Introduction
Senescence-accelerated mice prone (SAMP) strains,
which have been developed through the selective inbreeding of the
AKR/J strain, present a senescence-related phenotype involving a
short lifespan and rapid progression of senescence. Together with
SAMP strains, senescence-accelerated mice resistant (SAMR) strains
have been developed as their corresponding controls. Of the nine
SAMP sub-strains, the mice of P8 strain (SAMP8) exhibit Alzheimer's
disease (AD)-like pathologies, such as abundant expression of
amyloid precursor protein (APP) and production of amyloid-beta, tau
hyperphosphorylation and increased oxidative stress (1–4). In
addition, the neural network activity and number of neurons in the
brain of SAMP8 spontaneously reduce; this relates to their
cognitive dysfunction (1,2). To identify the mechanisms of AD and
develop effective therapeutic strategies, appropriate animal models
are essential. Although studies using SAMP8 are expanding, as SAMP8
mimics the non-familial form of AD compared to transgenic AD
models, the relationship between tau phosphorylation and metabolic
dysfunction in SAMP8 remains unclear.
Insulin and insulin-like growth factor-1 (IGF-1)
signaling are associated with aging and control vital growth,
survival, longevity and energy metabolism in the brain (5–7). A
defect of insulin/IGF-1 signaling is associated with
neurofibrillary tangles (NFTs) and amyloid plaque in AD (5,8) and
administration of insulin can improve memory and cognitive function
in patients with AD (9).
Dysregulation of insulin/IGF-1 signaling may be mediated by insulin
receptor substrate type 1 (IRS-1), which is a key mediator of the
signaling pathway, and IRS-1 is dysregulated in the AD brain. In
fact, reduced levels of IRS-1 have been detected in neurons in the
medial temporal cortex of AD, and the inhibitory phosphorylation of
IRS-1 at Ser312 and Ser616 is increased in neurons with NFTs
(10). The relationship between
impaired insulin actions and AD pathogenesis is widely accepted
(11).
AMP-activated protein kinase (AMPK), a key regulator
of cellular energy homeostasis, serves a critical role in neuronal
survival (12,13). Previous studies have indicated that
the pathogenesis of neurodegenerative diseases such as AD may
involve the alteration of AMPK signaling. For example, the
activation of AMPK by pharmacological activators increases Aβ
production and tau hyper-phosphorylation (14–16).
Conversely, studies demonstrating that AMPK activation inhibits Aβ
abundance and tau phosphorylation at several sites have been
reported (17–21). Moreover, AMPK activation may be
beneficial to reduce the neurotoxicity of glucose deprivation, Aβ
or α-synuclein (22–24). Collectively, the role of AMPK in
the pathogenesis of AD, such as tau phosphorylation and Aβ-mediated
neurotoxicity, remains controversial.
Previous studies indicated that impaired glucose
homeostasis is observed in the SAMP8 strain along with higher
levels of glucose, insulin and free fatty acids in serum (25,26).
Moreover, inhibited activity of AKT and expression of glucose
transporter 4 in the skeletal muscle in the SAMP8 strain, when
compared to the SAMR1 control strain, has been reported (26). In a previous SAMP8 study of the
authors, there was observation of AMPK activity upregulation in the
cortex of young SAMP8 (pre-symptomatic age), when compared with
SAMR1 mice; a change attributable to the inhibition of tau
phosphorylation at Ser396 (27).
However, the chronological relationship between metabolic
regulators and AD pathogenesis, particularly in tau
hyper-phosphorylation in SAMP8, has not been fully elucidated.
However, it has been reported that AD pathologies are developed in
SAMP8 as early as 4–5 months of age (3).
The aim of the present study was to characterize the
tau phosphorylation and the expression of proteins related to
energy metabolism in order to understand tau-related pathogenesis
and energy metabolism. In order to accomplish this, the authors
evaluated chronological changes in the expression of phosphorylated
tau and intracellular metabolic regulators, including AMPK, GSK3β,
sirtuin1 (Sirt1) and IRS-1, in the brains of SAMP8 across different
ages [young (2-month-old), middle (5-month-old) and old
(10-month-old)] to examine both pre-symptomatic and symptomatic
stages of AD.
Materials and methods
Animal care
The care of animals was conducted according to the
Guide for Care and Use of Laboratory Animals of the National
Institutes of Health (NIH, Bethesda, MD, USA). All experiments were
conducted in accordance with procedures and protocols approved by
the Animal Care and Handling Committee of Inha University (Incheon,
Korea). Male SAMR1 (n=30; body weight: 25–29 g) and SAMP8 (n=30;
body weight: 24–29 g) (all six-weeks-old) were obtained from
Central Laboratory Animal, Inc. (Seoul, Korea). Mice were housed
individually in a temperature- (22±2°C) and humidity-controlled
(45–55%) room under a 12 h light-dark cycle (7:00 a.m.-7:00 p.m.),
with free access to a standard rodent diet and water. Mice (n=10,
each strain) were sacrificed at 2, 5 and 10 months of age (n=20, at
each time point).
Isolation of the hippocampus and
cerebral cortex
Under intraperitoneal ketamine (80 mg/kg) and
xylazine (8 mg/kg)-induced anesthesia, the skin was flipped over
the eyes to free the skull, and an incision was made at the top of
the skull, beginning at the caudal part through the anterior part
of the skull, in order to expose the brain. The brain was quickly
isolated and the cerebral hemispheres were separated by a sagittal
midline incision following removal of the cerebellum. Using
forceps, the diencephalon was carefully removed to expose the
medial side of the hippocampus. From the remaining hemisphere
containing the cortex and hippocampus, the C-shaped hippocampus was
dissected out using fine scissors and a spatula. The whole cerebral
cortex and dissected hippocampus were immediately frozen in liquid
nitrogen, and stored at −80°C until use.
