Introduction
Previous studies using cell lines and animal models
have suggested that endothelial cells are the first to be affected
by heat stress injury and that severe heat stroke is prominently
characterized by damaged endothelial cells (1,2). In
addition, a previous study demonstrated that, in the course of the
acute phase response to heat stress, significant endothelial cell
apoptosis may be induced (3).
Although endothelial cell apoptosis appears to be important in heat
stroke, the molecular mechanism underlying the induction of
endothelial cell apoptosis by heat stress remains poorly
understood.
Protease-activated receptor 1 (PAR1), a G
protein-coupled transmembrane receptor, was first described as a
high-affinity thrombin receptor (4). PAR1 is expressed on the surface of
almost all cell types in the blood vessel wall, including
endothelial cells, smooth muscle cells, platelets, neutrophils and
macrophages. Following activation of PAR1 via proteolytic cleavage
of its extracellular N-terminus by the serine proteinase thrombin,
it induces platelet activation, cell proliferation, vascular
development, cell apoptosis and angiogenesis (4–6). Our
previous study suggested the involvement of PAR1 in endothelial
hyperpermeability in heat stress (7). However, whether PAR1 is involved in
heat stress-induced endothelial cell apoptosis remains to be
determined.
Nuclear factor (NF)-κB signaling is associated with
the transcriptional regulation of various genes involved in
inflammatory responses, cell growth, survival and apoptosis
(8,9). Our previous study demonstrated that
NF-κB signaling is crucial in preventing heat stress-induced
apoptosis of human umbilical vein endothelial cells (HUVECs)
(10). In addition, it has been
reported that stimulation of PAR1, via the activation of the NF-κB
signaling pathway, has a vital function in prostate cancer cell
survival (11). However, whether
PAR1 serves a role in NF-κB activation in heat stressed endothelial
cells remains unclear. A signal-transducing transcription factor of
the activating protein 1 (AP-1) family, c-Jun has been previously
implicated in cell cycle progression, differentiation and cell
transformation, and has been linked to apoptosis (12,13).
Heat stress-induced AP-1 activation in HeLa cells has been
demonstrated in earlier studies (14). However, whether crosstalk between
PAR, NF-κB and c-Jun occurs and affects endothelial cell apoptosis
remains poorly understood.
The current study demonstrated that increased
expression of heat stress-induced PAR1 may result in induction of
B-cell lymphoma 2 (Bcl-2) associated X (Bax) expression and
inhibition of myeloid cell leukemia 1 (Mcl-1) expression, which
activated caspase-3 to induce apoptosis in HUVECs. The present
study further clarifies the role of PAR1 in the regulation of NF-κB
and c-Jun activation induced by heat stress treatment. In addition,
the current study demonstrated that c-Jun expression is the
critical event in heat stress-induced apoptosis.
Materials and methods
Cell culture and treatment
HUVECs were purchased from the Shanghai Institute of
Biochemistry and Cell Biology, Chinese Academy of Sciences
(Shanghai, China). HUVECs were cultured in Dulbecco's Modified
Eagle's medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum
(FBS; Invitrogen; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin (Invitrogen; Thermo Fisher Scientific, Inc.) and 100
µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.), at
37°C in a humidified atmosphere of 5% CO2. To induce
heat stress, culture dishes were placed in a circulating water bath
at 37±0.5°C for the control group or at 43±0.5°C for the heat
stress group, for 90 min. Culture media were replaced with fresh
media and the cells were further incubated at 37°C for the
indicated times. HUVECs (~1×106) were pretreated with
DMSO, 40 µM TFLLR-NH2 (TF; cat. no. 1464; Tocris Bioscience,
Bristol, UK) for 10 min or 150 nM SCH79797 (SCH; cat. no. 1592;
Tocris Bioscience) for 1 h at 37°C prior to incubation at 37°C
(control) or 43°C (heat stress) for 90 min, followed by a 6 or 12 h
recovery period at 37°C.
Hoechst 33342 staining
HUVECs were washed with PBS three times for 2 min,
stained with Hoechst 33342 (1:1,000) at 37°C for 20 min, and
subsequently washed three times with PBS for 2 min. Images were
acquired using a fluorescence microscope.
Western blot analysis
HUVECs were pretreated at 37°C or 43°C for 90 min,
and further incubated for 0, 2, 6 or 12 h. The pre-cleaned HUVECs
were homogenized in radioimmunoprecipitation buffer with
phenylmethylsulfonyl fluoride (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). Following centrifugation at 14,000 × g at 4°C
for 10 min, the supernatants were used for western blot analysis.
Protein concentration was determined using a Bicinchoninic Acid
Protein assay kit (Thermo Fisher Scientific, Inc.). Proteins (20
µg/well) were separated by SDS-PAGE using 10% SDS polyacrylamide
gels and transferred onto polyvinylidene difluoride membranes.
Membranes were blocked with blocking solution (5% skimmed milk
diluted with PBS) at room temperature for 2 h, followed by
incubation with primary antibodies. The following primary
antibodies were used: GAPDH (rabbit antibodies; dilution, 1:2,000;
cat. no. ab70699; Abcam, Cambridge, MA, USA), PAR1 (rabbit
antibodies; dilution, 1:2,000; cat. no. ab30961; Abcam), Mcl-1
(rabbit antibodies; dilution, 1:2,000; cat. no. ab53709; Abcam),
Bax (rabbit antibodies; dilution, 1:2,000; cat. no. ab32503;
Abcam), phosphorylated (p)-c-Jun (rabbit antibodies; dilution,
1:2,000; cat. no. 8222S; Cell Signaling Technology, Inc., Danvers,
MA, USA), c-Jun (rabbit antibodies; dilution, 1:2,000; cat. no.
