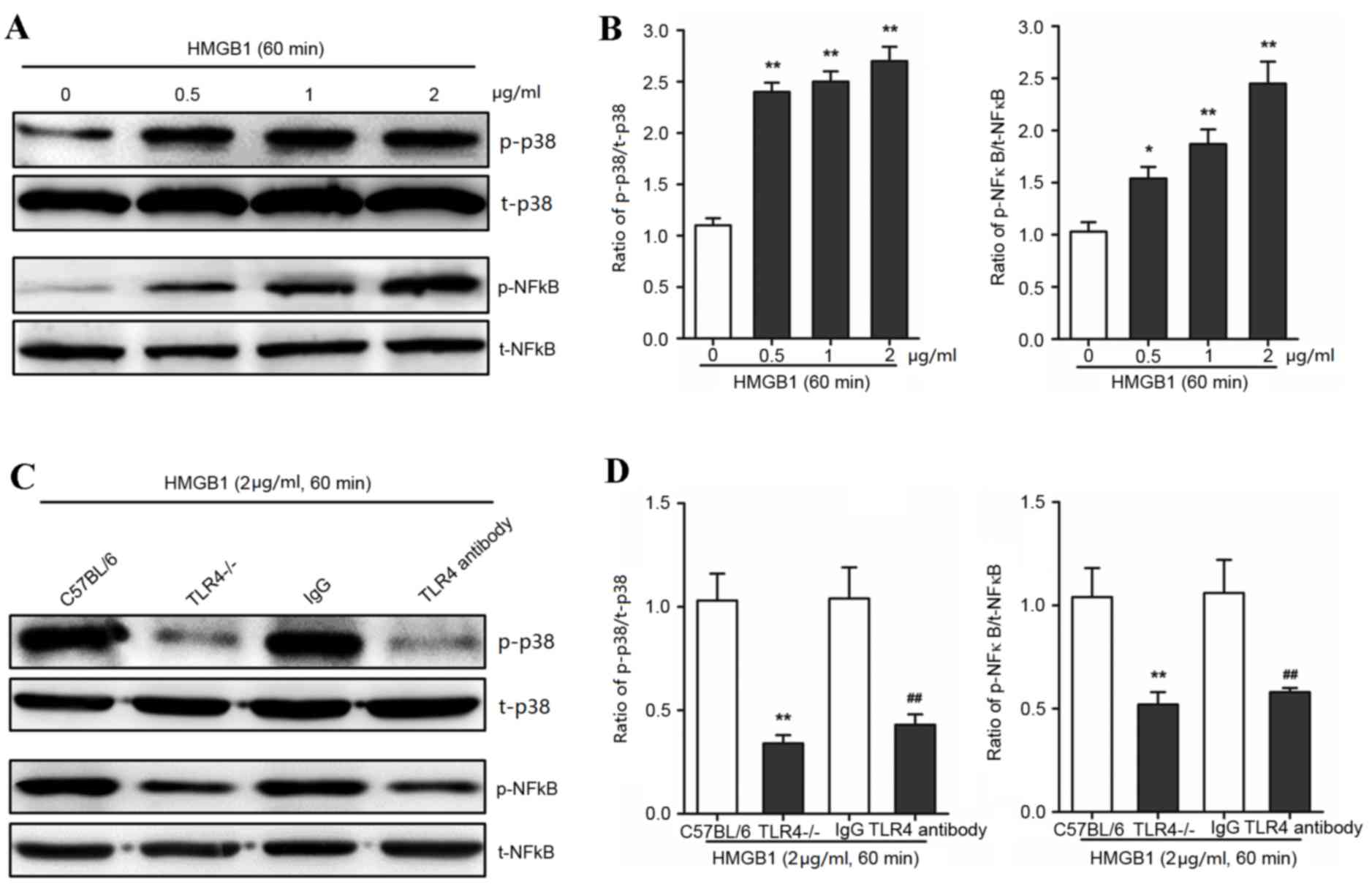 | Figure 4.HMGB1 activates p38 and NF-κB in AVICs
via TLR4 signaling. Primary AVICs were incubated with 0.5, 1 and 2
µg/ml HMGB1 for 60 min. The group without HMGB1 treatment was used
as a control. Primary AVICs derived from TLR4−/− mice or
C57BL/6 mice, and C57BL/6-derived AVICs treated with a specific
anti-TLR4 antibody (or mouse IgG as the control), were treated with
2 µg/ml HMGB1 for 60 min to determine the activation of p38 or
NF-κB. The phosphorylation of p38 and NF-κB was detected by (A and
C) western blotting and (B and D) quantified by densitometry in 3
independent experiments. *P<0.05 and **P<0.01 vs. 0 µg/ml
HMGB1 control. The results are expressed as relative units (p38 or
NF-κB phosphorylated protein/total protein). **P<0.01 vs. the
C57BL/6 group; ##P<0.01 vs. the IgG group. HMGB1,
high mobility group box 1; AVICs, aortic valve interstitial cells;
p-p38, phosphorylated-p38; t-p38, total-p38; p-NF-κB,
phosphorylated-nuclear factor κB; t-NF-κB, total-NF-κB; TLR4,
toll-like receptor 4. |
Discussion
Chronic inflammation is involved in CAVD and may
induce AVIC calcification (25).