Immunoblotting
To prepare tissue lysates, the collected hippocampi
or cerebral cortices of SAMR1 and SAMP8 mice were ground in
radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) using a pellet pestle followed by
homogenization under sonication. The debris of the homogenates was
removed by centrifugation at 13,000 × g for 15 min at 4°C, and
total protein content of the supernatant was quantified using a
bicinchoninic acid assay protein assay kit (Pierce; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Samples were prepared with
dithiothreitol (Sigma-Aldrich; Merck KGaA) and denatured by heating
at 95°C for 3 min. Proteins (10–20 µg) were separated by 4–20%
SDS-PAGE gels and transferred to polyvinylidene difluoride
membranes in a transfer buffer containing 25 mM Tris-HCl, 192 mM
glycine and 10% methanol. Membranes were blocked with 5% BSA or 5%
non-fat dry milk in TBS with 0.1% Tween-20 (Sigma-Aldrich; Merck
KGaA) and incubated with specific primary antibodies against AMPK
(2532; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
p-AMPK at Thr172 (p-AMPK; 2535; 1:1,000; Cell Signaling Technology,
Inc.), phosphorylated Acetyl-CoA carboxylase at Ser79 (p-ACC; 3661;
1:1,000; Cell Signaling Technology, Inc.), insulin receptor
substrate 1 (IRS-1; 06-248; 1:1,000; Upstate, Biotechnology, Inc.,
Lake Placid, NY, USA), tau (Tau5; SIG-39413; 1:1,000; Covance,
Inc., Princeton, NJ, USA), phosphorylated tau at Ser262
(p-tauS262; sc-32828; 1:500; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), phosphorylated tau at Ser396
(p-tauS396; sc-12414; 1:1,000; Santa Cruz Biotechnology,
Inc.), sirtuin-1 (Sirt1; 9475; 1:1,000; Cell Signaling Technology,
Inc.), α-synuclein (C-20; sc-7011; 1:1,000; Santa Cruz
Biotechnology, Inc.), phosphorylated GSK3β at Ser9
(p-GSK3βS9; 9336, 1:1,000; Cell Signaling Technology,
