Introduction
Aldose reductase (AR; EC.1.1.1.21) belongs to the
reduced coenzyme II-dependent aldo-keto reductase superfamily and
it is the first enzyme in the polyol pathway of glucose metabolism
(1). AR catalyzes the conversion
of glucose to sorbitol with the aid of NADPH, and sorbitol
dehydrogenase subsequently converts sorbitol to fructose, which can
easily pass through cell membranes. AR was first discovered in the
seminal vesicles. It is widely distributed in the brain, peripheral
nerves, retina, heart, blood vessels, kidneys, and other tissues
and organs (2). Alongside its
extensive distribution, it exhibits a wide range of roles in
various physiological processes, as has been demonstrated in in
vivo models. For example, AR participates in energy production
in sperm cells (1), serves a
cytoprotective role during hyperosmotic stress (3), and is involved in detoxification
mechanisms (4).
It has previously been reported that the polyol
pathway, which involves AR, participates in the development of
diabetic complications, in tissues severely affected by diabetes,
such as the kidney, heart, retina and blood vessels (5). To the best of our knowledge, very few
studies have explored the functions of AR in liver tissue. Under
physiological circumstances, relatively little AR expression has
been detected in the liver (6,7);
however, its expression is altered in disease states. Previous
studies have demonstrated significant increases in AR expression
levels in liver tissue from type II diabetic mice with hepatic
steatosis (8) and mice with
methionine-choline deficient diet-induced nonalcoholic fatty liver
disease (9). The AR inhibitor
zopolrestat significantly mitigated the steatosis and liver
inflammation in type II diabetic mice (8) and methionine-choline deficient
diet-induced mice (10). In
addition, AR expression has been revealed to be significantly
elevated in the liver tissue of patients with ethanol-induced liver
disease (11,12). However, the detailed mechanisms
underlying the involvement of AR in the development of
ethanol-induced liver disease have yet to be elucidated. The aim of
the present study was to evaluate the effects of the AR inhibitor
zopolrestat on ethanol-induced steatosis in HepG2 cells and to
investigate the possible underlying molecular mechanisms.
Materials and methods
Cell culture
Human hepatoma HepG2 cells were obtained from the
Chinese Academy of Sciences Committee Type Culture Collection Cell
Bank/CAS Shanghai Institute for Biologic Science Cell Resource
Center (Shanghai, China) and cultured in Dulbecco's modified
Eagle's medium supplemented with 10% fetal bovine serum (Hyclone;
GE Healthcare Life Sciences, Logan, UT, USA), 100 U/ml penicillin
and 100 µg/ml streptomycin. The cells were maintained in an
incubator at 37°C in a 5% CO2 atmosphere.
Oil red O staining
HepG2 cells were plated in 6-well plates at a
density of ~4×105 cells/well. The following day, cells
were stimulated with 100 mM ethanol for 48 h, and simultaneously
treated with 50 µM zopolrestat (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). A total of 48 h post-treatment, cells were
washed with ice-cold PBS, fixed with 10% formalin, and stained with
Oil Red O to detect lipid droplets in cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TriPure
Isolation Reagent (Roche Applied Science, Mannheim, Germany),
according to the manufacturer's protocol. Total RNA was reverse
transcribed into cDNA with the RevertAid First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's instructions. The primer sequences
for hepatic tumor necrosis factor (TNF)-α, transforming growth
factor (TGF)-β1, interleukin (IL)-6, sterol regulatory element
binding protein (SREBP)-1c, fatty acid synthase (FAS), peroxisome
proliferator-activated receptor (PPAR)α, acyl-CoA oxidase (ACO) and
carnitine palmitoyltransferase (CPT)-1 are presented in Table I. qPCR was conducted using the
FastStart Universal SYBR-Green Master (Rox; Roche Applied Science).
The reaction was run at 95°C for 10 min, followed by 35 cycles at
95°C for 15 sec, 53–60°C for 30 sec, 72°C for 30 sec and a final
extension at 72°C for 10 min. The relative expression of each gene
was normalized to GAPDH and was calculated using the comparative Cq
method (ΔΔCq) (13).
 | Table I.Primer sequences used for mRNA
amplification by reverse transcription quantitative polymerase
chain reaction. |
Table I.