9165p; Cell Signaling Technology, Inc.) overnight at 4°C. A
horseradish peroxidase-conjugated anti-rabbit IgG antibody was used
as the secondary antibody (dilution, 1:5,000; cat. no. TA130023;
OriGene Technologies, Inc., Beijing, China) for incubation at room
temperature for 2 h. Antibodies were detected with Enhanced
Chemiluminescence Western Blot Detection reagent (Pierce; Thermo
Fisher Scientific, Inc.). Membranes were exposed to light-sensitive
film and quantified using ImageJ software (version 1.3.4.67;
National Institutes of Health, Bethesda, MD, USA).
Measurement of caspase 3 activity in
vitro
Caspase 3 activity was determined using a caspase 3
activity assay kit, according to the manufacturer's instructions
(Biovision, Inc., Milpitas, CA, USA). Briefly, the cells were lysed
in caspase 3 sample lysis buffer (Biovision, Inc.). The homogenates
were then centrifuged at 10,000 × g and 4°C for 10 min and the
supernatant was collected for protein estimation using
bicinchoninic acid for the caspase 3 assay. The cell lysates were
then exposed to the DEVD substrate conjugate provided in the kit
for 1 h at 37°C. The sample was measured in an automatic microplate
reader (SpectraMax M5; Molecular Devices, LLC, Sunnyvale, CA, USA)
at an excitation of 400 nm and emission of 505 nm.
Flow cytometric analysis of cell
apoptosis using annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) staining
Cell apoptosis was analyzed with an Annexin V-FITC
Apoptosis kit (Lianke Biological Engineering Co., Ltd., Zheijiang,
China) according to the manufacturer's protocol. HUVECs
(~1×106) were collected, washed with ice-cold PBS and
resuspended in binding buffer containing 5 µl annexin V-FITC and 10
µl PI. Following incubation at room temperature for 15 min, samples
were analyzed using a flow cytometer (FACSCanto™ II; BD
Biosciences, San Jose, CA, USA).
Small interfering (si)RNA
transfection
PAR1 siRNA, c-Jun siRNA and negative control siRNA
were designed and synthesized by Shanghai GenePharma Co., Ltd.
(Shanghai, China). The sequences for each gene and the negative
control are presented in Table I.
A total of 24 h prior to transfection, HUVECs were seeded onto a
6-well plate (Nest Biotechnology Co., Ltd., Wuxi, China) at 30–50%
confluence. HUVECs were subsequently transfected with 1 µM siRNAs
using siRNAMate Transfection reagent (Shanghai GenePharma Co.,
Ltd.), according to the manufacturer's protocol. After 48–72 h,
cells were collected for further experiments.
 | Table I.Oligonucleotide sequences used in
small interfering RNA experiments. |
Table I.
Oligonucleotide sequences used in
small interfering RNA experiments.
| Target gene | Primer | Sequence (5′-3′) |
|---|
| PAR1 | Sense |
GAACCCUGCUCGAAGGCUACUATT |
|
| Antisense |
UUCUCCGAACGUGUCACGUTT |
| c-Jun | Sense |
GCAAACCUCAGCAACUUCATT |
|
| Antisense |
UGAAGUUGCUGAGGUUUGCTT |
| Negative control | Sense |
UUCUCCGAACGUGUCACGUTT |
|
| Antisense |
ACGUGACACGUUCGGAGAATT |
Adenoviral infection
An adenovirus (Ad) constitutively overexpressing
PAR1 and empty adenovirus were constructed by ViGene Biosciences,
Inc. (Rockville, MD, USA). 1×105 cells/well were
infected with 100 MOI Ad in serum-free DMEM for 6 h, following
which the media was replaced with DMEM supplemented with 10%
FBS.
Measurement of NF-κB DNA-binding
capacity by enzyme-linked immunosorbent assay (ELISA)
Nuclear extracts were prepared from treated and
control HUVECs using a nuclear extraction kit (Active Motif,
Carlsbad, CA USA; cat. no. 40410) and were performed according to
the manufacturer's instructions. These extracts were then assayed
determine the ability of nuclear NF-κB p65 to bind a DNA consensus
sequence provided by an ELISA-based TransAM NF-κB p65 transcription
factor assay kit (Active Motif; cat. no. 40096), according to the
manufacturer's protocol.
Statistical analysis
All data were analyzed for statistical significance
using SPSS software version 13.0 (SPSS, Inc., Chicago, IL, USA).