Inc.), GSK3β (sc-9166, 1:1,000; Santa Cruz Biotechnology, Inc.) at
4°C for overnight, or β-actin (a1978, 1:10,000, Sigma-Aldrich;
Merck KGaA) at room temperature for 2 h, and then incubated with
appropriate secondary antibodies at room temperature for 2 h
(peroxidase conjugated anti-rabbit IgG; NCI1460KR; 1:5,000 and
peroxidase conjugated anti-mouse IgG; NCI1430KR; 1:5,000; both from
Thermo Fisher Scientific Inc.). Immunoreactive bands were
visualized by an enhanced chemiluminescence detection system
(15,159; Pierce; Thermo Fisher Scientific, Inc.) and quantified
using the image analysis software Quantity One (version 4.6.6;
Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Results are expressed as the mean ± the standard
error of the mean, unless otherwise stated. P<0.05 was deemed
statistically significant using GraphPad Prism version 5.0
(GraphPad Software Inc., La Jolla, CA, USA). The Mann-Whitney
nonparametric test was used to compare groups.
Results
Characteristics of tau phosphorylation
in the cerebral cortex of SAMP8
In order to employ the SAMP8 strain as an
appropriate experimental model for understanding the precise
molecular mechanisms of AD, a profiling analysis through a time
course study of proteins related to AD pathogenesis was necessary.
To characterize AD-related tau phosphorylation, the authors
initially investigated the expression levels of tau phosphorylation
at the positions of both the KXGS motif (i.e., Ser262;
p-tauS262) and the proline-directed motif (i.e., Ser396;
p-tauS396) in the cerebral cortices and hippocampi
obtained from SAMP8 or SAMR1 at young (2-months-old), middle
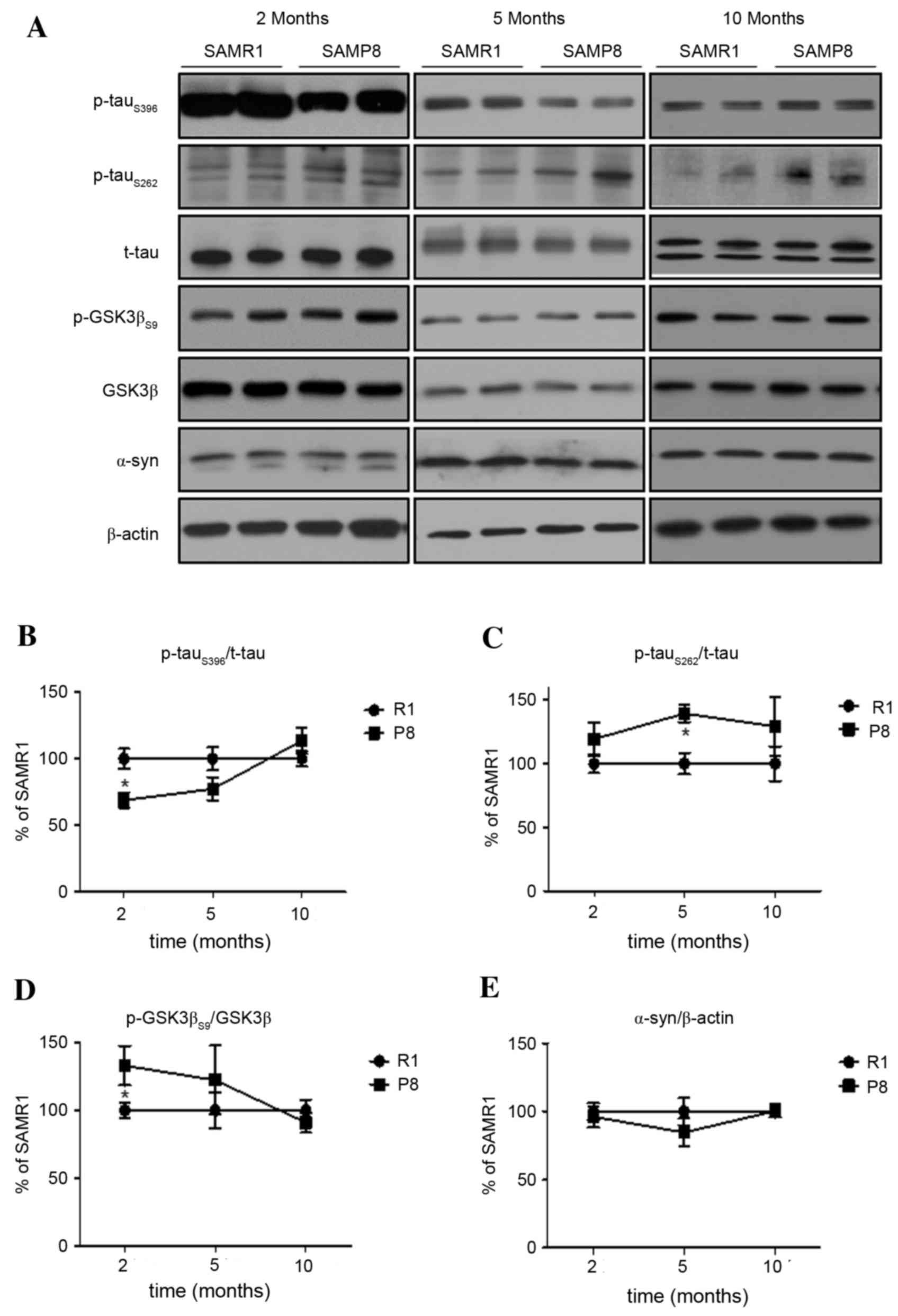
(5-months-old) and old (10-months-old) ages. In the cerebral cortex
of SAMP8, differential levels of p-tauS262 and
p-tauS396 were observed, compared to those in SAMR1. The
