Introduction
Limb-girdle muscular dystrophy [LGMD (MIM 253600 and
MIM 159000)] refers to a long list of Mendelian disorders
characterized by a progressive deterioration of proximal limb
muscles (1). Men and women are
equally affected, and the age of onset is usually between 5 and 30
years. The clinical course and expressivity can be variable,
ranging from severe forms with rapid onset and progression to mild
forms, in which the affected individual has relatively normal life
span and levels of activity (2).
The comprehensive analysis of clinical, electrophysiological and
physiological findings, imaging, and biochemical data, may
contribute to the clinical diagnosis of LGMD. However, genetic
techniques are the most efficient tools for the identification of
diagnosis and classification of LGMD. LGMD is divided into
autosomal dominant LGMD1 and autosomal recessive LGMD2 based on the
inheritance mode, and the appended letter refers to the order of
identification for different chromosomal loci, followed by the
inheritance mode (3). LGMD2Q is
mutation specific to the plectin (PLEC1) gene at chromosome 8q24.3.
The most common plectin-associated disorder is epidermolysis
bullosa simplex (EBS-MD), a rare autosomal-recessive skin
blistering disorder with late onset muscular dystrophy. In addition
to EBS-MD, Plectin mutations cause EBS-MD with a myasthenic
syndrome (EBS-MDMyS), EBS with pyloric atresia (EBS-PA; OMIM
612138) and EBS-Ogna (OMIM #131950), which affect the skin
exclusively (4). The PLEC mutation
has been identified in four patients from a consanguineous Turkish
family as a homozygous deletion (c.1_9del) in exon 1f of the gene,
which was the first report of plectin-associated LGMD2Q without
skin involvement or myasthenic syndrome (5).
In the present study, targeted sequencing using a
muscle disease gene panel was performed in a family with muscular
dystrophy. The novel compound heterozygous mutations, c.5995C>T
(p.Arg1999Trp) and c.9940T>A (p.Phe3314 Ile), in the PLEC gene
were identified as the genetic cause of LGMD2Q in this family.
Furthermore, defects in the plectin protein within the
gastrocnemius were determined using immunocytochemistry. To the
best of our knowledge, this is the second report of