HMGB1, as an inflammatory cytokine, may contribute to chronic
inflammatory responses (12–14).
The results of the current study demonstrated that the expression
of HMGB1 was increased in the calcification region of CAVD aortic
valves, which suggests that HMGB1 may contribute to the
calcification of AVICs. AVICs were then incubated with different
doses of HMGB1, and analysis of ALP activity and alizarin red S
staining levels demonstrated a dose-dependent increase. Wang et
al (26) determined that
diabetes accelerated saphenous vein graft calcification by ≥2 years
following coronary artery bypass grafting surgery; however, HMGB1
mediated the high-glucose-induced calcification in vascular smooth
muscle cells of saphenous veins (24). Lai et al (27) reported that HMGB1 was involved in
aortic aneurysms induced by calcium chloride in mice. These results
demonstrate that HMGB1 may function as a key factor that enhances
AVIC calcification. The results of the present study demonstrated
that HMGB1 promoted the expression of Runx2, Msx2, BMP2 and OPN in
a dose-dependent manner. Runx2, Msx2, BMP2 and OPN are important
genes associated with calcification. Runx2 is an osteogenic and
chondrogenic transcription factor regulated by post-translational
modifications, sub-nuclear localization, and modulatory
protein-protein interactions (28). A previous study determined that the
expression level of Runx2 was increased in atherosclerotic
calcification and endochondral mineralization programs (29). Hydrogen peroxide was observed to
activate osteogenic Runx2 (30)
and Msx2/Wnt (31) signaling, thus
enhancing mineralization. A previous study indicated that BMP2
increased the secretion of OPN through upregulation of ALP, which
led to the degradation of tissue pyrophosphate (32). Runx2 promoted OPN expression in
cardiovascular calcification (33). The present study revealed that
HMGB1 was expressed in calcified valves, and that HMGB1 treatment
induced calcification of AVICs and osteogenic protein expression.
Therefore, the present findings emphasized the importance of HMGB1
in the calcification of CAVD.
The results of the present study demonstrated that
TLR4, as a receptor of HMGB1, mediated the extent of the
inflammatory response and participated in the pathology of CAVD.
Zeng et al (18,34) determined that AVICs from stenotic
valves expressed higher levels of BMP2 in response to TLR4
stimulation and NF-κB activation (17,33).
In addition, Deng et al (16) demonstrated that adult AVICs exhibit
greater inflammatory and osteogenic responses to TLR4 stimulation
(34). Together, these results
suggest that the HMGB1-induced mineralization of AVICs may be
regulated by TLR4. The results of the present study indicated that
the calcification region of aortic valves expressed higher levels
of TLR4 and HMGB1 when compared with control valves. ALP activity
and the level of alizarin red S staining were increased by
incubating cells with HMGB1; however, these effects were
significantly reduced in TLR4−/− mice and when anti-TLR4
antibodies were used to inhibit TLR4. These findings demonstrate
that TLR4 may mediate HMGB1-induced AVIC calcification.
Furthermore, the protein expression levels of Runx2, Msx2, BMP2 and
OPN were significantly reduced following TLR4 knockout or
inhibition. Therefore, TLR4 altered calcification and the
expression of a various of osteogenic proteins involved in
HMGB1-induced AVIC mineralization.
CAVD is a chronic inflammatory disease where the
inflammatory response accelerates calcification (6,7).
HMGB1 may induce phosphorylation of p38 and NF-κB, thus activating
an inflammatory response via TLR4 (15). The present study incubated AVICs
with different doses of HMGB1 for 60 min, and the activation of p38
and NF-κB increased in a dose-dependent manner. Additionally, p38
and NF-κB phosphorylation was inhibited following TLR4 knockout or
inhibition. These results demonstrated that TLR4 mediated
HMGB1-induced p38 and NF-κB phosphorylation, thereby increasing the
inflammatory response in AVICs. In addition, this suggests that the
p38/NF-κB signaling pathway may be a promoter of inflammatory
calcification that participates in the pathology of CAVD.