Data are expressed as the mean ± standard deviation from at least
three independent experiments performed in duplicate. One-way
analysis of variance was performed for multiple comparisons
followed by Fisher's least significant difference post hoc
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of heat stress on cell
apoptosis and the expression levels of apoptosis-associated
proteins in HUVECs
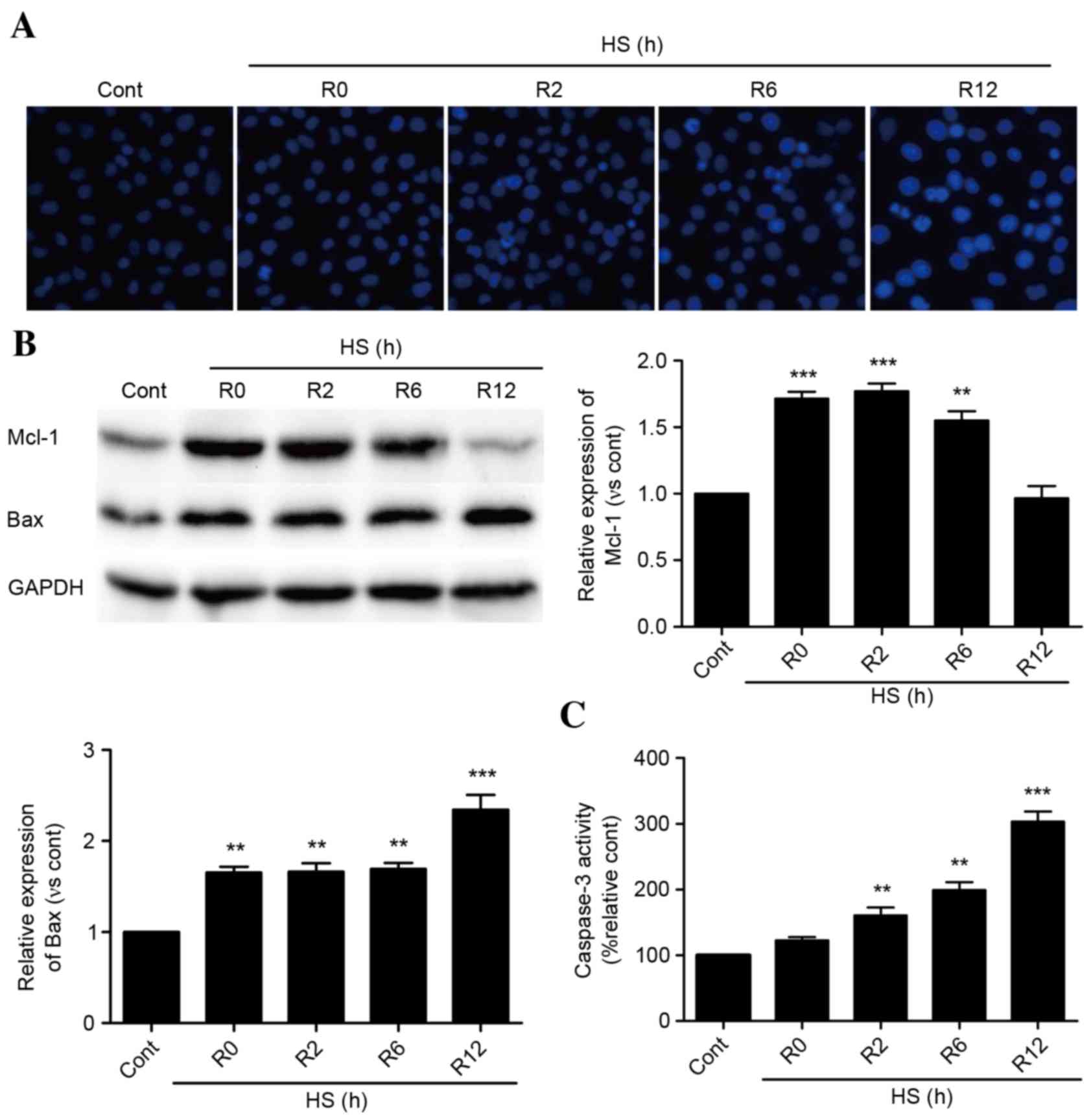
As presented in Fig.
1A, time-dependent apoptosis of HUVECs occurred following
exposure to heat stress for 90 min and incubation for 0–12 h. Heat
stress increased the protein expression levels of Mcl-1 rapidly,
and these levels were sustained for 6 h. Following a 12-h recovery
period, Mcl-1 protein expression levels decreased to baseline
levels (Fig. 1B). Bax protein
expression levels were increased immediately following heat stress,
and experienced a further significant increase at 12 h (Fig. 1B). Caspase-3 activity levels
followed a similar pattern (Fig.
1C).
Role of PAR1 in activating heat
stress-induced apoptosis in HUVECs
Our previous study indicated that heat stress may
increase PAR1 expression, a key step in disrupting endothelial
barrier function in response to heat stress (7). To identify whether PAR1 engages in
heat stress-induced cell apoptosis, PAR1 siRNA- or adenovirus
expressing PAR1-transfected HUVECs were subjected to heat stress
treatment prior to a recovery period of 12 h. The knockdown and
overexpression of PAR1 were confirmed by western blot analysis
(Fig. 2A). Subsequently, flow
cytometry using annexin V-FITC/PI staining was conducted to analyze
the levels of apoptosis. There were increased levels of heat
stress-induced apoptosis in cells overexpressing PAR1 and decreased
levels following PAR1 knockdown (Fig.
2B). Following pretreatment of cells with the PAR1 agonist
TFLLR-NH2 or the inhibitor SCH79797, a similar pattern was observed
(Fig. 2C). These data suggested a
pro-apoptotic role for PAR1 in heat stress-induced HUVEC
apoptosis.
