Introduction
It is well-known that atherosclerosis preferentially
occurs at sites where the blood flow is slow or disturbed, and
where the wall shear stress is low or oscillatory (1). Studies have demonstrated that low
shear stress (LSS) or oscillatory flow [0-4 Dyne
(dyn)/cm2), promotes an atherogenic endothelial
phenotype with increased endothelial cell proliferation and
apoptosis (2), which destroys the
endothelium barrier, and initiates an inflammatory response,
causing oxidized low-density lipoprotein accumulation in the artery
wall and therefore, progression of atherosclerosis (3). The causative association of LSS with
atherosclerosis has been demonstrated (4). LSS is a well-established risk factor
resulting in atherosclerosis, and LSS is critically important in
regulating the vascular physiology and pathology of the vessel
walls, by modulating the endothelial cell function (5). However, the detailed molecular
mechanisms underlying LSS-induced atherosclerosis remain
unclear.
Autophagy is a highly regulated process, that may be
involved in the turnover of long-lived proteins and organelles, and
may help cells survive in an unfavorable environment (6). Parts of the cytoplasm and
intracellular organelles are sequestered within characteristic
double-membrane autophagic vacuoles (known as autophagosomes) and
are ultimately delivered to lysosomes for bulk degradation.
Previously, increasing evidence revealed that autophagy is involved
in the pathogenesis of atherosclerosis, stimulated by oxidized
lipids, inflammation or metabolic stress (7,8).
However, it is unclear whether autophagy participates in the
molecular mechanism underlying LSS-induced atherosclerosis.
Furthermore, the role of autophagy, either protective or
detrimental, in human umbilical vein endothelial cell (HUVEC) death
induced by LSS is also poorly understood. In the present study, it
was examined whether LSS was able to induce activation of autophagy
in HUVECs, and the contribution of autophagy to cell apoptosis and
survival under LSS was evaluated.
Materials and methods
Reagents
Antibodies against MAP1 light chain 3-like protein
(LC3; cat. no. L7543), rapamycin (cat. no. V900930), chloroquine
(CQ; cat. no. C6628) and DAPI (cat. no. D9542) were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Antibodies against
apoptosis regulator Bcl-2 (Bcl-2; cat. no. sc7382), apoptosis
regulator BAX (Bax; cat. no. sc70408), Beclin-1 (cat. no. sc48381)
and β-actin (cat. no. sc47778) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Antibody against p62 (cat.
no. 5114 s) was purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). IR-Dye 680 (cat. no. 926-32220) or 800cw (cat.
no. 926-32211) labeled secondary antibodies were purchased from
Li-Cor Biosciences (Lincoln, NE, USA). The HUVECs were provided by
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). High-glucose Dulbecco's modified Eagle's medium
(DMEM) and fetal bovine serum (FBS) were purchased from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Annexin-V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
Apoptosis Detection kits were purchased from BD Biosciences
(Franklin Lakes, NJ, USA).
Cell culture
The HUVECs were cultured in high-glucose DMEM
supplemented with 10% FBS, in a 95% humidified incubator, with 5%
CO2 at 37°C. For all of the experiments, HUVECs in
passage 3 were used.
Shear stress experiment
The flow experiments were performed as previously
described (9). A parallel-plate
flow system was used to impose a laminar shear stress of 1.5
dyn/cm2. The system was maintained at 37°C and
ventilated with 95% humidified air containing 5%
CO2.
Treatment of cells with rapamycin and
CQ
At 70–80% confluency, the cells were treated with 5
nM rapamycin or 20 µM CQ for 24 h, followed by treatment with LSS
(1.5 dyn/cm2) for an additional 0.5, 1, 2 or 3 h,
respectively. The samples under static conditions (no flow) were
used as the control.
Flow cytometry analysis of
apoptosis
Apoptosis in the HUVECs was measured with the
Annexin V-FITC/PI Apoptosis Detection kit, according to the
manufacturer's protocol. The stained cells were analyzed by flow
cytometry (BD FACSAria III; BD Biosciences, Franklin Lakes, NJ,
USA). Data analysis was performed using FlowJo version 7.6.1 (Tree
Star, San Carlos, CA, USA).