Primer sequences used for mRNA
amplification by reverse transcription quantitative polymerase
chain reaction.
| Gene | Forward primer | Reverse primer |
|---|
| GAPDH |
5′-ACCCACTCCTCCACCTTTG-3′ |
5′-CTCTTGTGCTCTTGCTGGG-3′ |
| SREBP-1c |
5′-CGACATCGAAGACATGCTTCAG-3′ |
5′-GGAAGGCTTCAAGAGAGGAGC-3′ |
| FAS |
5′-GACATCGTCCATTCGTTTGTG-3′ |
5′-CGGATCACCTTCTTGAGCTCC-3′ |
| IL-6 |
5′-AAAAGTCCTGATCCAGTTC-3′ |
5′-GAGATGAGTTGTCATGTCC-3′ |
| TGF-β1 |
5′-CCGAGAAGCGGTACCTGAAC-3′ |
5′-GAGGTATCGCCAGGAATTGTTG-3′ |
| TNF-α |
5′-CCAGACCAAGGTCAACCTC-3′ |
5′-CCAGATAGATGGGCTCATACC-3′ |
| PPARα |
5′-GCAGAAACCCAGAACTCAGC-3′ |
5′-ATGGCCCAGTGTAAGAAACG-3′ |
| ACO |
5′-TCTGTTGACCTTGTTCGAGCAA-3′ |
5′-CAAGCACAGAGCCAAGTGTCAC-3′ |
| CPT-1 |
5′-ACAGTCGGTGAGGCCTCTTATGAA-3′ |
5′-TCTTGCTGCCTGAATGTGAGTTGG-3′ |
Western blot analysis
Whole cell extracts were prepared by dissolution of
cell pellets in ice-cold radioimmunoprecipitation assay lysis
buffer (1% Triton X-100, 50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM
EDTA, 10% glycerin, 10 mM Na4P2O7,
20 mM glycerophosphate, 10 mM NaF, 10 mM sodium orthovanadate and
proteinase inhibitor mixture) until the cells were completely
lysed. Following centrifugation at 12,000 × g, 4°C for 5 min,
supernatants were collected and stored at −80°C before use. The
protein concentrations of the extracts were measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology, Haimen, China). Equal amounts of extracted protein
samples (40 µg) were separated by 12% SDS-PAGE and transferred onto
a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). Membranes were blocked with 5% non-fat milk in 0.1% Tween-20
TBS for 1 h at room temperature and incubated with primary
antibodies in TBS-0.1% Tween-20 with 5% non-fat milk at 4°C
overnight. Primary antibodies used were: 5′ adenosine
monophosphate-activated protein kinase α (AMPKα; cat. no. #2532;
1:1,000 dilution; Cell Signaling Technology, Inc., Danvers, MA,
USA), phosphorylated (p)-AMPKα (cat. no. #2535; 1:1,000 dilution;
Cell Signaling Technology, Inc.), AR (cat. no. sc-17735; 1:500
dilution; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
β-actin (cat. no. A2228; 1:2,000 dilution; Sigma-Aldrich; Merck
KGaA). After several washes with TBS-0.1% Tween-20, the membranes
were incubated with horseradish peroxidase-conjugated anti-rabbit
or anti-goat immunoglobulin G secondary antibodies (dilution
1:2,000; cat. nos. AP307P and AB324P, respectively; Merck KGaA) in
TBS-0.1% Tween-20 with 5% non-fat milk. The bands were visualized
using the SuperSignal Chemiluminescent Substrate kit (Beyotime
Institute of Biotechnology). Densitometric analysis of protein
bands was performed using ImageJ v. 1.40 software (National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The statistical significance of the difference
between groups was assessed by Student's t-test for pair-wise
comparisons or one-way analysis of variance, followed by a post hoc
Bonferroni's test for multiple comparisons. Data are expressed as
the mean ± standard error of the mean. P<0.05 was considered to
indicate a statistically significant difference. The analysis was
performed using GraphPad Prism software version 5.0 (GraphPad
Software, Inc., La Jolla, CA, USA).
Results
AR is induced following ethanol
stimulation, whereas AR inhibition attenuates ethanol-induced
hepatic steatosis
Previous studies have demonstrated that stimulating
cells with ethanol may induce hepatic steatosis (14,15).