lower level of p-tauS396 in the cortex of 2-month-old
SAMP8 compared to SAMR1 controls increased in a time-dependent
manner up to 10-months-old, at which point it reached levels
comparable the SAMR1 controls (P<0.05; Fig. 1A and B). Meanwhile, the level of
p-tauS262 in young SAMP8 was comparable to that in
SAMR1; however, these levels significantly increased in the cortex
of middle-aged SAMP8 and were sustained in old SAMP8, when compared
to those of age-matched SAMR1 (P<0.05; Fig. 1A and C). The chronological changes
in the cortical level of p-tauS396 were negatively
correlated with the expression of phosphorylated GSK3β (P<0.05;
Fig. 1A and D). In addition, the
pattern of changes in the level of active (phosphorylated at
Thr172) AMPK, an upstream kinase of GSK3β and ACC, was similar to
GSK3βS9 and p-tauS396 (Fig. 3A and E). There are several reports
that tau pathology can be accelerated by overexpression of
α-synuclein (28,29). However, the expression level of
α-synuclein in the cortex of SAMP8 was not significantly different
from that of SAMR1 (P>0.05; Fig. 1A
and E) at all ages.
Characteristics of hippocampal tau
phosphorylation of SAMP8
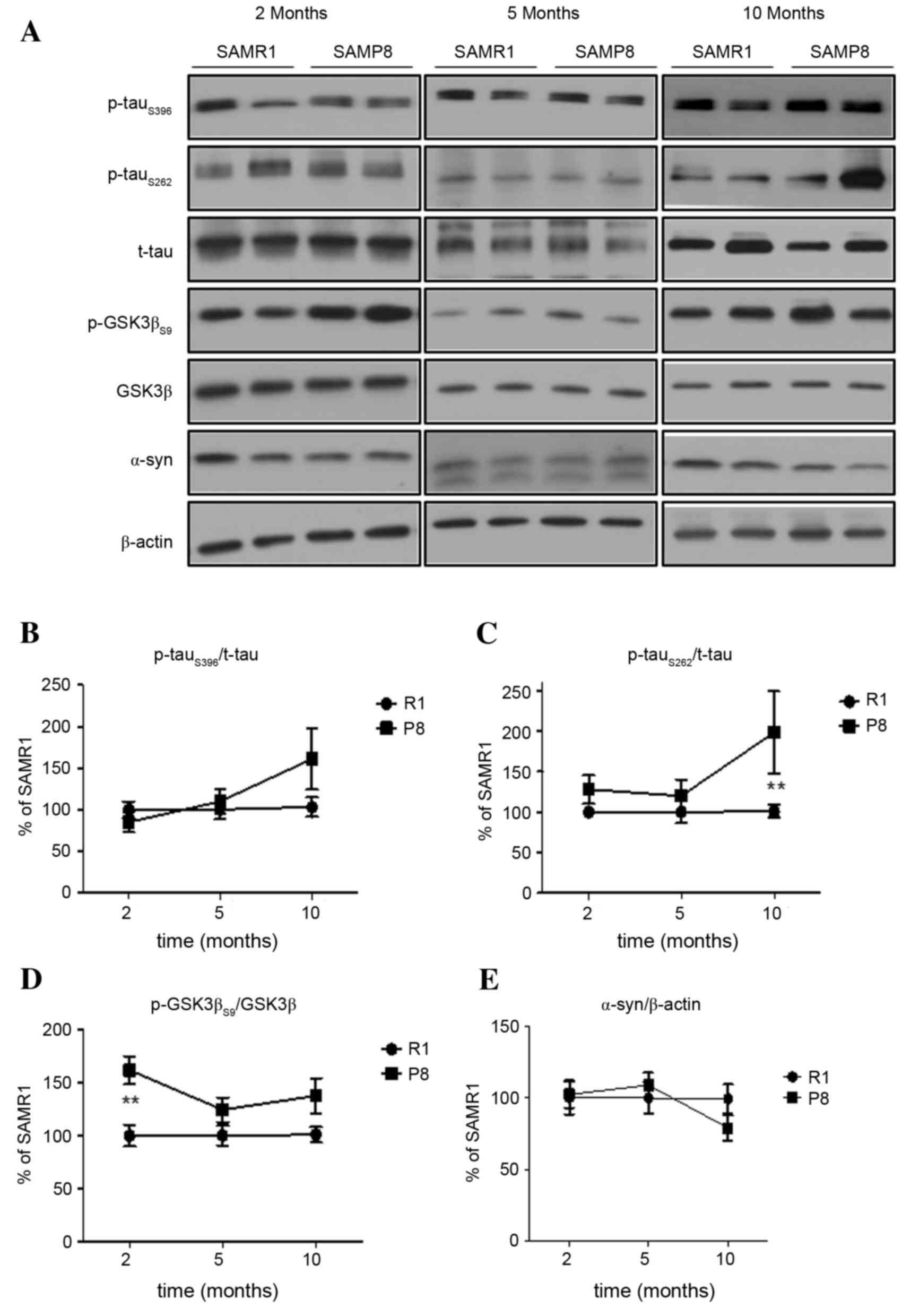
In the hippocampus, p-tauS396 and
p-tauS262 were observed to increase as SAMP8 increased
in age (Fig. 2A-C). In young
SAMP8, a significant difference was not observed between the
expressions of p-tau at both sites (P>0.05), however, the levels
of hippocampal p-tau in aged SAMP8 were higher than age-matched
SAMR1 (p-tauS262, P<0.01; Fig. 2A and C). The level of hippocampal
p-GSK3βS9 expression in young SAMP8 was significantly
higher than in SAMR1 (P<0.01; Fig.
2D), but the pattern of changes in expression levels of
p-tauS396 and p-AMPK was not similar. Similar to the
cortex, the expression levels of α-synuclein and total tau in the