plectin-associated LGMD2Q without other symptoms, however, a novel
genotype was found.
Patients and methods
Patients
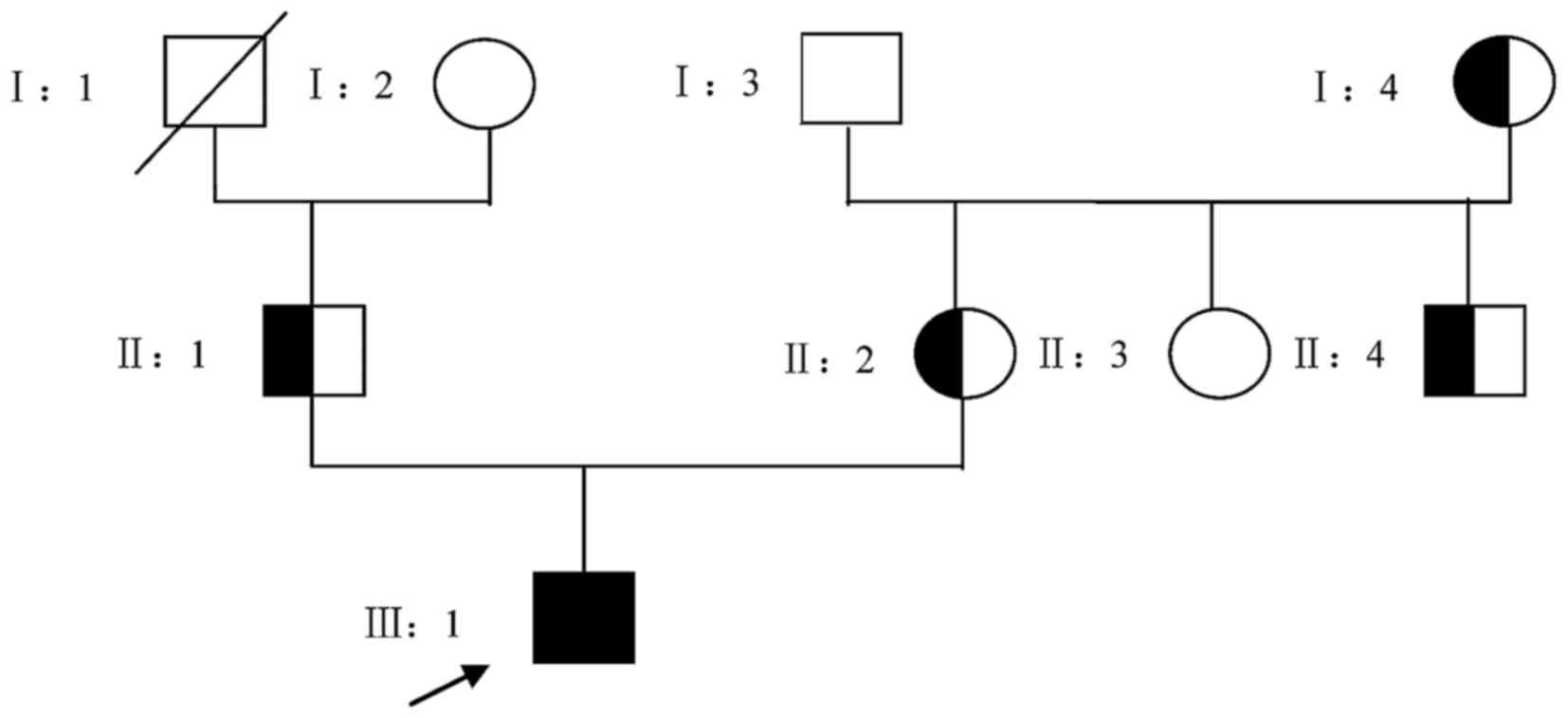
The patient examined in the present study was from a
three-generation, non-consanguineous Chinese family (Fig. 1). All the available individuals
were subjected to a thorough neurological examination. Individuals
were considered to be affected if they demonstrated progressive
proximal pelvic and/or shoulder girdle muscle weakness. Other
information, including the age at onset, symptoms at onset,
creatine kinase (CK) level, electromyography, muscle atrophy and
ultrasonic cardiogram, were obtained. A total of 200 unrelated
ethnically matched normal controls (male/female:100/100) without
diagnostic features of muscular dystrophy were recruited from
mainland China. Venous blood samples were obtained from all alive
members of the family. Genomic DNA was extracted according to a
standard phenol-chloroform extraction method. As Duchenne muscular
dystrophy is the most common form of inherited muscular dystrophy
(6), the dystrophin gene was
screened by multiplex ligation-dependent probe amplification
(MLPA). If no mutation was identified by MLPA, subsequent targeted
sequencing using a muscle disease gene panel was performed. Sanger
sequencing was used to confirm the mutation in the proband and the
other family members.
Exome capture
To systematically identify the disease-causing gene,
targeted sequencing using a muscle disease gene panel, containing
169 known MD genes, was performed in an affected individual from
the family. The muscle disease panel was a complete kit designed by
Zhongguancun Huakang Gene Institute (Beijing, China) and
synthetized using the Agilent SureSelect Target Enrichment
technique. Next-generation sequencing was performed on an
IlluminaNextSeq500 platform (Illumina, San Diego, CA, USA). A 4 µg
sample of genomic DNA from the proband was used to construct the
exome library. The genomic DNA was sheared into 150–250 bp by
sonication and hybridized for enrichment, according to the
manufacturer's protocol. The library enriched for the target region
was sequenced on the Illumina NextSeq500 platform to obtain
single-end reads with a read length of 75 bp.