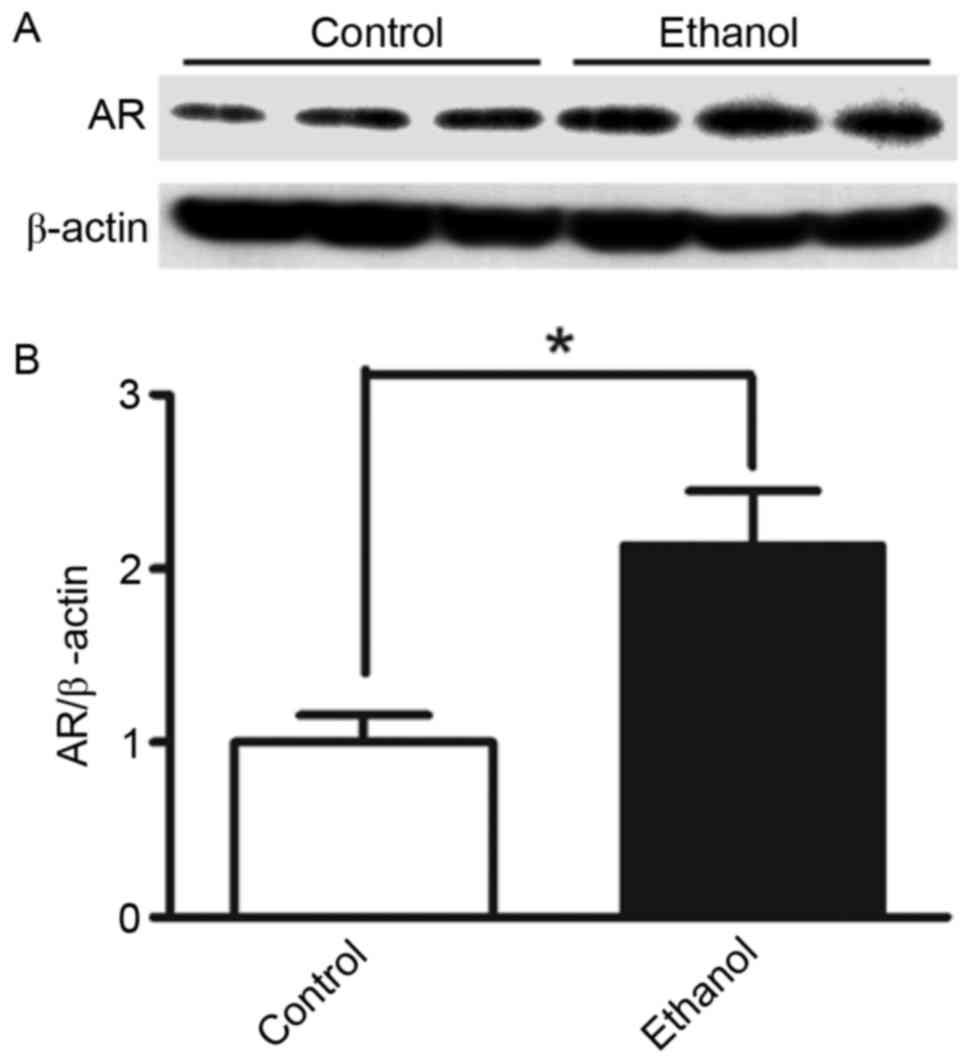
In the present study, protein expression levels of AR were assessed
in HepG2 cells stimulated with 100 mM ethanol. AR protein
expression in cells stimulated with ethanol for 36 h was ~2.1x
higher (P<0.05) compared with in control cells without ethanol
stimulation (Fig. 1). To further
investigate the role of AR in the development of ethanol-induced
steatosis, AR activity was inhibited via treatment with the AR
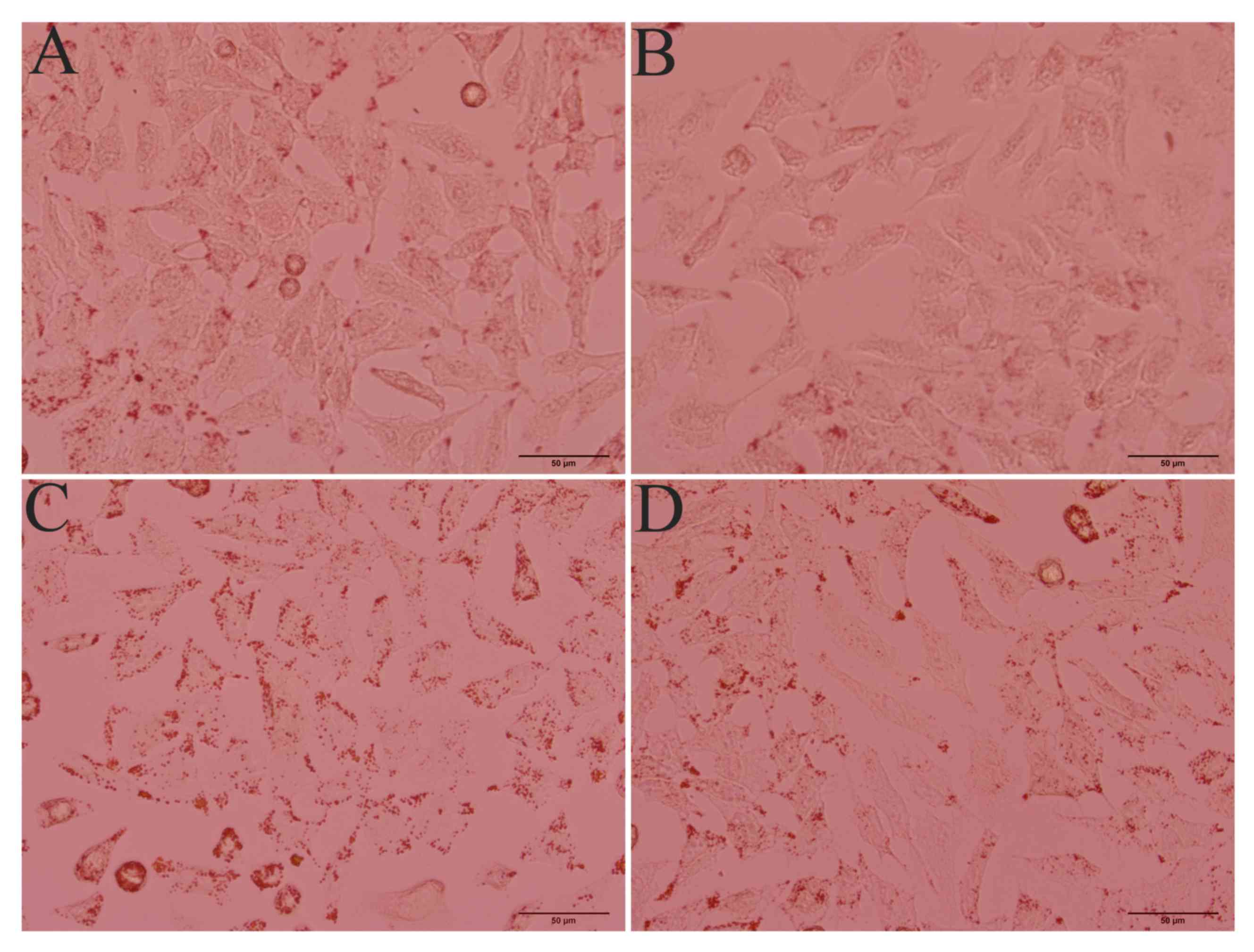
inhibitor zopolrestat (50 µM). Oil Red O staining revealed a marked
lipid accumulation in ethanol-stimulated HepG2 cells, whereas
control cells without ethanol stimulation did not exhibit steatosis
(Fig. 2). When AR activity was
inhibited, lipid accumulation in ethanol-stimulated cells was
markedly attenuated. These findings suggested that AR is involved
in the development of ethanol-induced steatosis in hepatocytes.
AR inhibition activates AMPK and
mitigates the ethanol-induced elevation of SREBP-1c and FAS mRNA
expression
AMPK inactivation has been reported during the
development of ethanol-induced hepatic steatosis (16,17).
SREBP-1c is a transcription factor regulated by AMPK, which
modulates the expression of several lipogenic enzymes, including
FAS (18,19). To investigate the role AR serves in