hippocampus of SAMP8 were not significantly different from that in
the hippocampus of SAMR1 (P>0.05; Fig. 2A and E).
Changes in the expression of proteins
related to aging and cellular metabolism in the cortex of
SAMP8
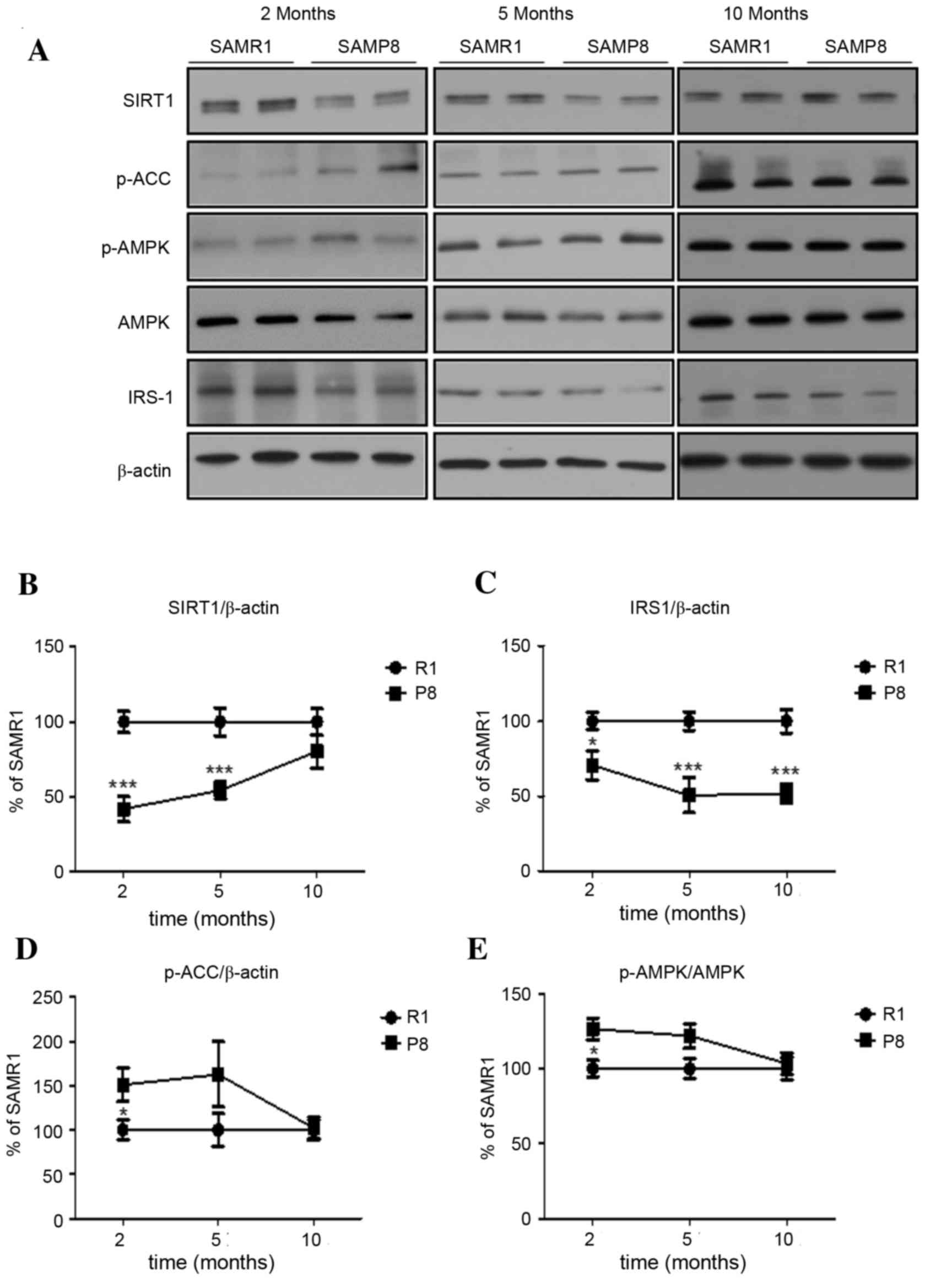
The authors demonstrated the chronological
expression pattern of proteins related to senescence (Sirt1) and
intracellular energy metabolism (AMPK and IRS-1) in the brains of
SAMP8. In the cerebral cortex of SAMP8, the level of Sirt1
expression at 2-months-old was significantly lower than in SAMR1
controls (P<0.001), which persisted until 5-months-old and
disappeared in mice at 10-months-old (Fig. 3A and B). Interestingly, the level
of IRS-1 expression in the cortex of young SAMP8 was significantly
lower than in SAMR1, which persisted until 10-months-old (P<0.05
at 2 months, P<0.001 at 5 and 10 months; Fig. 3A and C). In addition, the levels of
active p-AMPK and of inhibitory phosphorylation of ACC at Ser79, a
well-known AMPK substrate, in the cerebral cortex of young SAMP8
were significantly higher than those in SAMR1 (both P<0.05),
with the increase disappearing in middle- and old-aged mice
(Fig. 3A, D and E).
Changes in the expression of proteins
related to aging and cellular metabolism in the SAMP8
hippocampus
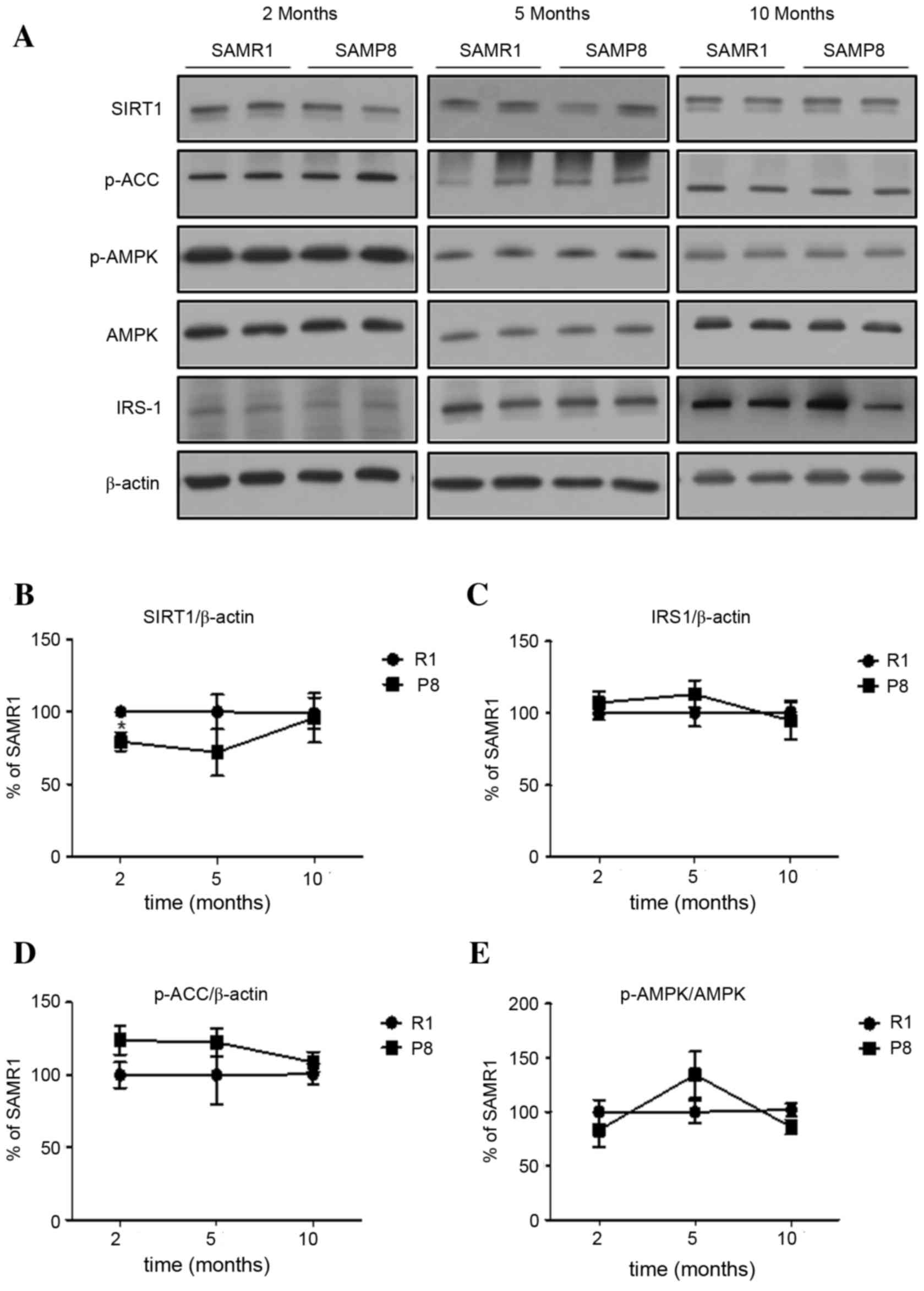
In the hippocampus of SAMP8, the level of Sirt1
expression at young age was significantly lower, when compared with
levels in SAMR1 (P<0.05; Fig. 4A
and B). However, the levels of Sirt1 expression in old SAMP8
and SAMR1 were not significantly different. Finally, the levels of
other proteins (IRS-1, p-AMPK and p-ACCS79) in SAMP8
were not significantly different from those in SAMR1 (P>0.05;
Fig. 4A, C-E) across all ages,
which was different from the pattern of changes of these proteins
in the cortex.