Read mapping and variant analysis
The human reference genome was obtained from the
University of California Santa Cruz database (build 37.1; version
hg19; http://genome.ucsc.edu/), and sequence
alignment was performed using the Burrows-Wheeler Alignment tool
(version 0.7.10; http://bio-bwa.sourceforge.net/). High-quality
alignment was required to guarantee variant calling accuracy
(>0). Picard (version 1.119; http://sourceforge.net/projects/picard/) was used to
mark duplicates resulting from polymerase chain reaction
amplification. The Genome Analysis Tool kit (GATK; version 3.2–2;
https://software.broadinstitute.org/gatk/)
IndelRealigner and GATK RealignerTargetCreator were used to perform
realignment around the Indels. GATK BaseRecalibrator was used to
perform base quality score recalibration. GATK Variant Filtration
was performed to render the raw callsets suitable for meaningful
analysis. SAMtools (version 1.0; http://www.htslib.org/) was used to perform variant
calling and identify single nucleotide polymorphisms (SNPs) or
Indels. Following obtaining of the analysis-ready BAM alignment
result, the ANNOVAR tool (version 2014-11-12; http://annovar.openbioinformatics.org)
was performed to annotate SNPs and Indels. All candidate mutations
were filtered against the SNP database (dbSNP138; http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi),
International Hapmap Project (http://hapmap.ncbi.nlm.nih.gov/) and 1000 Genomes
Project (2012 April release; http://www.1000genomes.org/) to remove the
polymorphism loci. Polymorphism Phenotyping version 2 (PolyPhen-2),
sorting intolerant from tolerant (SIFT) and MutationTaster
(7) were used to predict whether
an amino acid substitution affected the function of the protein.
Sanger sequencing was used to validate the identified potential
disease-causing variant in the family members.
Muscle pathology
Biopsy of the right gastrocnemius was performed in
the patient. The morphology was observed under a microscope via
hematoxylin and eosin staining. Immunohistochemistry was used to
evaluate the plectin protein expression. Sections were incubated
with the anti-rabbit monoclonal antibody targeting plectin (cat.
no. ab32528; 1:100; Abcam, Cambridge, UK), and subsequently
incubated with the Supervision™ Universal (Anti-Mouse/Rabbit)
Detection Reagent (horseradish peroxidase conjugated; cat. no.
D-3004; Lab Vision Corporation; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The operation was performed according to the
manufacturer's protocol.
Results
Patients
The proband in the present study was a 7-year-old
boy with delayed independent walking at 2 years of age. Thereafter,
he developed additional signs of weakness, with occasional falls
and difficulties in climbing stairs. At his final follow-up, at 7
years of age, he demonstrated a Gowers sign, with proximal muscle
strength of 4/5 on the Medical Research Council scale (8). There were no fluctuations of symptoms
throughout the day. Muscle hypertrophy was observed. Serum CK at
the age of 7 years was 2,408 U/l (normal<174 U/l), CK-MB was 350
U/l (normal<25 U/l), alanine aminotransferase30 U/l
(normal<60 U/l), aspartate aminotransferase was75 U/l
(normal<60 U/l). Echocardiogram showed normal anatomy and heart
function. Pulmonary function tests showed no restriction. There
were no ocular or bulbar signs. Neuropsychological assessment was
normal. No mutation was identified by MLPA of DMD gene, and
subsequent targeted sequencing using a muscle disease gene panel
was performed.
Genetics
In the present study, a total of 2,840,000 reads of
75-bp single-end read sequence were obtained from the patient, with
2,830,000 reads (99.93%) aligned to the human reference sequence
and 193 Mb mapped to the targeted region. A sequence depth of
~112.24X provided sufficient data to obtain 99.9% coverage of the
target exome region. A total of 112 genetic variants were
identified in the coding regions or the splice sites. Known
variants, which were identified in dbSNP138, International Hapmap
Project and 1,000 Genome Project, were excluded. Synonymous
mutations were also excluded. PolyPhen-2, SIFT and MutationTaster
were used to predicted the possible effect of amino acid
substitutions and the functions of proteins. Applying the above
strategy, >96% candidate genes were reduced. Among the 169
selected genes, no candidate homozygous mutation was identified.