the development of ethanol-induced steatosis, the effect of AR
inhibition on AMPK activity and on SREBP-1c and FAS mRNA expression
was assessed in ethanol-stimulated HepG2 cells. Treatment with 100
mM ethanol resulted in a significant reduction in AMPK
phosphorylation in HepG2 cells; the reduction in p-AMPK levels was
markedly attenuated when ethanol-stimulated cells were treated
withzopolrestat (Fig. 3A). In
addition, treatment with ethanol caused a significant increase in
SREBP-1c mRNA expression levels compared with in untreated cells;
the increase was significantly attenuated by zopolrestat treatment
(P<0.05). FAS mRNA expression levels were significantly reduced
in ethanol-stimulated cells treated with zopolrestat compared with
in cells that didn't receive the AR inhibitor (Fig. 3B). Furthermore, the effect of AR
inhibition on PPARα, a transcription factor that regulates several
enzymes involved in fatty acid oxidation, such as ACO and CPT-1,
was investigated (20,21). AR inhibition did not alter the mRNA
expression levels of PPARα, ACO or CPT-1 (Fig. 3C). Taken together, the present
results suggested that the inhibition of AR may ameliorate
ethanol-induced steatosis in HepG2 cells through activating AMPK
and inhibiting SREBP-1c-regulated lipogenesis.