Discussion
Multiple mechanisms have been implicated in the
development of AD, including accumulation of Aβ, oxidative stress,
increased synaptic damage, hyperphosphorylation of tau, defective
neurogenesis and alterations in the signal transduction pathways
associated with neuronal survival (1–4).
Therefore, a number of studies have been conducted to augment the
information of SAMP8 as a model of AD using the aforementioned
targets (2,30). Apart from these mechanisms, the
present study investigated whether the brains of SAMP8 that mimic
the AD brain present dysregulation of molecules involved in both
energy metabolism and tau phosphorylation (i.e., AMPK and GSK3β),
and whether the alteration of AMPK-GSK3β activity is related to the
hyperphosphorylation of tau.
In terms of energy metabolism, the brain largely
relies on circulating glucose as a primary source of fuel because
it stores only small sources of energy as glycogen; therefore,
understanding glucose metabolism in the brain is important
(11,13). Moreover, type 2 diabetes increases
the risk of sporadic AD, and AD leads to insulin resistance, which
is associated with dysfunction of IRS-1 and Sirt1 in the brain
(10,31). Consistent with previous studies
(32), the current study observed
an early and significant decrease in both IRS-1 and Sirt1
expression in the cortex of SAMP8. A double knockout of IRS-1 and
−2 in the skeletal muscle has demonstrated the activation of AMPK
(33), and Sirt1 and AMPK also
serves roles in the energy-sensing network that controls energy
expenditure and protects against metabolic imbalance in various
cells including neurons (34). The
authors identified significantly reduced levels of IRS-1 and Sirt1
in the cortex of SAMP8 mice, which was accompanied by increased
AMPK activity at young ages. Since the Sirt1 pathway is closely
related to AMPK signaling as a sensor of energy availability
(35,36), the authors speculated that the
reduction of IRS-1 and Sirt1 expression may involve the
compensatory activation of AMPK in the cortex of young SAMP8 mice.
It is not clear why the association of IRS-1 and Sirt1 expression
with AMPK disappeared in older SAMP8; it may be that the prolonged
suppression of insulin signaling pathways disturbed the
compensatory activation of AMPK (35,36).
AMPK has recently been proposed as a novel
therapeutic target for regulating metabolism-related pathogenesis
in AD, as well as a target in Aβ- and tau-related pathogenesis.
AMPK inhibits the activity of GSK3β through increasing the
inhibitory phosphorylation of GSK3β at Ser9 (37). GSK3β is a well-known tau kinase
that generates pathological phosphor-epitopes at proline-directed
sites, such as p-tauS396, in tau, which has been
described as a reliable marker of AD (37–39).
Accordingly, the authors hypothesized that AMPK activation may
inhibit GSK3β activity, resulting in a reduction of
p-tauS396 in young SAMP8. This hypothesis was supported
by evidence that the pattern of p-AMPK in the cortex of SAMP8 at 2,
5 and 10 months of age was correlated with the levels of
p-GSK3βS9 and negatively correlated with the levels of
p-tauS396. Moreover, a previous study of the authors
indicated that 5-aminoimidazole-4-carboxamide
ribonucleotide-induced AMPK activation reduces GSK3β activity and
levels of p-tauS396 in differentiated SH-SY5Y cells
(27). Thus, these data suggested
that the activity of AMPK in the cerebral cortex of young SAMP8 was
associated with decreased levels of p-tauS396 via a
reduction of GSK3β activity. In addition, a significant increase to
p-tauS262 levels in the cortex of middle-aged SAMP8 mice
was observed, when compared to those of age-matched SAMR1 mice.
However, the pattern of changes in cortical levels of
p-tauS262 were not similar to the changes in activity of
AMPK and GSK3β. Neither were the chronological changes in levels of
p-tauS396 and p-tauS262 in the hippocampus
similar to the changes in the activity of AMPK and GSK3β.
Collectively, the role of GSK3β and AMPK in tau phosphorylation may
be dependent on the phosphor-epitopes (e.g., p-tauS396
and p-tauS262) in tau or the region (e.g., cortex and
hippocampus) of SAMP8 brain. In neuronal cells, AMPK impairs
dendritic growth and branching of primary hippocampal neurons
(40). Several studies have
recently revealed that AMPK activation protects neuronal cells from
cell death triggered by abnormalities in brain energy metabolism or
accumulation of Aβ or α-synuclein (23,24,41).
Therefore, whether AMPK activation is beneficial or harmful at the
preclinical stage of AD should be further evaluated in various
in vivo models and in clinical studies.