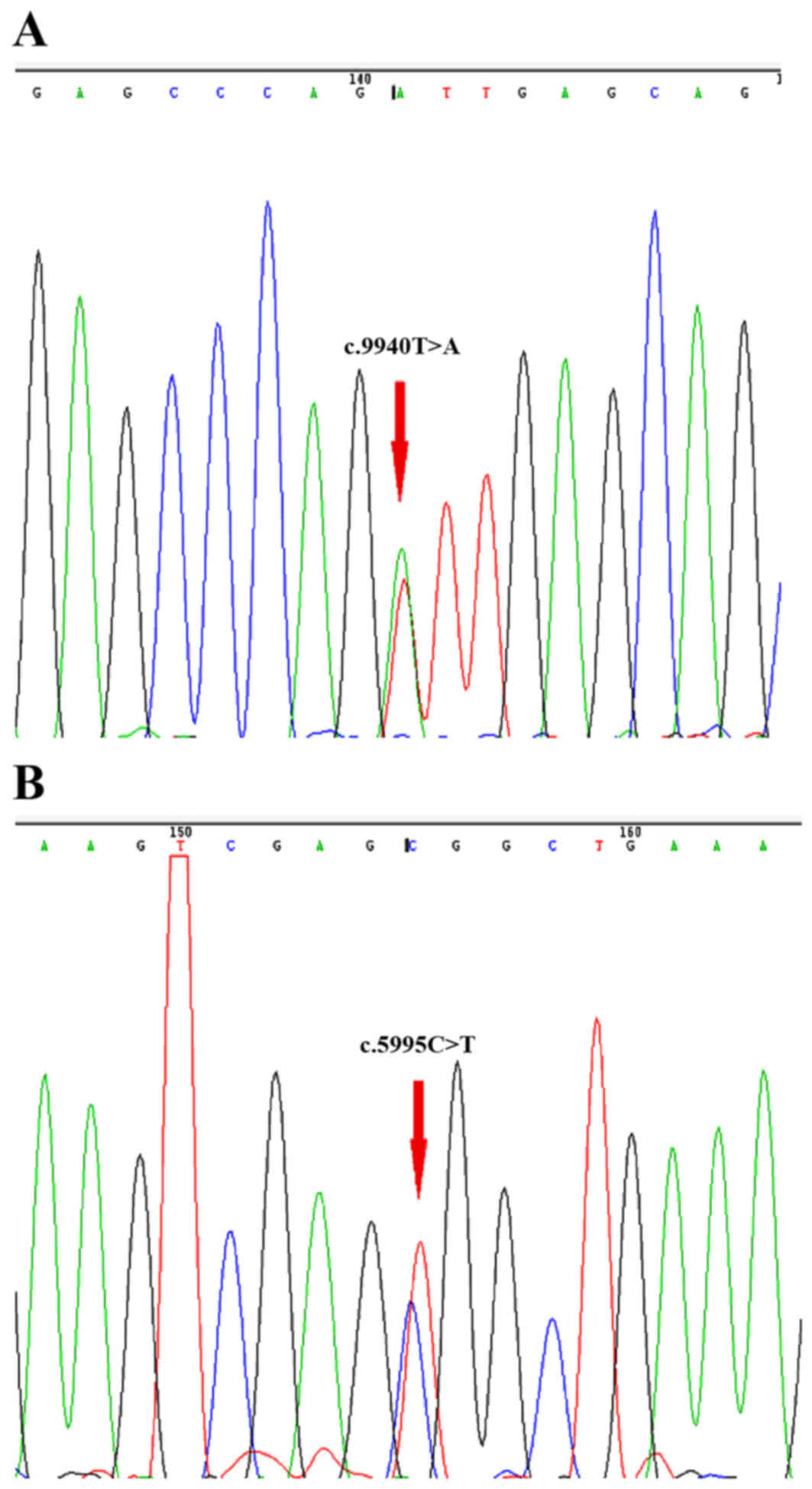
Novel compound heterozygous mutations, c.5995C>T (p.Arg1999Trp)
and c.9940T>A (p.Phe3314 Ile) in the PLEC gene, and
c.24646A>G (p.Ile8216Val) and c.18530T>G (p.Leu6177Arg) in
the TTN gene, were identified (Table
I; Fig. 3A and B). All
mutations were missense mutations, which can affect protein
function. The proband's father was identified with the same
mutations in the TTN gene, but without the LGMD phenotype.
Therefore, it was possible to exclude the heterozygous
c.24646A>G (p.Ile8216Val) and c.18530T>G (p.Leu6177Arg)
mutations as potential causes of the LGMD phenotype in the family.