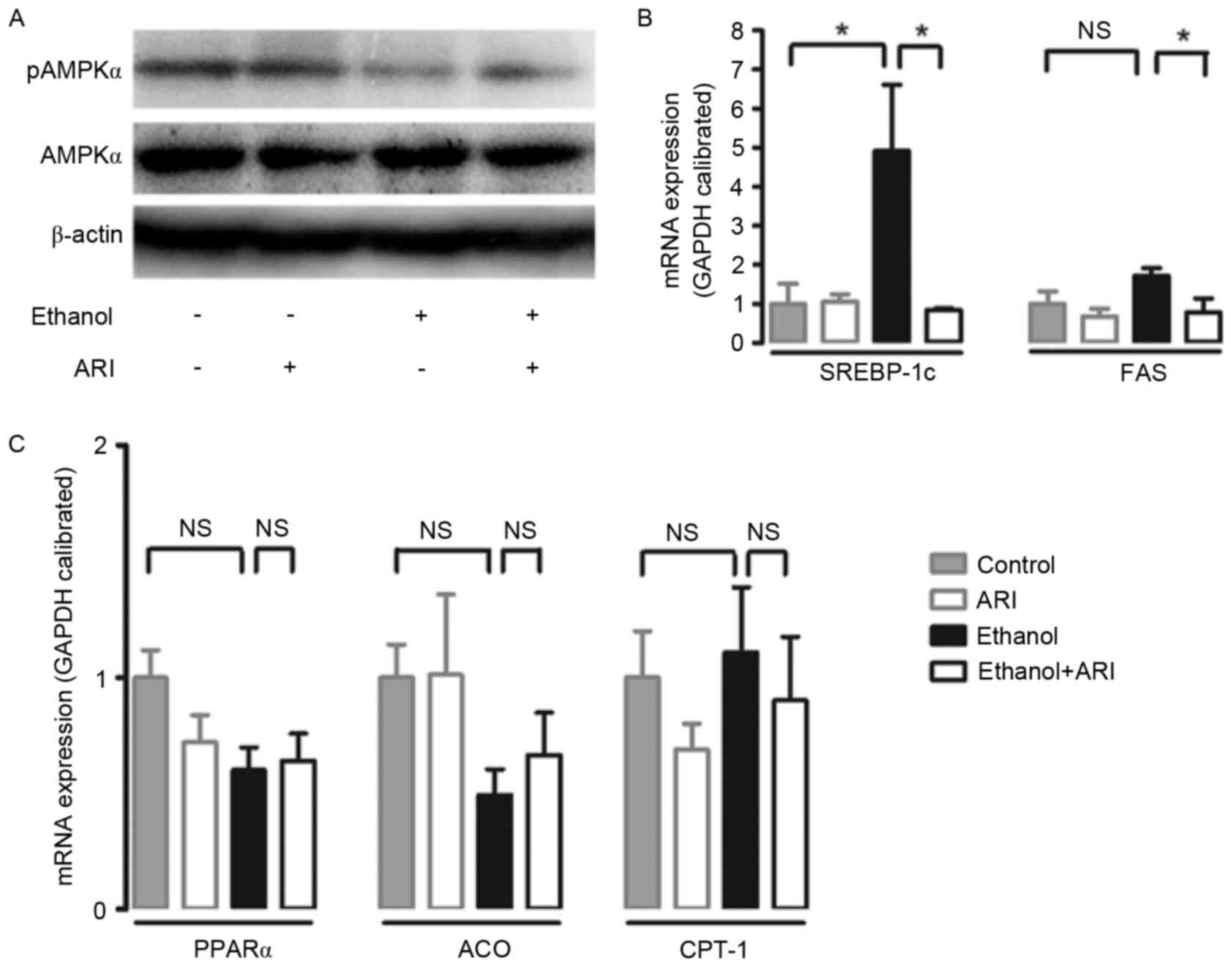 | Figure 3.Effect of AR inhibition on AMPK
activation and mRNA expression of lipid metabolism genes in
ethanol-stimulated HepG2 cells. (A) Representative western blot
demonstrates the activation of hepatic AMPK following AR inhibition
in ethanol-stimulated cells. mRNA expression levels of (B) SREBP-1c
and FAS, and (C) PPARα, ACO and CPT-1 were assessed by reverse
transcription-quantitative polymerase chain reaction, standardized
against an internal control (GAPDH), and are presented as fold
change over the control cells (n=3). Data are expressed as the mean
± standard error of the mean. *P<0.05. AR, aldose reductase;
AMPK, 5′ adenosine monophosphate-activated protein kinase; p,
phosphorylated; SREBP, sterol regulatory element binding protein;
FAS, fatty acid synthase; PPAR, peroxisome proliferator-activated
receptor; ACO, acyl-CoA oxidase; CPT, carnitine
palmitoyltransferase; NS, not significant. |
AR inhibition attenuates the
ethanol-induced elevation of TNF-α mRNA expression
TNF-α has been reported to serve a pivotal role in
the development of ethanol-induced hepatic steatosis (22,23).