In conclusion, the present study demonstrated the
expression of HMGB1 and TLR4 in the calcification region of CAVD
arterial valves. Using an in vitro model it was revealed
that this activation was functionally and physically associated
with AVIC calcification. The clinical implication of the present
study was that deactivation of HMGB1 and TLR4 may effectively
attenuate the pathogenesis of CAVD.
References
|
1
|
Freeman RV and Otto CM: Spectrum of
calcific aortic valve disease: Pathogenesis, disease progression,
and treatment strategies. Circulation. 111:3316–3326. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iung B, Baron G, Butchart EG, Delahaye F,
Gohlke-Bärwolf C, Levang OW, Tornos P, Vanoverschelde JL, Vermeer
F, Boersma E, et al: A prospective survey of patients with valvular
heart disease in Europe: The Euro heart survey on valvular heart
disease. Eur Heart J. 24:1231–1243. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rajamannan NM, Evans FJ, Aikawa E,
Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS,
Mathieu P, O'Brien KD, et al: Calcific aortic valve disease: Not
simply a degenerative process: A review and agenda for research
from the National heart and lung and blood institute aortic
stenosis working group. Executive summary: Calcific aortic valve
disease-2011 update. Circulation. 124:1783–1791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jian B, Narula N, Li QY, Mohler ER III and
Levy RJ: Progression of aortic valve stenosis: TGF-beta1 is present
in calcified aortic valve cusps and promotes aortic valve
interstitial cell calcification via apoptosis. Ann Thorac Surg.
75:457–466. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galeone A, Brunetti G, Oranger A, Greco G,
Di Benedetto A, Mori G, Colucci S, Zallone A, Paparella D and Grano
M: Aortic valvular interstitial cells apoptosis and calcification
are mediated by TNF-related apoptosis-inducing ligand. Int J
Cardiol. 169:296–304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cotè N, Mahmut A, Bosse Y, Couture C, Pagé
S, Trahan S, Boulanger MC, Fournier D, Pibarot P and Mathieu P:
Inflammation is associated with the remodeling of calcific aortic
valve disease. Inflammation. 36:573–581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Brien KD: Epidemiology and genetics of
calcific aortic valve disease. J Investig Med. 55:284–291. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andersson U, Erlandsson-Harris H, Yang H
and Tracey KJ: HMGB1 as a DNA-binding cytokine. J Leukoc Biol.
72:1084–1091. 2002.PubMed/NCBI
|
|
9
|
Ueda T, Chou H, Kawase T, Shirakawa H and
Yoshida M: Acidic C-tail of HMGB1 is required for its target
binding to nucleosome linker DNA and transcription stimulation.
Biochemistry. 43:9901–9908. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamada S and Maruyama I: HMGB1, a novel
inflammatory cytokine. Clin Chim Acta. 375:36–42. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Ling Y, Huang M, Yin T, Gou SM, Zhan
NY, Xiong JX, Wu HS, Yang ZY and Wang CY: Heparin inhibits the
inflammatory response induced by LPS and HMGB1 by blocking the
binding of HMGB1 to the surface of macrophages. Cytokine. 72:36–42.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nativel B, Marimoutou M, Thon-Hon VG,
Gunasekaran MK, Andries J, Stanislas G, Planesse C, Da Silva CR,
Césari M, Iwema T, et al: Soluble HMGB1 is a novel adipokine
stimulating IL-6 secretion through RAGE receptor in SW872
preadipocyte cell line: Contribution to chronic inflammation in fat
tissue. PLoS One. 8:e760392013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hou C, Kong J, Liang Y, Huang H, Wen H,
Zheng X, Wu L and Chen Y: HMGB1 contributes to allergen-induced
airway remodeling in a murine model of chronic asthma by modulating
airway inflammation and activating lung fibroblasts. Cell Mol
Immunol. 12:409–423. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Min HJ, Kim SJ, Kim TH, Chung HJ, Yoon JH
and Kim CH: Level of secreted HMGB1 correlates with severity of
inflammation in chronic rhinosinusitis. Laryngoscope.
125:E225–E330. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lotze MT and Tracey KJ: High-mobility
group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal.