Animal models are extremely useful to predict the
outcome of drug candidates for prevention or therapy of AD, as well
as to study mechanistic hypotheses for AD pathogenesis. However, no
animal model recapitulates the entirety of AD pathophysiology in
humans. Therefore, it is important to characterize the advantages
and limitations of particular animal models. Among the animal
models of AD, the sequence of pathologies and clinical
characteristics of SAMP8 is similar to that observed in human
patients with AD (2). In the
present study, the data provided basic information on SAMP8 that
included the relationship between alterations in energy metabolism
and AD-related tau hyper-phosphorylation in the brain. Based on
these data, the authors have suggested that the SAMP8 strain of
mice has pathologic similarities to AD, and is therefore an
excellent model for advancing the knowledge for AD-related
metabolic dysfunction through age-related neurodegenerative
processes. However, it should be noted that selected sites of the
phosphor-epitope in tau may be differentially regulated by the
AMPK-GSK3β-mediated pathway, and particular regions of the brain
may be a factor in studies targeting tau phosphorylation in the
SAMP8 AD model.
In conclusion, the presented results demonstrated
early deteriorations in energy metabolism and concurrent
alterations of tau hyper-phosphorylation dependent on the region of
the brain and phosphor-tau epitopes. In addition, these findings
provided basic information on the chronological changes in the
expression of metabolic proteins and AD-related tau phosphorylation
in SAMP8, and this model may allow researchers to further
investigate preventive and therapeutic targets for AD.
Acknowledgements
This study was financially supported by Mid-career
Researcher Program (grant no. 2013R1A2A2A01008223) and Medical
Research Center (grant no. 2014009392) through the National
Research Foundation of Korea (NRF).
References
|
1
|
Takeda T: Senescence-accelerated mouse
(SAM): A biogerontological resource in aging research. Neurobiol
Aging. 20:105–110. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morley JE, Armbrecht HJ, Farr SA and Kumar
VB: The senescence accelerated mouse (SAMP8) as a model for
oxidative stress and Alzheimer's disease. Biochim Biophys Acta.
1822:650–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morley JE, Kumar VB, Bernardo AE, Farr SA,
Uezu K, Tumosa N and Flood JF: Beta-amyloid precursor polypeptide
in SAMP8 mice affects learning and memory. Peptides. 21:1761–1767.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Canudas AM, Gutierrez-Cuesta J, Rodriguez
MI, Acuña-Castroviejo D, Sureda FX, Camins A and Pallàs M:
Hyperphosphorylation of microtubule-associated protein tau in
senescence-accelerated mouse (SAM). Mech Ageing Dev. 126:1300–1304.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de la Monte SM and Tong M: Brain metabolic
dysfunction at the core of Alzheimer's disease. Biochem Pharmacol.
88:548–559. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aberg ND, Brywe KG and Isgaard J: Aspects
of growth hormone and insulin-like growth factor-I related to
neuroprotection, regeneration, and functional plasticity in the
adult brain. ScientificWorldJournal. 6:53–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kleinridders A, Ferris HA, Cai W and Kahn
CR: Insulin action in brain regulates systemic metabolism and brain
function. Diabetes. 63:2232–2243. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cole GM and Frautschy SA: The role of
insulin and neurotrophic factor signaling in brain aging and
Alzheimer's Disease. Exp Gerontol. 42:10–21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reger MA, Watson GS, Frey WH II, Baker LD,
Cholerton B, Keeling ML, Belongia DA, Fishel MA, Plymate SR,
Schellenberg GD, et al: Effects of intranasal insulin on cognition
in memory-impaired older adults: modulation by APOE genotype.
Neurobiol Aging. 27:451–458. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moloney AM, Griffin RJ, Timmons S,
O'Connor R, Ravid R and O'Neill C: Defects in IGF-1 receptor,
insulin receptor and IRS-1/2 in Alzheimer's disease indicate
possible resistance to IGF-1 and insulin signalling. Neurobiol
Aging. 31:224–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Biessels GJ and Reagan LP: Hippocampal
insulin resistance and cognitive dysfunction. Nat Rev Neurosci.
16:660–671. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hardie DG: AMPK-sensing energy while
talking to other signaling pathways. Cell Metab. 20:939–952. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weisová P, Dávila D, Tuffy LP, Ward MW,
Concannon CG and Prehn JH: Role of 5′-adenosine
monophosphate-activated protein kinase in cell survival and death
responses in neurons. Antioxid Redox Signal. 14:1863–1876. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma
T, Thompson RC, Zhao Y, Smith L, Gasparini L, et al: Antidiabetic
drug metformin (GlucophageR) increases biogenesis of Alzheimer's
amyloid peptides via up-regulating BACE1 transcription. Proc Natl
Acad Sci USA. 106:3907–3912. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vingtdeux V, Davies P, Dickson DW and
Marambaud P: AMPK is abnormally activated in tangle- and
pre-tangle-bearing neurons in Alzheimer's disease and other
tauopathies. Acta Neuropathol. 121:337–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thornton C, Bright NJ, Sastre M, Muckett
PJ and Carling D: AMP-activated protein kinase (AMPK) is a tau
kinase, activated in response to amyloid beta-peptide exposure.
Biochem J. 434:503–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vingtdeux V, Chandakkar P, Zhao H,
d'Abramo C, Davies P and Marambaud P: Novel synthetic
small-molecule activators of AMPK as enhancers of autophagy and
amyloid-β peptide degradation. FASEB J. 25:219–231. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai Z, Li B, Li K and Zhao B:
Down-regulation of amyloid-β through AMPK activation by inhibitors
of GSK-3β in SH-SY5Y and SH-SY5Y-AβPP695 cells. J Alzheimers Dis.