In the present study, mRNA expression levels of proinflammatory
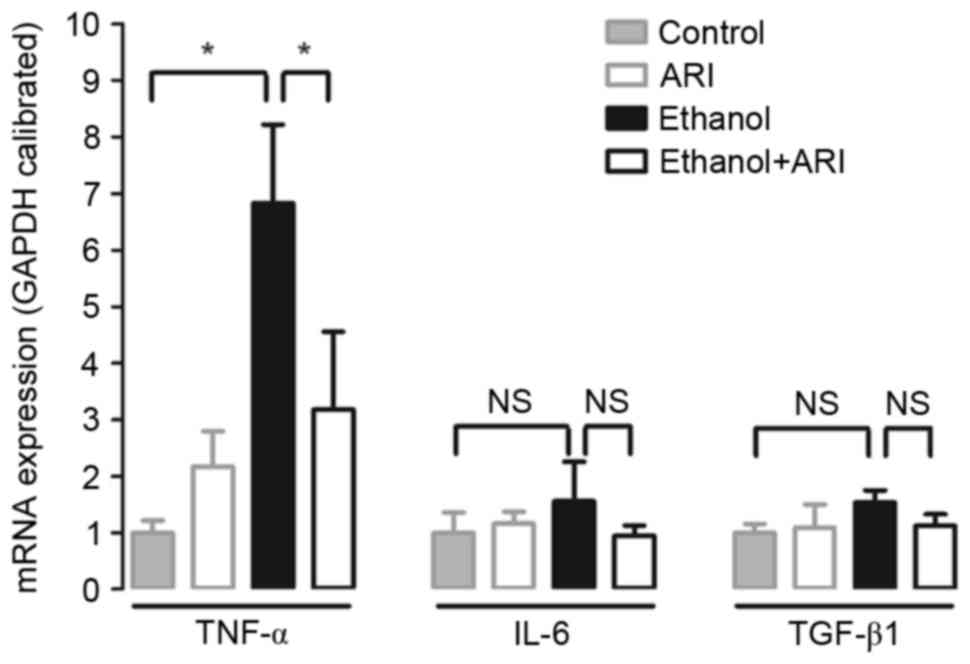
cytokines, including TNF-α, IL-6 and TGF-β1, were investigated.
Cells stimulated with ethanol exhibited significantly elevated
TNF-α mRNA expression levels compared with in unstimulated cells
(Fig. 4), whereas treatment with
zopolrestat significantly attenuated the ethanol-induced increase
in TNF-α mRNA expression (P<0.05). Conversely, AR inhibition had
no effect on IL-6 and TGF-β1 expression. These results suggested
that AR inhibition may affect the production of proinflammatory
cytokines, such as TNF-α, to prevent ethanol-induced steatosis in
HepG2 cells.
Discussion
Previous studies have suggested that AR may be
involved in the development of non-alcoholic fatty liver disease,
and that its inhibition may ameliorate hepatic steatosis (8–10).
The present study demonstrated that ethanol induced an increase in
AR expression, whereas the inhibition of AR markedly reduced lipid
droplet accumulation in HepG2 cells. These results suggested that
AR may be involved in the development of alcoholic fatty liver
disease, whereas its inhibition may ameliorate ethanol-induced
hepatic steatosis.