Nat Rev Immunol. 5:331–342. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deng XS, Meng X, Zeng Q, Fullerton D,
Mitchell M and Jaggers J: Adult aortic valve interstitial cells
have greater responses to toll-like receptor 4 stimulation. Ann
Thorac Surg. 99:62–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng X, Ao L, Song Y, Babu A, Yang X, Wang
M, Weyant MJ, Dinarello CA, Cleveland JC Jr and Fullerton DA:
Expression of functional Toll-like receptors 2 and 4 in human
aortic valve interstitial cells: Potential roles in aortic valve
inflammation and stenosis. Am J Physiol Cell Physiol. 294:C29–C35.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng Q, Song R, Ao L, Weyant MJ, Lee J, Xu
D, Fullerton DA and Meng X: Notch1 promotes the pro-osteogenic
response of human aortic valve interstitial cells via modulation of
ERK1/2 and nuclear factor-κB activation. Arterioscler Thromb Vasc
Biol. 33:1580–1590. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mathieu P, Boulanger MC and Bouchareb R:
Molecular biology of calcific aortic valve disease: Towards new
pharmacological therapies. Expert Rev Cardiovasc Ther. 12:851–862.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mohler ER III, Chawla MK, Chang AW,
Vyavahare N, Levy RJ, Graham L and Gannon FH: Identification and
characterization of calcifying valve cells from human and canine
aortic valves. J Heart Valve Dis. 8:254–260. 1999.PubMed/NCBI
|
|
21
|
Mathieu P, Voisine P, Pépin A, Shetty R,
Savard N and Dagenais F: Calcification of human valve interstitial
cells is dependent on alkaline phosphatase activity. J Heart Valve
Dis. 14:353–357. 2005.PubMed/NCBI
|
|
22
|
Osman L, Yacoub MH, Latif N, Amrani M and
Chester AH: Role of human valve interstitial cells in valve
calcification and their response to atorvastatin. Circulation.
114:(1 Suppl). I547–I552. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Towler DA: Molecular and cellular aspects
of calcific aortic valve disease. Circ Res. 113:198–208. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ji Y, Liu J, Wang Z and Liu N: Angiotensin
II induces inflammatory response partly via toll-like receptor
4-dependent signaling pathway in vascular smooth muscle cells. Cell
Physiol Biochem. 23:265–276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pasipoularides A: Calcific Aortic valve
disease: Part 1-molecular pathogenetic aspects, hemodynamics, and
adaptive feedbacks. J Cardiovasc Transl Res. 9:102–118. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Shan J, Yang W, Zheng H and Xue S:
High mobility group box 1 (HMGB1) mediates high-glucose-induced
calcification in vascular smooth muscle cells of saphenous veins.
Inflammation. 36:1592–1604. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lai CH, Shi GY, Lee FT, Kuo CH, Cheng TL,
Chang BI, Ma CY, Hsu FC, Yang YJ and Wu HL: Recombinant human
thrombomodulin suppresses experimental abdominal aortic aneurysms
induced by calcium chloride in mice. Ann Surg. 258:1103–1110. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Steitz SA, Speer MY, Curinga G, Yang HY,
Haynes P, Aebersold R, Schinke T, Karsenty G and Giachelli CM:
Smooth muscle cell phenotypic transition associated with
calcification: Upregulation of Cbfa1 and downregulation of smooth
muscle lineage markers. Circ Res. 89:1147–1154. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tyson KL, Reynolds JL, McNair R, Zhang Q,
Weissberg PL and Shanahan CM: Osteo/chondrocytic transcription
factors and their target genes exhibit distinct patterns of
expression in human arterial calcification. Arterioscler Thromb
Vasc Biol. 23:489–494. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Byon CH, Javed A, Dai Q, Kappes JC,
Clemens TL, Darley-Usmar VM, McDonald JM and Chen Y: Oxidative
stress induces vascular calcification through modulation of the
osteogenic transcription factor Runx2 by AKT signaling. J Biol
Chem. 283:15319–15327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lai CF, Shao JS, Behrmann A, Krchma K,
Cheng SL and Towler DA: TNFR1-activated reactive oxidative species
signals up-regulate osteogenic Msx2 programs in aortic
myofibroblasts. Endocrinology. 153:3897–3910. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Steitz SA, Speer MY, McKee MD, Liaw L,
Almeida M, Yang H and Giachelli CM: Osteopontin inhibits mineral
deposition and promotes regression of ectopic calcification. Am J
Pathol. 161:2035–2046. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sowa AK, Kaiser FJ, Eckhold J, Kessler T,
Aherrahrou R, Wrobel S, Kaczmarek PM, Doehring L, Schunkert H,
Erdmann J and Aherrahrou Z: Functional interaction of osteogenic
transcription factors Runx2 and Vdr in transcriptional regulation
of Opn during soft tissue calcification. Am J Pathol. 183:60–68.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zeng Q, Song R, Ao L, Xu D, Venardos N,
Fullerton DA and Meng X: Augmented osteogenic responses in human
aortic valve cells exposed to oxLDL and TLR4 agonist: A mechanistic
role of Notch1 and NF-κB interaction. PLoS One. 9:e954002014.
View Article : Google Scholar : PubMed/NCBI
|