The compound heterozygous variants in the PLEC gene were
co-segregated within the family. Heterozygous variants were
identified in other unaffected family members. PolyPhen-2 and SIFT
predicted that the mutations were possibly damaging and damaging
respectively. MutationTaster predicted that the alteration was
disease causing, with a probability value of 0.935 and 1.
Additionally, these variants were absent in the 200 normal controls
from exome sequencing of the Ensembl database.
 | Table I.List of candidate heterozygote
variants. |
Table I.
List of candidate heterozygote
variants.
| Variant | Gene | Classification | Protein | Exon |
PolyPhen/SIFT/MutationTaster | Mutation | Co-segr-egation |
|---|
| c.995C>T | PLEC | Coding | p.Arg1999Trp | 31 | Possibly
damaging/damaging/disease-causing | Missense | Yes |
| c.940T>A | PLEC | Coding | p.Phe3314 Ile | 32 | Possibly
damaging/damaging/disease-causing | Missense | Yes |
| c.24646A>G | TTN | Coding | p.Ile8216Val | 85 | Possibly
damaging/damaging/disease-causing | Missense | No |
| c.18530T>G | TTN | Coding | p.Leu6177Arg | 64 | Possibly
damaging/damaging/disease-causing | Missense | No |
Muscle pathology
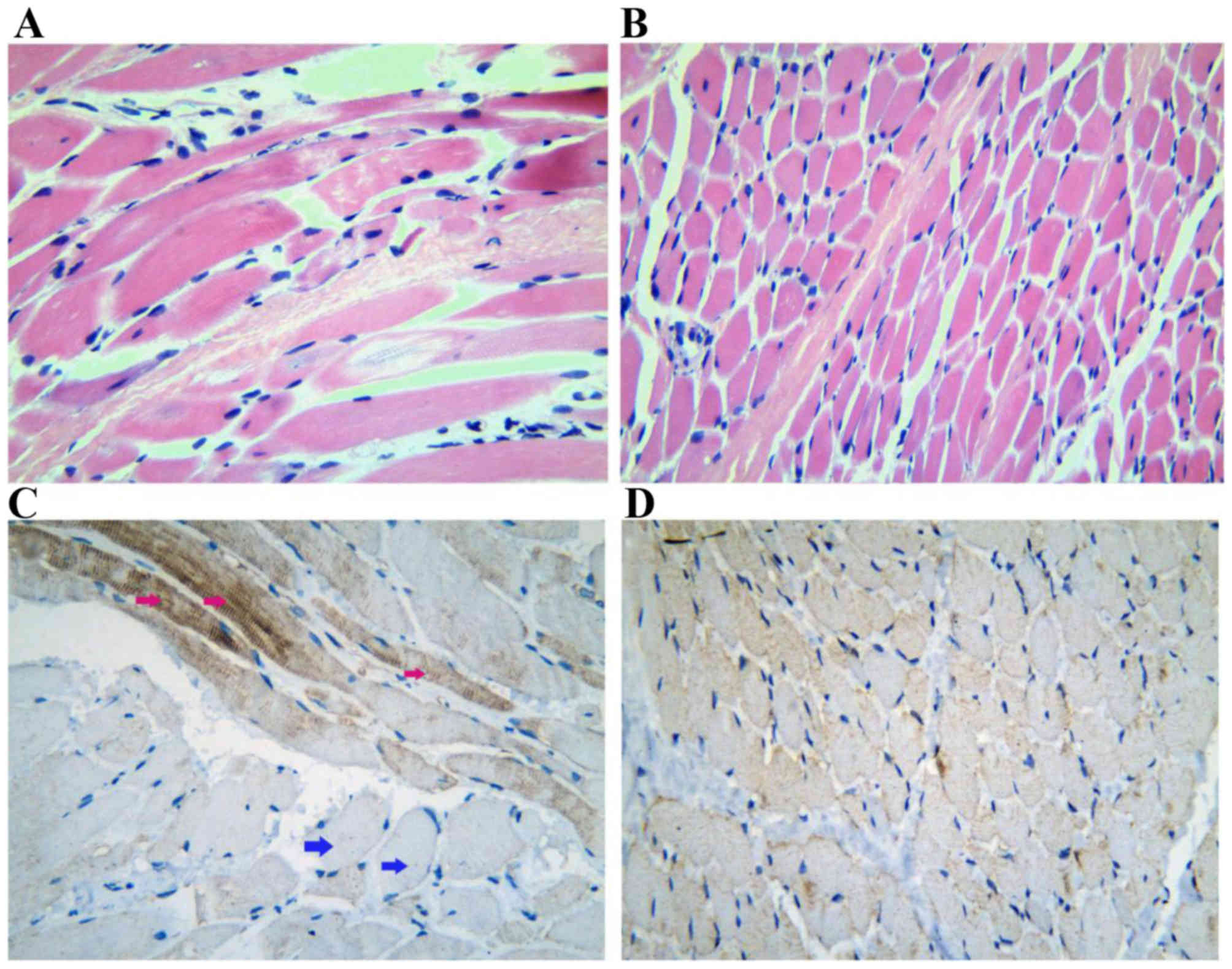
Muscle biopsy showed that significant dystrophic
features were present in muscular fibers, which had structure
distortion. The muscular fibers were also unequal in size, and
scattered necrotic fibers were found (Fig. 2A and B). To determine the
immunolocalization and alteration of plectin (Fig. 2C and D) in the skeletal muscle of
the patient, immunohistochemical staining of the muscle tissue was
performed. Plectin showed marked sarcoplasmic staining in certain
fibers, however, staining was irregular and faint in other fibers,
and loss of sarcolemmal staining was observed (Fig. 2C and D).
Discussion
PLEC is a multifunctional cytolinker (>500 kDa),
which is expressed in a wide variety of cell and tissues, including
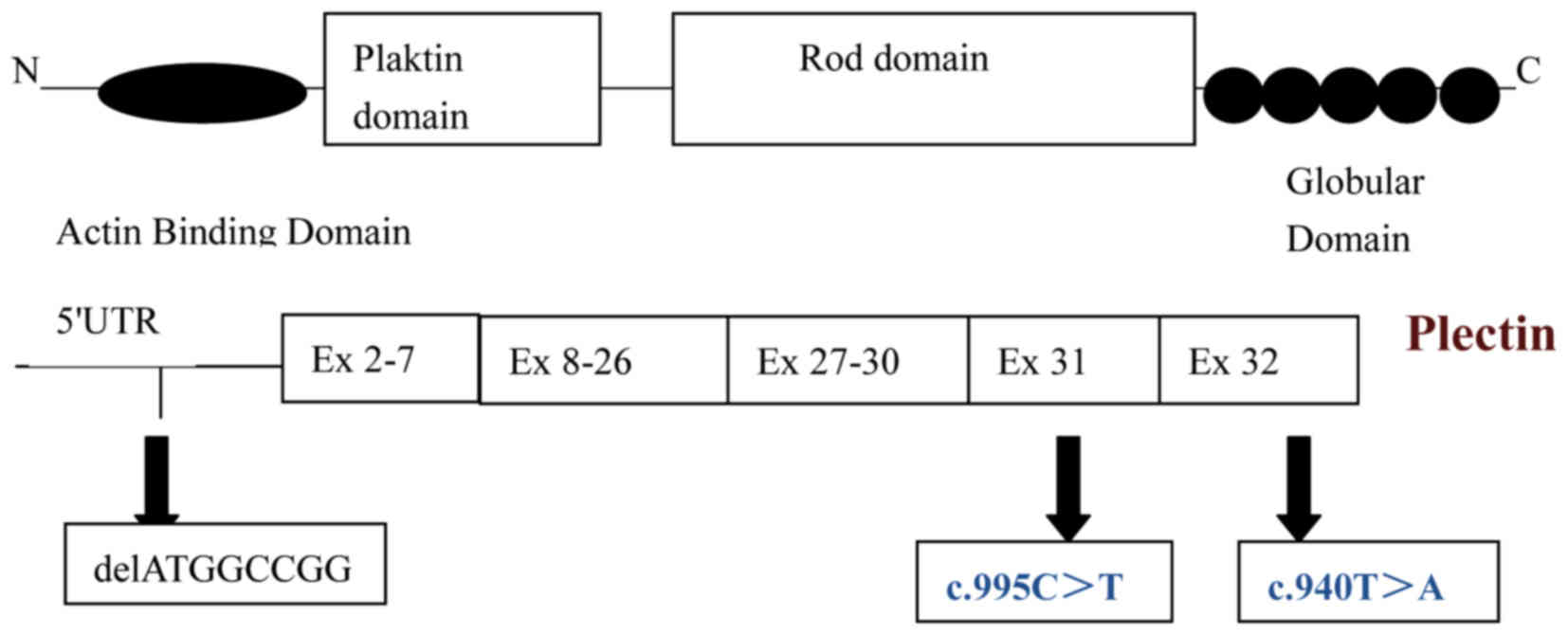
skin and muscle (9,10). The PLEC gene comprises a complex
organization of 32 exons located on chromosome 8q24. It has eight
variable first exons (A-H), which are each spliced to a common set
of downstream constant second exons to generate diverse functional
mRNAs (11,12). In 1996, EBS-MD (OMIM #226670) was
reported as being caused by mutations in the PLEC gene (13,14).