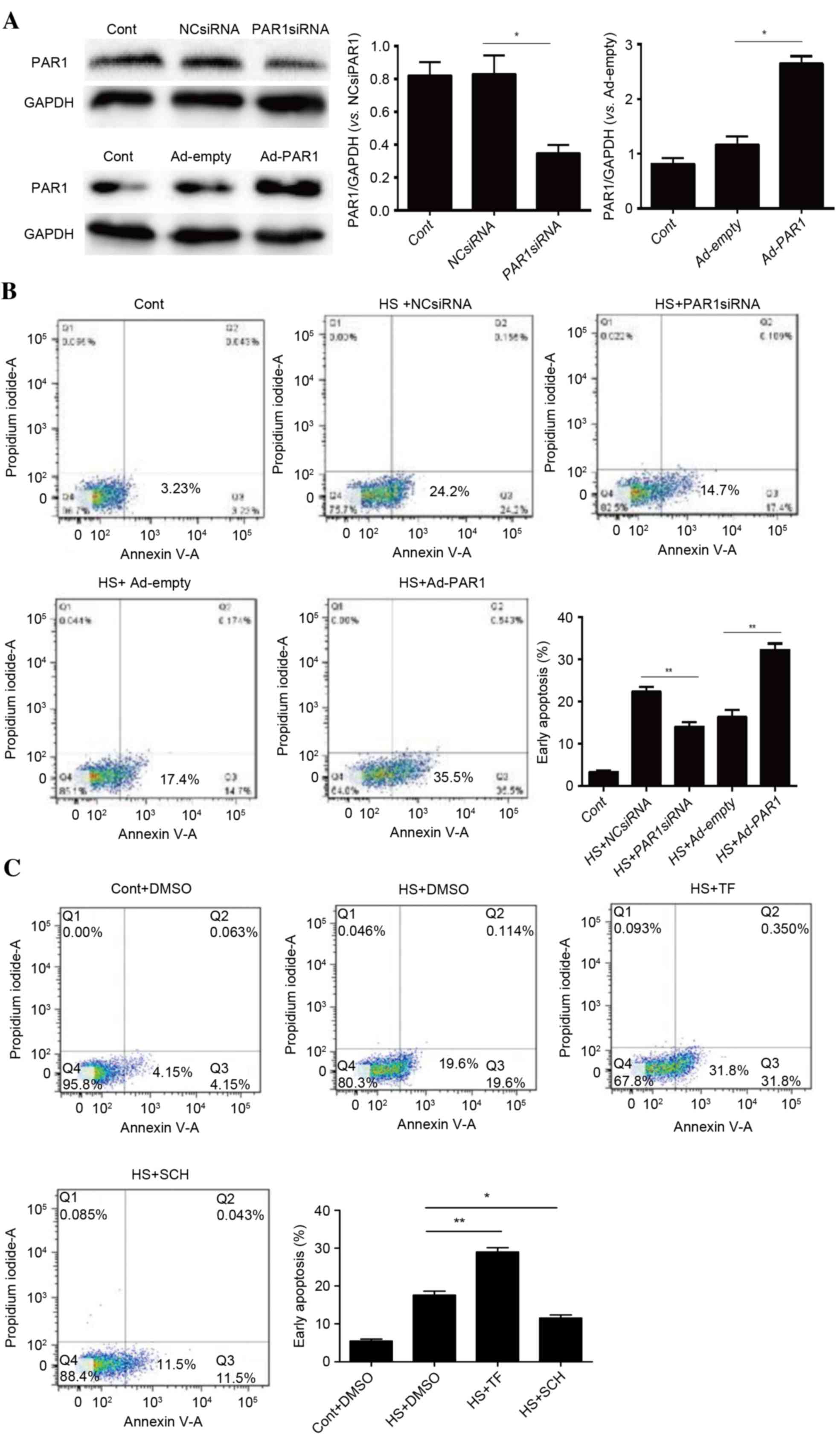 | Figure 2.Role of PAR1 in heat stress-induced
apoptosis in HUVECs. (A) NC siRNA, PAR1 siRNA, Ad-empty or Ad-PAR1
were transfected into HUVECs, thus achieving knockdown and
overexpression of PAR, as confirmed by western blot analysis. (B)
Transfected HUVECs were incubated at 37°C (control) or 43°C (heat
stress) for 90 min, followed by a 12-h recovery period at 37°C.
Apoptosis of HUVECs was analyzed using annexin V and propidium
iodide, and the percentage of early apoptotic cells (lower right
quadrant) was calculated. (C) Untransfected HUVECs were pretreated
with DMSO, 40 µM TF for 10 min or 150 nM SCH for 1 h prior to
incubation at 37°C (control) or 43°C (heat stress) for 90 min,
followed by a 12-h recovery period at 37°C. Apoptosis of HUVECs was
analyzed using annexin V and propidium iodide, and the percentage
of early apoptotic cells (lower right quadrant) was calculated.
Data are presented as the mean ± standard deviation of three
separate experiments. *P<0.05; **P<0.01. PAR1,
protease-activated receptor 1; HUVECs, human umbilical vein
endothelial cells; NC, negative control; siRNA, small interfering
RNA; Ad, adenovirus; DMSO, dimethyl sulfoxide; TF, TFLLR-NH2; SCH,
SCH79797; HS, heat stress; Cont, control. |
Influence of PAR1 on the expression
levels of apoptosis-associated proteins
Following the demonstration that PAR1 is involved in
the apoptosis of heat stress-induced HUVECs, subsequent experiments
further investigated whether PAR1 influenced the protein expression
levels of Mcl-1 and Bax, and caspase-3 activity. When compared with
the heat stressed Ad-empty group, PAR1 overexpression significantly
decreased the heat stress-induced high protein expression levels of
Mcl-1 and significantly increased those of Bax at 6 h (Fig. 3A). In addition, PAR1 knockdown
induced the opposite effect (Fig.
3B). Caspase-3 activity was significantly increased by PAR1
overexpression and was significantly decreased by PAR1 knockdown
(Fig. 3C), when compared with the
heat stressed Ad-empty group and NC siRNA group. Pretreatment of
cells with TF significantly decreased Mcl-1 and significantly
increased Bax protein expression levels (Fig. 3D). In addition, it also
significantly increased caspase-3 activity at 6 h when compared to
the heat stressed DMSO group (Fig.
3E). Pretreatment of cells with SCH had the opposite
effect.