29:89–98. 2012.PubMed/NCBI
|
|
19
|
Greco SJ, Sarkar S, Casadesus G, Zhu X,
Smith MA, Ashford JW, Johnston JM and Tezapsidis N: Leptin inhibits
glycogen synthase kinase-3beta to prevent tau phosphorylation in
neuronal cells. Neurosci Lett. 455:191–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim J, Park YJ, Jang Y and Kwon YH: AMPK
activation inhibits apoptosis and tau hyperphosphorylation mediated
by palmitate in SH-SY5Y cells. Brain Res. 1418:42–51. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim B, Figueroa-Romero C, Pacut C, Backus
C and Feldman EL: Insulin resistance prevents AMPK-induced tau
dephosphorylation through Akt-mediated increase in AMPKSer-485
phosphorylation. J Biol Chem. 290:19146–19157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Culmsee C, Monnig J, Kemp BE and Mattson
MP: AMP-activated protein kinase is highly expressed in neurons in
the developing rat brain and promotes neuronal survival following
glucose deprivation. J Mol Neurosci. 17:45–58. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chan KH, Lam KS, Cheng OY, Kwan JS, Ho PW,
Cheng KK, Chung SK, Ho JW, Guo VY and Xu A: Adiponectin is
protective against oxidative stress induced cytotoxicity in
amyloid-beta neurotoxicity. PLoS One. 7:e523542012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dulovic M, Jovanovic M, Xilouri M,
Stefanis L, Harhaji-Trajkovic L, Kravic-Stevovic T, Paunovic V,
Ardah MT, El-Agnaf OM, Kostic V, et al: The protective role of
AMP-activated protein kinase in alpha-synuclein neurotoxicity in
vitro. Neurobiol Dis. 63:1–11. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cuesta S, Kireev R, Garcia C, Rancan L,
Vara E and Tresguerres JA: Melatonin can improve insulin resistance
and aging-induced pancreas alterations in senescence-accelerated
prone male mice (SAMP8). Age (Dordr). 35:659–671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu HW, Chan YC, Wang MF, Wei CC and Chang
SJ: Dietary (−)-epigallocatechin-3-gallate supplementation
counteracts aging-associated skeletal muscle insulin resistance and
fatty liver in senescence-accelerated mouse. J Agric Food Chem.
63:8407–8417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim HS, Moon S, Paik JH, Shin DW, Kim LS,
Park CS, Ha J and Kang JH: Activation of the 5′-AMP-activated
protein kinase in the cerebral cortex of young
senescence-accelerated P8 mice and association with GSK3β- and
PP2A-dependent inhibition of p-tau396 expression. J
Alzheimers Dis. 46:249–259. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo JL, Covell DJ, Daniels JP, Iba M,
Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ and
Lee VM: Distinct α-synuclein strains differentially promote tau
inclusions in neurons. Cell. 154:103–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giasson BI, Forman MS, Higuchi M, Golbe
LI, Graves CL, Kotzbauer PT, Trojanowski JQ and Lee VM: Initiation
and synergistic fibrillization of tau and alpha-synuclein. Science.
300:636–640. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morley JE, Farr SA, Kumar VB and Armbrecht
HJ: The SAMP8 mouse: A model to develop therapeutic interventions
for Alzheimer's disease. Curr Pharm Des. 18:1123–1130. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de la Monte SM: Brain insulin resistance
and deficiency as therapeutic targets in Alzheimer's disease. Curr
Alzheimer Res. 9:35–66. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pallàs M, Pizarro JG, Gutierrez-Cuesta J,
Crespo-Biel N, Alvira D, Tajes M, Yeste-Velasco M, Folch J, Caudas
AM, Sureda FX, et al: Modulation of SIRT1 expression in different
neurodegenerative models and human pathologies. Neuroscience.
154:1388–1397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Long YC, Cheng Z, Copps KD and White MF:
Insulin receptor substrates Irs1 and Irs2 coordinate skeletal
muscle growth and metabolism via the Akt and AMPK pathways. Mol
Cell Biol. 31:430–441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Burkewitz K, Zhang Y and Mair WB: AMPK at
the nexus of energetics and aging. Cell Metab. 20:10–25. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM,
Saha AK, Lan F and Ido Y: AMPK and SIRT1: A long-standing
partnership? Am J Physiol Endocrinol Metab. 298:E751–E760. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ruderman NB, Carling D, Prentki M and
Cacicedo JM: AMPK, insulin resistance, and the metabolic syndrome.
J Clin Invest. 123:2764–2772. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park H, Kam TI, Kim Y, Choi H, Gwon Y, Kim
C, Koh JY and Jung YK: Neuropathogenic role of adenylate kinase-1
in Aβ-mediated tau phosphorylation via AMPK and GSK3β. Hum Mol
Genet. 21:2725–2737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu YY, He SS, Wang X, Duan QH,
Grundke-Iqbal I, Iqbal K and Wang J: Levels of nonphosphorylated
and phosphorylated tau in cerebrospinal fluid of Alzheimer's
disease patients: An ultrasensitive bienzyme-substrate-recycle
enzyme-linked immunosorbent assay. Am J Pathol. 160:1269–1278.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanger DP, Anderton BH and Noble W: Tau
phosphorylation: The therapeutic challenge for neurodegenerative
disease. Trends Mol Med. 15:112–119. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ramamurthy S, Chang E, Cao Y, Zhu J and
Ronnett GV: AMPK activation regulates neuronal structure in
developing hippocampal neurons. Neuroscience. 259:13–24. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin CL, Cheng YS, Li HH, Chiu PY, Chang
YT, Ho YJ and Lai TJ: Amyloid-β suppresses AMP-activated protein
kinase (AMPK) signaling and contributes to α-synuclein-induced
cytotoxicity. Exp Neurol. 275:84–98. 2016. View Article : Google Scholar : PubMed/NCBI
|