An increasing number of diseases, including LGMD2Q, EBS-MD-MyS,
EBS-PA and EBS-Ogna, have subsequently been identified by mutations
within this gene. The majority of PLEC mutations have been
associated with EBS and progressive muscular dystrophy. Few studies
have reported mutations in PLEC as being associated with muscular
dystrophy without a skin disorder.
In the present study, targeted sequencing using a
muscle disease gene panel was performed in the affected individual
of the family examined. Following the exclusion of SNPs and
synonymous mutations, PolyPhen-2, SIFT and MutationTaster were used
to predict the function of mutations. The novel compound
heterozygous mutations, c.5995C>T (p.Arg1999Trp) and
c.9940T>A (p.Phe3314 Ile), were identified in the PLEC gene in
the Chinese family with LGMD2Q. To acquire a more accurate
diagnosis and an improved understanding of the clinical spectrum,
immunocytochemistry was used to visualize and localize specific
proteins within the gastrocnemius. A defect in the plectin protein
was found in myofibers. The proband exhibited the first signs of
muscle weakness at 2 years old. Neither epidermolysis bullosa nor
any other dermatologic disorder or symptoms, for example myasthenic
syndrome, were found until the proband's final follow-up. Gundesli
et al (5) reported on a
consanguineous Turkish family containing four individuals suffering
from muscle weakness. They showed early-onset LGMD symptoms with
progressivity, but no myasthenic features, oculo-bulbar weakness or
no skin disorders. The individuals were diagnosed with LGMD2Q due
to a homozygous 9 bp deletion in plectin isoform 1f using GeneChip
Mapping 250K NspI SNP arrays from Affymetrix (5). The tissue-specific expression of the
plectin isoform 1f was suggested as an explanation for the
observation of an MD phenotype without skin abnormality. In the
present study, the proband exhibited the same LGMD2Q phenotype as
the previous study, however, the genotype differed from that of the
prior study. The c.5995C>T (p.Arg1999Trp) and c.9940T>A
(p.Phe3314 Ile) compound heterozygous mutations in the PLEC gene
were located within C-terminal globular domains and were identified
in all PLEC isoforms. Exon31, 32 encodes the central Rod domain
where dimerization occurs (Fig.
4). Previously, it was shown that Exon 31, 32 predominantly
harbors mutations in patients with EBS-MD (4). Rouan et al (15) reported on a patient with EBS-MD
with compound heterozygous mutations (6013G>T and 13378A>T)
in Exon31, 32. These mutations are nonsense mutations, which result
in downstream premature termination of codons and a non-functional
truncated peptide chain. However, the heterozygous mutations in the
present study were missense mutations. These can alter amino acid
sequences and lead to an abnormal peptide chain, which affects
protein features and function. This may explain the lack of
dermatologic disorders in the proband, despite mutations being in
the same exon.