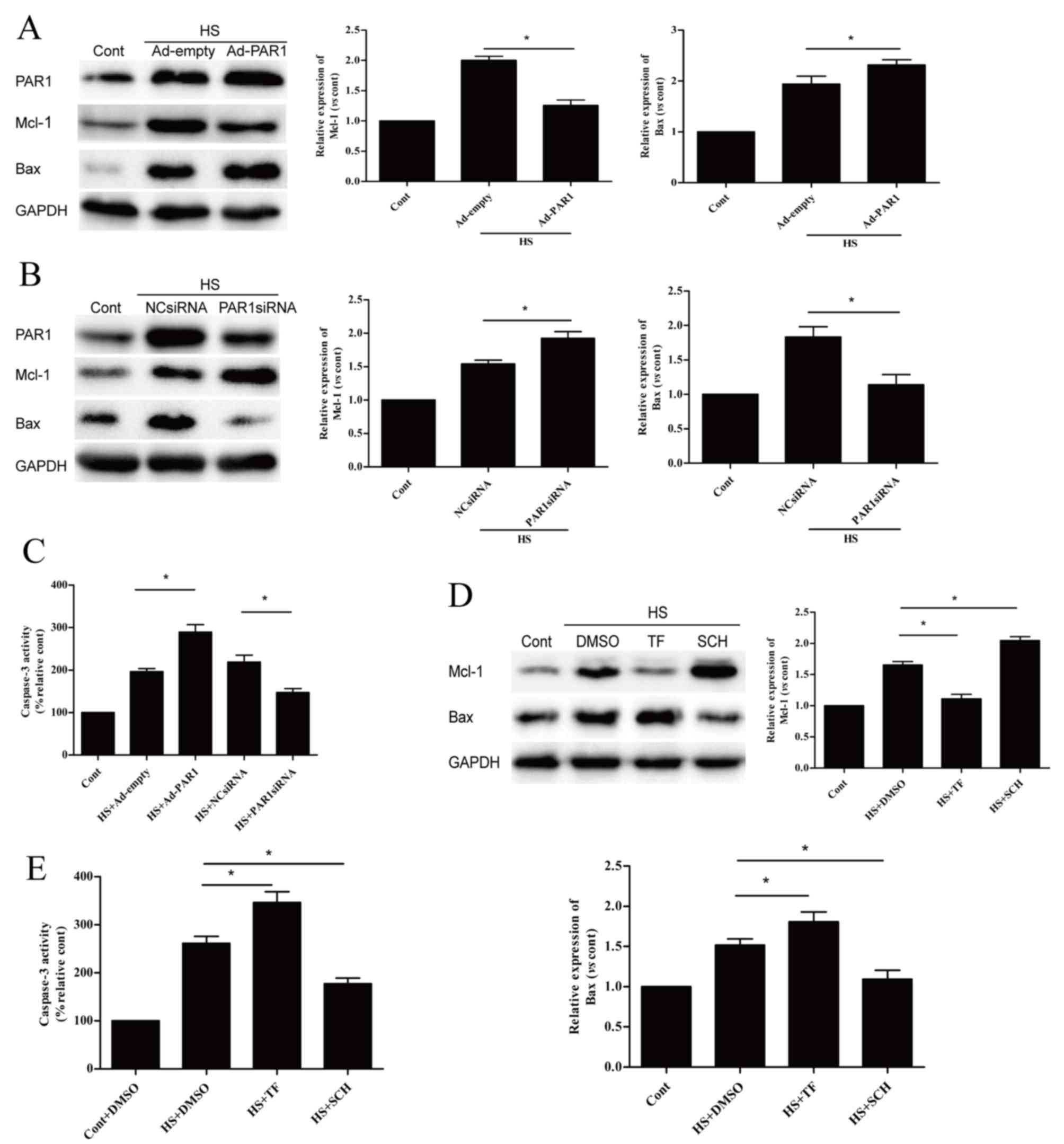 | Figure 3.Effects of PAR1 on
apoptosis-associated proteins following heat stress in HUVECs.
Protein expression levels of Mcl-1 and Bax were detected by western
blot analysis following transfection with (A) Ad-empty or Ad-PAR1,
or (B) NC siRNA or PAR1 siRNA for >48 h. HUVECs were incubated
at 37°C (control) or 43°C (heat stress) for 90 min, followed by a
6-h recovery period at 37°C. (C) Caspase-3 enzymatic activity was
measured in the cell lysates. Untransfected cells were pretreated
with DMSO or 40 µM TF for 10 min or 150 nM SCH for 1 h prior to
incubation at 37°C (control) or 43°C (heat stress) for 90 min,
followed by a 6-h recovery period at 37°C. (D) Mcl-1 and Bax
proteins were identified by western blotting analysis. (E)
Enzymatic activity of caspase-3 was measured in the cell lysates.
Data are presented as the mean ± standard deviation of three
independent experiments. *P<0.05; **P<0.01; ***P<0.001.
PAR1, protease-activated receptor 1; HUVECs, human umbilical vein
endothelial cells; Ad, adenovirus; NC, negative control; siRNA,
small interfering RNA; Mcl-1, myeloid cell leukemia 1; Bax, B-cell
lymphoma 2 associated X; TF, TFLLR-NH2; SCH, SCH79797; DMSO,
dimethyl sulfoxide; HS, heat stress; Cont, control. |
Influence of PAR1 on NF-κB and c-Jun
activation in heat stress-induced HUVECs
Our previous study reported that NF-κB signaling is
crucial in preventing heat stress-induced early apoptosis (10). The present study revealed that PAR1
siRNA and its inhibitor SCH increased heat stress-induced
activation of NF-κB (Fig. 4A).
This suggested that PAR1 exerts pro-apoptotic effects via
suppressing the activation of NF-κB.