AMPK is an enzyme that serves a role in cellular
energy homeostasis, as its activation stimulates fatty acid
oxidation and suppresses lipogenesis (24). AMPK activators have been reported
to reduce the expression of SREBP-1c, a gene mainly associated with
fat synthesis (25). In addition,
SREBP-1c has been reported to act as a key regulator of hepatic
lipid metabolism that modulates the transcription of various genes
involved in hepatic triglyceride and fatty acid synthesis (18). Overexpression of SREBP-1c and one
of its downstream genes, FAS, results in hepatic fat accumulation
(18). Fatty acid synthesis
appeared significantly potentiated in transgenic mice
overexpressing SREBP-1c, and hepatic triglyceride levels were much
increased (26). Acetaldehyde,
which is a product of the hepatic metabolism of ethanol, was
demonstrated to stimulate the overexpression of SREBP-1c in
hepatocytes (27). Ethanol-induced
SREBP-1c activation is one of the main causes of hepatic
triglyceride accumulation in alcoholic fatty liver disease
(28,29). The results of the present study
demonstrated that AR inhibition ameliorated the ethanol-induced
AMPK inactivation and suppressed the ethanol-induced mRNA
overexpression of SREBP-1c and FAS, thereby reducing hepatic lipid
formation and mitigating hepatic steatosis. These results suggested
that the AR-mediated dysregulation of the AMPK/SREBP-1c signaling
pathway may contribute to the development of ethanol-induced
hepatic steatosis.
The development of alcoholic liver diseases is
closely associated with overexpression of several inflammatory
cytokines, among which TNF-α holds a prominent role (23). Previous studies have reported that
TNF-α expression is potentiated in patients with alcoholic liver
diseases (23,30). TNF-α induces lipid accumulation and
promotes inflammatory and apoptotic processes in hepatocytes
(31). The present study revealed
that AR inhibition significantly reduced TNF-α mRNA expression
levels. These results suggested that the amelioration of
ethanol-induced hepatic steatosis achieved via AR inhibition may be
partially attributed to the inhibition of ethanol-induced TNF-α
overexpression.
In conclusion, in the present study, AR inhibition
significantly mitigated the ethanol-induced lipid dysregulation in
HepG2 cells, through the activation of AMPK and suppression of
SREBP-1c and FAS. In addition, the AR inhibitor suppressed
ethanol-induced TNF-α overexpression in hepatocytes. These results
suggested that AR inhibition may improve ethanol-induced hepatic
steatosis, whereas AR inhibitors may have potential as alternative
therapeutic strategies for the treatment of alcoholic fatty liver
diseases.
Acknowledgements
The present study was partly supported by the
Training Program of Fujian Excellent Talents in University (grant
no. MJR201558) and the Science Planning Program of Longyan
University (grant no. LG2014012).
References
|
1
|
Hers HG: Aldose reductase. Biochim Biophys
Acta. 37:120–126. 1960.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clements RS Jr: The polyol pathway. A
historical review. Drugs. 32:(Suppl 2). S3–S5. 1986. View Article : Google Scholar
|
|
3
|
Ferraris JD, Williams CK, Jung KY, Bedford
JJ, Burg MB and García-Pérez A: ORE, a eukaryotic minimal essential
osmotic response element. The aldose reductase gene in hyperosmotic
stress. J Biol Chem. 271:18318–18321. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yabe-Nishimura C: Aldose reductase in
glucose toxicity: A potential target for the prevention of diabetic
complications. Pharmacol Rev. 50:21–33. 1998.PubMed/NCBI
|
|
5
|
Nishimura-Yabe C: Aldose reductase in the
polyol pathway: A potential target for the therapeutic intervention
of diabetic complications. Nihon Yakurigaku Zasshi. 111:137–145.
1998.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clements RS Jr, Weaver JP and Winegrad AI:
The distribution of polyol: NADP oxidoreductase in mammalian
tissues. Biochem Biophys Res Commun. 37:347–353. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Markus HB, Raducha M and Harris H: Tissue
distribution of mammalian aldose reductase and related enzymes.
Biochem Med. 29:31–45. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qiu L, Lin J, Xu F, Gao Y, Zhang C, Liu Y,
Luo Y and Yang JY: Inhibition of aldose reductase activates hepatic
peroxisome proliferator-activated receptor-α and ameliorates
hepatosteatosis in diabetic db/db mice. Exp Diabetes Res.
2012:7897302012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qiu L, Lin J, Ying M, Chen W, Yang J, Deng
T, Chen J, Shi D and Yang JY: Aldose reductase is involved in the
development of murine diet-induced nonalcoholic steatohepatitis.