Until now, no clear genotype/phenotype correlation
has been identified from the positions of mutations in the PLEC
gene. The phenotype is variable due to the different genotype in
the PLEC gene. The present study described a pure LGMD2Q, which was
caused by novel compound heterozygous mutations in Exon31, 32 of
the PLEC gene. These findings improve current knowledge of the
mutation spectrum of the PLEC gene associated with LGMD2Q. Compound
heterozygous mutations in the central Rod domain caused LGMD2Q with
the absence of prominent skin involvement. Additional cases are
required for confirmation of these observations, and further
investigations are required to understand the pathologic mechanism
of dystrophies by detecting downstream and upstream signaling
pathways.
Acknowledgements
This study was supported by the Hospital Starting
Fund for Study Abroad Returnees (grant no. 20100001) and the Fund
of Prevention and Treatment of Critical Illness for Children.
References
|
1
|
Narayanaswami P, Weiss M, Selcen D, David
W, Raynor E, Carter G, Wicklund M, Barohn RJ, Ensrud E, Griggs RC,
et al: Evidence-based guideline summary: Diagnosis and treatment of
limb-girdle and distal dystrophies: Report of the guideline
development subcommittee of the American Academy of Neurology and
the practice issues review panel of the American Association of
Neuromuscular & Electrodiagnostic Medicine. Neurology.
83:1453–1463. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nigro V, Aurino S and Piluso G: Limb
girdle muscular dystrophies: Update on genetic diagnosis and
therapeutic approaches. Curr Opin Neurol. 24:429–436. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bushby KM: Diagnostic criteria for the
limb-girdle muscular dystrophies: Report of the ENMC consortium on
limb-girdle dystrophies. Neuromuscul Disord. 5:71–74. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Winter L and Wiche G: The many faces of
plectin and plectinopathies: Pathology and mechanisms. Acta
Neuropathol. 125:77–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gundesli H, Talim B, Korkusuz P,
Balci-Hayta B, Cirak S, Akarsu NA, Topaloglu H and Dincer P:
Mutation in exon 1f of PLEC, leading to disruption of plectin
isoform 1f, causes autosomal-recessive limb-girdle muscular
dystrophy. Am J Hum Genet. 87:834–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Emery AE: Population frequencies of
inherited neuromuscular diseases-a world survey. Neuromuscul
Disord. 1:19–29. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scott OM, Hyde SA, Goddard C and Dubowitz
V: Quantitation of muscle function in children: A prospective study
in Duchenne muscular dystrophy. Muscle Nerve. 5:291–301. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wiche G: Plectin: General overview and
appraisal of its potential role as a subunit protein of the
cytomatrix. Crit Rev Biochem Mol Biol. 24:41–67. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wiche G, Krepler R, Artlieb U, Pytela R
and Denk H: Occurrence and immunolocalization of plectin in
tissues. J Cell Biol. 97:887–901. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang T, Haws P and Wu Q: Multiple
variable first exons: A mechanism for cell- and tissue-specific
gene regulation. Genome Res. 14:79–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fuchs P, Zörer M, Rezniczek GA, Spazierer
D, Oehler S, Castañón MJ, Hauptmann R and Wiche G: Unusual 5′
transcript complexity of plectin isoforms: Novel tissue-specific
exons modulate actin binding activity. Hum Mol Genet. 8:2461–2472.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gache Y, Chavanas S, Lacour JP, Wiche G,
Owaribe K, Meneguzzi G and Ortonne JP: Defective expression of
plectin/HD1 in epidermolysis bullosa simplex with muscular
dystrophy. J Clin Invest. 97:2289–2298. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McLean WH, Pulkkinen L, Smith FJ, Rugg EL,
Lane EB, Bullrich F, Burgeson RE, Amano S, Hudson DL, Owaribe K, et
al: Loss of plectin causes epidermolysis bullosa with muscular
dystrophy: cDNA cloning and genomic organization. Genes Dev.
10:1724–1735. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rouan F, Pulkkinen L, Meneguzzi G, et al:
Epidermolysis bullosa: novel and de novo premature termination
codon and deletion mutations in the plectin gene predict late-onset
muscular dystrophy. J Invest Dermatol. 114:381–387. 2000.
View Article : Google Scholar : PubMed/NCBI
|