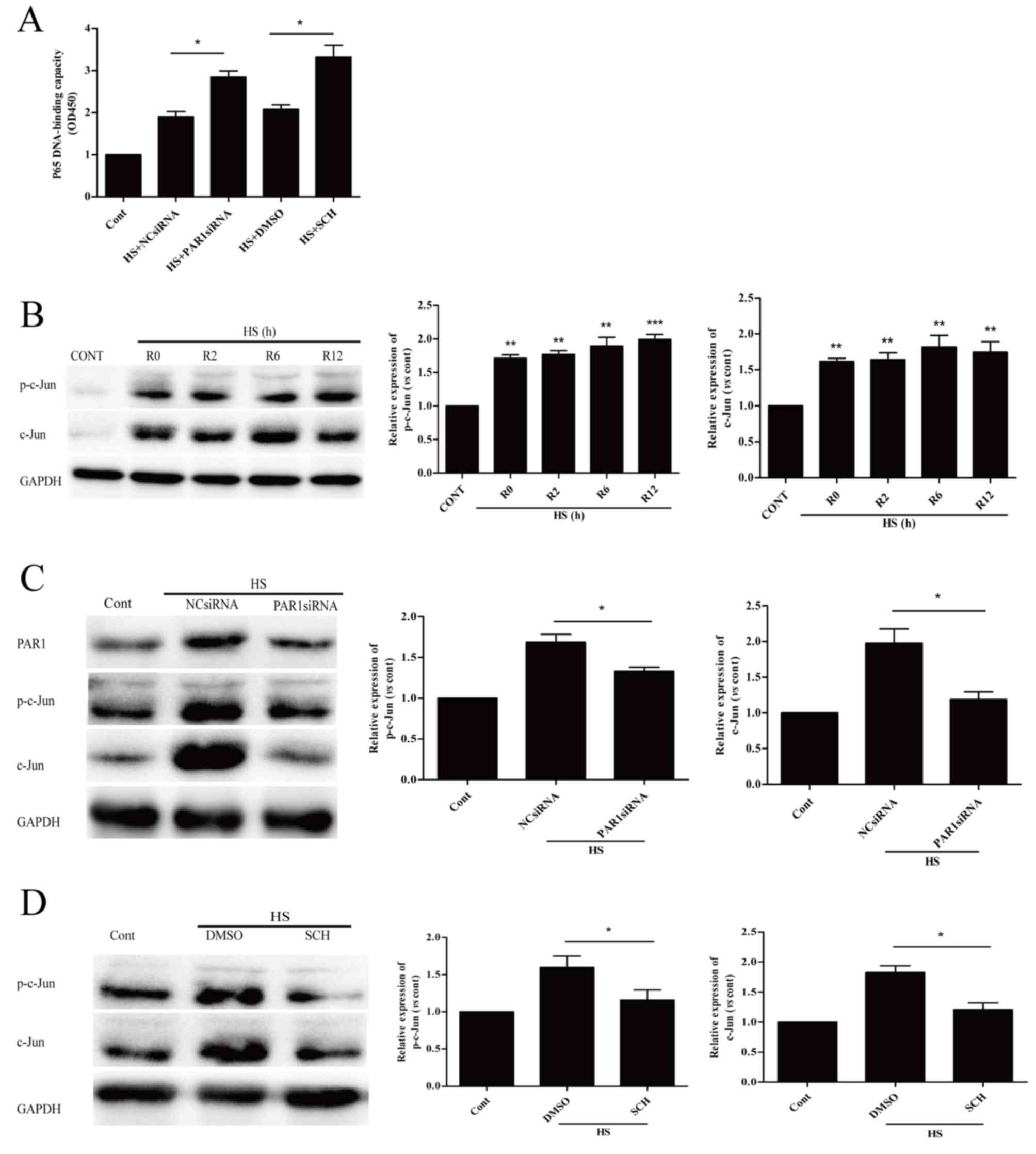 | Figure 4.PAR1 is involved in NF-κB and c-Jun
activation in heat stressed HUVECs. (A) After 48 h transfection
with NC siRNA or PAR1 siRNA, or an 1 h pretreatment with DMSO or
150 nM SCH, HUVECs were incubated at 37°C (control) or 43°C (heat
stress) for 90 min, followed by a 6-h recovery period at 37°C.
NF-κB binding to DNA was quantified. (B) HUVECs underwent an
incubation period at 37°C (control) or 43°C (heat stress) for 90
min, followed by a recovery period at 37°C for 0, 2, 6 or 12 h.
Protein expression levels of p-c-Jun and c-Jun were detected in
HUVECs by western blotting. **P<0.01 and ***P<0.001 vs.
control. (C) After 48 h transfection with NC siRNA or PAR1 siRNA,
or (D) an 1 h pretreatment with DMSO or 150 nM SCH, HUVECs were
incubated at 37°C (control) or 43°C (heat stress) for 90 min,
followed by a 6-h recovery period at 37°C. Expression of p-c-Jun
and c-Jun were detected by western blotting. Data are expressed as
the mean ± standard deviation of three independent experiments.
*P<0.05; **P<0.01; ***P<0.001. PAR1, protease-activated
receptor 1; NF-κB, nuclear factor-κB; HUVECs, human umbilical vein
endothelial cells; NC, negative control; siRNA, small interfering
RNA; SCH, SCH79797; DMSO, dimethyl sulfoxide; HS, heat stress;
Cont, control; R, recovery time; p, phosphorylated. |
In Fig. 4B, a rapid
increase in the protein expression levels of p-c-Jun and c-Jun was
observed and these high levels were sustained for >12 h. The
effects of PAR1 on p-c-Jun protein and c-Jun expression levels in
heat stressed HUVECs were additionally examined. PAR1-depleted
HUVECs significantly decreased the heat stress-induced c-Jun
phosphorylation and c-Jun expression levels at 6 h following heat
stress, when compared with heat stressed NC siRNA group (Fig. 4C). Pretreatment with SCH reduced
protein expression levels of p-c-Jun and c-Jun at 6 h following
heat stress (Fig. 4D). These
results suggested that PAR1 contributes to c-Jun activation in
response to heat stress.