PLoS One. 8:e735912013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen T, Shi D, Chen J, Yang Y, Qiu M, Wang
W and Qiu L: Inhibition of aldose reductase ameliorates
diet-induced nonalcoholic steatohepatitis in mice via modulating
the phosphorylation of hepatic peroxisome proliferator-activated
receptor α. Mol Med Rep. 11:303–308. 2015.PubMed/NCBI
|
|
11
|
Brown KE, Broadhurst KA, Mathahs MM,
Kladney RD, Fimmel CJ, Srivastava SK and Brunt EM: Immunodetection
of aldose reductase in normal and diseased human liver. Histol
Histopathol. 20:429–436. 2005.PubMed/NCBI
|
|
12
|
O'Connor T, Ireland LS, Harrison DJ and
Hayes JD: Major differences exist in the function and
tissue-specific expression of human aflatoxin B1 aldehyde reductase
and the principal human aldo-keto reductase AKR1 family members.
Biochem J 343 Pt. 2:487–504. 1999. View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Keegan A, Martini R and Batey R:
Ethanol-related liver injury in the rat: A model of steatosis,
inflammation and pericentral fibrosis. J Hepatol. 23:591–600. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Altamirano J and Bataller R: Alcoholic
liver disease: Pathogenesis and new targets for therapy. Nat Rev
Gastroenterol Hepatol. 8:491–501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sid B, Verrax J and Calderon PB: Role of
AMPK activation in oxidative cell damage: Implications for
alcohol-induced liver disease. Biochem Pharmacol. 86:200–209. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
You M, Matsumoto M, Pacold CM, Cho WK and
Crabb DW: The role of AMP-activated protein kinase in the action of
ethanol in the liver. Gastroenterology. 127:1798–1808. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roder K, Zhang L and Schweizer M: SREBP-1c
mediates the retinoid-dependent increase in fatty acid synthase
promoter activity in HepG2. FEBS Lett. 581:2715–2720. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kong L, Ren W, Li W, Zhao S, Mi H, Wang R,
Zhang Y, Wu W, Nan Y and Yu J: Activation of peroxisome
proliferator activated receptor alpha ameliorates ethanol induced
steatohepatitis in mice. Lipids Health Dis. 10:2462011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kota BP, Huang TH and Roufogalis BD: An
overview on biological mechanisms of PPARs. Pharmacol Res.
51:85–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thiele DL: Tumor necrosis factor, the
acute phase response and the pathogenesis of alcoholic liver
disease. Hepatology. 9:497–499. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawaratani H, Tsujimoto T, Douhara A,
Takaya H, Moriya K, Namisaki T, Noguchi R, Yoshiji H, Fujimoto M
and Fukui H: The effect of inflammatory cytokines in alcoholic
liver disease. Mediators Inflamm. 2013:4951562013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Viollet B, Guigas B, Leclerc J, Hébrard S,
Lantier L, Mounier R, Andreelli F and Foretz M: AMP-activated
protein kinase in the regulation of hepatic energy metabolism: From
physiology to therapeutic perspectives. Acta Physiol (Oxf).
196:81–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tomita K, Tamiya G, Ando S, Kitamura N,
Koizumi H, Kato S, Horie Y, Kaneko T, Azuma T, Nagata H, et al:
AICAR, an AMPK activator, has protective effects on alcohol-induced
fatty liver in rats. Alcohol Clin Exp Res. 29:(12 Suppl).
S240–S245. 2005. View Article : Google Scholar
|
|
26
|
Shimano H, Horton JD, Shimomura I, Hammer
RE, Brown MS and Goldstein JL: Isoform 1c of sterol regulatory
element binding protein is less active than isoform 1a in livers of
transgenic mice and in cultured cells. J Clin Invest. 99:846–854.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lluis JM, Colell A, García-Ruiz C,
Kaplowitz N and Fernández-Checa JC: Acetaldehyde impairs
mitochondrial glutathione transport in HepG2 cells through
endoplasmic reticulum stress. Gastroenterology. 124:708–724. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
You M, Fischer M, Deeg MA and Crabb DW:
Ethanol induces fatty acid synthesis pathways by activation of
sterol regulatory element-binding protein (SREBP). J Biol Chem.
277:29342–29347. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
You M and Crabb DW: Recent advances in
alcoholic liver disease II. Minireview: Molecular mechanisms of
alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol.
287:G1–G6. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McClain CJ and Cohen DA: Increased tumor
necrosis factor production by monocytes in alcoholic hepatitis.
Hepatology. 9:349–351. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Endo M, Masaki T, Seike M and Yoshimatsu
H: TNF-alpha induces hepatic steatosis in mice by enhancing gene
expression of sterol regulatory element binding protein-1c
(SREBP-1c). Exp Biol Med (Maywood). 232:614–621. 2007.PubMed/NCBI
|