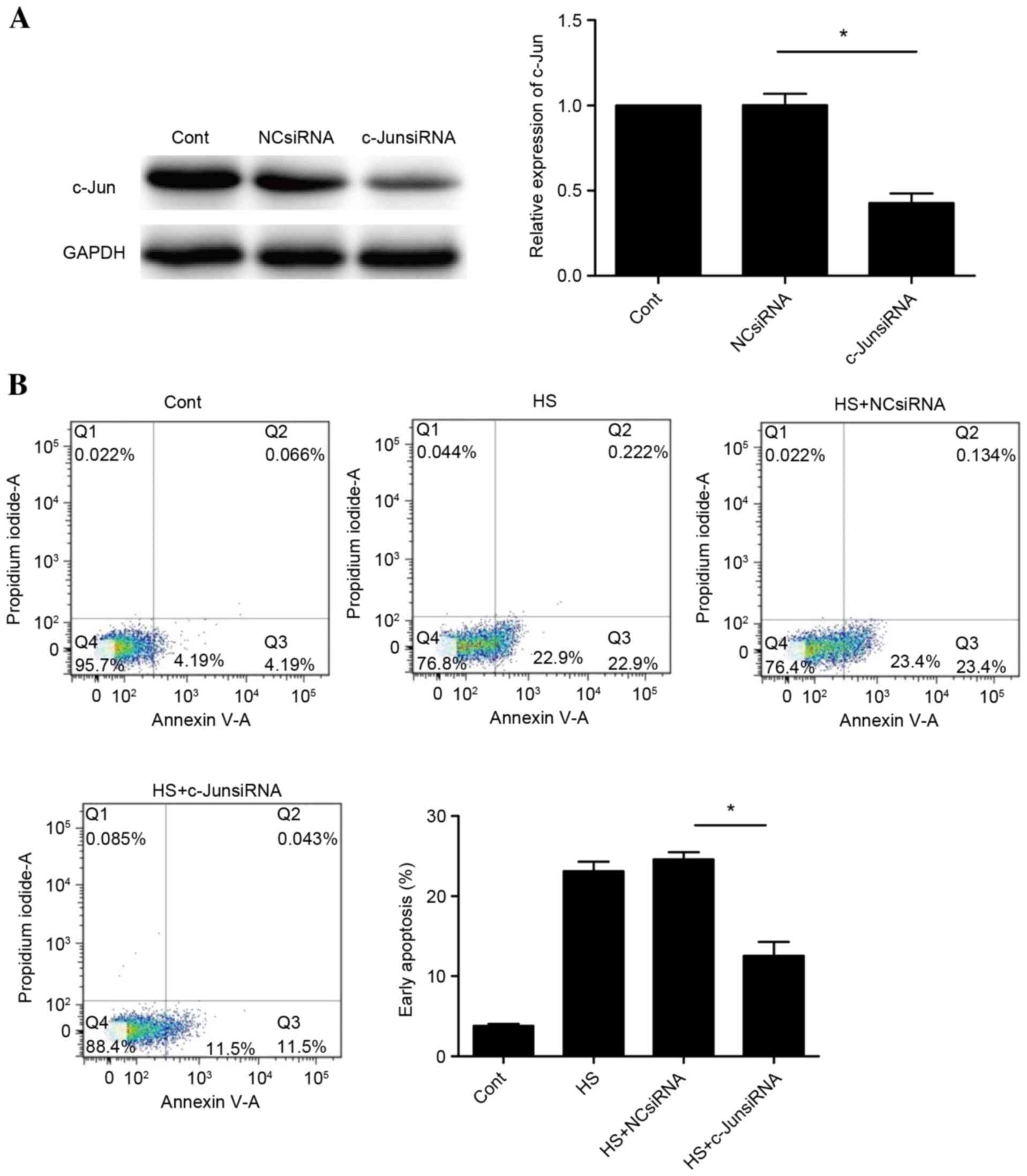
c-Jun is required for heat
stress-induced apoptosis of HUVECs
To determine whether c-Jun is involved in heat
stress-induced cell apoptosis, the effects of knockdown of c-Jun
with siRNA were assessed. Successful knockdown of c-Jun was
confirmed by western blot analysis (Fig. 5A). A significant suppression of
apoptosis was observed following c-Jun knockdown (P<0.05;
Fig. 5B). These observations
indicated that cell apoptosis induced by heat stress may be
mediated by c-Jun.
Discussion
Heat stroke, a life-threatening condition
characterized by a rapidly increasing core temperature to >40°C
and multiple organ dysfunction syndrome, is the primary cause of
morbidity and mortality in heat waves. An epidemiological study
reported that during heat waves in urban areas of the United
States, the incidence of heat stroke varied from 17.6 to 26.5 cases
per 100,000 population. The majority of people affected by classic
heat stroke are very young or elderly, poor, socially isolated and
do not have access to air conditioning (15). An important extracellular stimulus
for heat stroke is heat stress (16). Previous studies have demonstrated
that apoptosis may be significant in the pathophysiology of heat
stroke (17). The endothelial cell
is exposed early to heat stress injury; therefore, the mechanisms
underlying endothelial cell injury and cell death are relevant to
understanding the pathogenesis of heat stroke (3,16).
In the present study, the protein expression levels
of Mcl-1 and Bax, and caspase-3 activity, were increased by heat
stress in a time-dependent manner. This indicated the ability of
heat stress to induce apoptosis in HUVECs. A primary finding of the
current study suggested involvement of PAR1 in the apoptosis of
endothelial cells following heat stress. The presented data
suggested that blocking PAR1 with a specific inhibitor or siRNA
results in a reduction in HUVEC apoptosis, caspase-3 activity and
Bax expression, as well as an increase in Mcl-1 expression induced
by heat stress. In addition, following treatment with a PAR1
agonist or overexpression of PAR1, a significant increase in heat
stress-induced apoptosis, caspase-3 activity and the expression of
Bax was observed, accompanied by decreased protein expression
levels of Mcl-1. These results indicated that PAR1 is involved in
HUVEC apoptosis following exposure to heat stress. The Mcl-1
protein, which belongs to the Bcl-2 family of proteins, serves as
an anti-apoptotic factor (18). In
a previous study, a reduction of cytochrome c release and
caspase-9 activation was identified in cells containing reduced
levels of Bax, which suggested that HUVECs may be protected from
heat stress-induced apoptosis by decreases in Bax levels (19).
The importance of NF-κB signaling in regulating the
apoptotic program has been demonstrated in various cells (9). Our previous study suggested that the
NF-κB signaling pathway involving HSP27, ROS and MAPK, is activated
in response to heat stress, and this affords protection against
heat stress-induced HUVEC apoptosis (10). Previous studies have indicated that
c-Jun, a signal-transducing transcription factor of the AP-1
family, is associated with apoptosis (12). In the present study, PAR1 was
demonstrated to be involved in the regulation of the NF-кB
signaling pathway, and PAR1 functions upstream of c-Jun to modulate
its phosphorylation and protein accumulation. Furthermore, the
levels of cell apoptosis markedly decreased when c-Jun-targeting
siRNA inhibited c-Jun activation. These data suggested that a
pro-apoptotic pathway may be induced by PAR1 via inhibition of
NF-кB and c-Jun activation.
In conclusion, the current study provides, to the
best of our knowledge, the first demonstration of the potential
underlying mechanism by which PAR1 expression contributes to
apoptotic cell death induced by heat stress. It appears that the
interactions between PAR1, NF-κB and c-Jun are crucial for
apoptosis in HUVEC cells; the interaction between these three
proteins is worthy of further study. The results of the present
study suggested that an understanding of PAR1 regulation and the
underlying mechanism by which PAR1 induces cell apoptosis may lead
to the development of novel strategies for treating heat-associated
illness.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81471839) and the
project team of the Natural Science Foundation of Guangdong
Province (grant no. s2013030013217).
References
|
1
|
Bouchama A, Hammami MM, Haq A, Jackson J
and al-Sedairy S: Evidence for endothelial cell activation/injury
in heatstroke. Crit Care Med. 24:1173–1178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roberts GT, Ghebeh H, Chishti MA,
Al-Mohanna F, El-Sayed R, Al-Mohanna F and Bouchama A:
Microvascular injury, thrombosis, inflammation, and apoptosis in
the pathogenesis of heatstroke: A study in baboon model.
Arterioscler Thromb Vasc Biol. 28:1130–1136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brinton MR, Tagge CA, Stewart RJ, Cheung
AK, Shiu YT and Christensen DA: Thermal sensitivity of endothelial
cells on synthetic vascular graft material. Int J Hyperthermia.
28:163–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirano K and Kanaide H: Role of
protease-activated receptors in the vascular system. J Atheroscler
Thromb. 10:211–225. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Austin KM, Covic L and Kuliopulos A:
Matrix metalloproteases and PAR1 activation. Blood. 121:431–439.
2003. View Article : Google Scholar
|
|
6
|
Tressel SL, Kaneider NC, Kasuda S, Foley
C, Koukos G, Austin K, Agarwal A, Covic L, Opal SM and Kuliopulos
A: A matrix metalloprotease-PAR1 system regulates vascular
integrity, systemic inflammation and death in sepsis. EMBO Mol Med.
3:370–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu Q, Liu J, Wang Z, Guo X, Zhou G, Liu Y,
Huang Q and Su L: Heat stress-induced disruption of endothelial
barrier function is via PAR1 signaling and suppressed by Xuebijing
injection. PLoS One. 10:e01180572015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baldwin AS: Control of oncogenesis and
cancer therapy resistance by the transcription factor NF-kappaB. J
Clin Invest. 107:241–246. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Zhou G, Wang Z, Guo X, Xu Q, Huang
Q and Su L: NF-κB signaling is essential for resistance to heat
stress-induced early stage apoptosis in human umbilical vein
endothelial cells. Sci Rep. 5:135472015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tantivejkul K, Loberg RD, Mawocha SC, Day
LL, John LS, Pienta BA, Rubin MA and Pienta KJ: PAR1-mediated
NFkappaB activation promotes survival of prostate cancer cells
through a Bcl-xL-dependent mechanism. J Cell Biochem. 96:641–652.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bossy-Wetzel E, Bakiri L and Yaniv M:
Induction of apoptosis by the transcription factor c-Jun. EMBO J.
16:1695–1709. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Watson A, Eilers A, Lallemand D, Kyriakis
J, Rubin LL and Ham J: Phosphorylation of c-Jun is necessary for
apoptosis induced by survival signal withdrawal in cerebellar
granule neurons. J Neurosci. 18:751–762. 1998.PubMed/NCBI
|
|
14
|
Stein B, Baldwin AS Jr, Ballard DW, Greene
WC, Angel P and Herrlich P: Cross-coupling of the NF-kappa B p65
and Fos/Jun transcription factors produces potentiated biological
function. EMBO J. 12:3879–3891. 1993.PubMed/NCBI
|
|
15
|
Bouchama A and Knochel JP: Heat stroke. N
Engl J Med. 346:1978–1988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sohal RS, Sun SC, Colcolough HL and Burch
GE: Heat stroke. An electron microscopic study of endothelial cell
damage and disseminated intravascular coagulation. Arch Intern Med.
122:43–47. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakaguchi Y, Stephens LC, Makino M, Kaneko
T, Strebel FR, Danhauser LL, Jenkins GN and Bull JM: Apoptosis in
tumors and normal tissues induced by whole body hyperthermia in
rats. Cancer Res. 55:5459–5464. 1995.PubMed/NCBI
|
|
18
|
Morciano G, Giorgi C, Balestra D, Marchi
S, Perrone D, Pinotti M and Pinton P: Mcl-1 involvement in
mitochondrial dynamics is associated with apoptotic cell death. Mol
Biol Cell. 27:20–34. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gu ZT, Li L, Wu F, Zhao P, Yang H, Liu YS,
Geng Y, Zhao M and Su L: Heat stress induced apoptosis is triggered
by transcription independent p53, Ca(2+) dyshomeostasis and the
subsequent Bax mitochondrial translocation. Sci Rep. 5:114972015.
View Article : Google Scholar : PubMed/NCBI
|