Transmission electron microscopy
(TEM)
Cells were seeded at a density 2×105
cells/well and fixed in 2.5% PBS glutaraldehyde at 4°C for 1 h.
Post-fixation was performed in 1% OsO4, for 1 h. The
cells were dehydrated in an ethanol gradient and embedded in
Araldite (Huntsman Co., Ltd., Salt Lake City, UT, USA). Sections
(40–60 nm) were placed on a grid (200 mesh) and were double-stained
with uranylacetate and lead citrate. The sections were observed
under a Philips CM-120 TEM.
Immunofluorescence
The cells were fixed with 4% paraformaldehyde,
permeabilized with 0.2% Triton X-100, blocked with 5% non-fat milk
for 2 h at room temperature, incubated with LC3antibodies (1:100)
overnight at 4°C and stained with DAPI for 1 h, followed by
incubation with FITC-conjugated secondary antibody (1:80, cat. no.
ZF-0311, Beijing Zhongshan Golden Bridge Biotechnology Co. Ltd.,
Beijing, China), immunoglobulin G, for 2 h. The images of the cells
were captured using a fluorescence microscope (Leica TCS SP5). To
quantify autophagic cells, LC3 puncta were determined in triplicate
by counting >30 cells.
Western blotting (WB)
WB was performed as previously described (10). Briefly, cells were washed twice
with ice-cold PBS and lysed in ice-cold Western and IP cell protein
lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China)
containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100,
supplemented with 1% (v/v) protease inhibitor cocktail and
phenylmethanesulfonyl fluoride (Beyotime Institute of
Biotechnology). The extracts were incubated on ice for 30 min,
centrifuged at 12,000 × g for 10 min at 4°C and the supernatants
were collected. Protein concentrations were determined with a BCA
Protein Assay kit (Beyotime Institute of Biotechnology), and total
protein (20 µg) was separated by 10–12% SDS-PAGE, and transferred
to nitrocellulose membranes. The membranes were blocked with 5%
non-fat milk for 2 h at room temperature and incubated overnight at
4°C with antibodies specific for Beclin-1 (1:600), LC3 (1:1,000),
p62 (1:800), Bax (1:500), Bcl-2 (1:500), and β-actin (1:1,000),
followed by incubation with goat anti-mouse or goat anti-rabbit
IR-Dye 680 or 800cw labeled secondary antibodies (1:10,000) for 1 h
at room temperature. Membranes were scanned using a Li-Cor Odyssey
scanner. Each band of interest returned near-infrared fluorescent
values of raw intensity with intra-lane background subtracted using
Odyssey 3.0 analytical software (Li-Cor Biosciences).
Statistical analysis
All of the data were representative of at least
three independent experiments and were expressed as the mean ±
standard deviation. Statistical analyses were performed using
one-way analysis of variance, followed by the Student-Newman-Keuls
test. P<0.05 was considered to indicate a statistically
significant difference. The statistical analysis was performed
using SPSS software (version 18.0; SPSS, Inc., Chicago, Il,
USA).
Results
Cell morphological and viability
changes of HUVECs
Increased LSS-only treatment times led to a gradual
reduction in cell viability, with cell shrinkage and easy
detachment from the coverslip compared with static cells (Fig. 1A-E). Treatment with rapamycin alone
exhibited no effect on the morphology of HUVECs in static condition
(Fig. 1F), whereas treatment
combined with LSS effectively attenuated the cell injury induced by
LSS and preserved the shape of the HUVECs compared with LSS group
at the same time point (Fig.
1G-J). Treatment with CQ alone had no influence on the
morphology of HUVECs in static condition (Fig. 1K), whereas CQ+LSS treatment
exacerbated cell shrinkage and the cells detached more easily from
the coverslip compared with LSS-only group at the same time points
(Fig. 1L-O).
 | Figure 1.Effects of LSS on the morphology of
HUVECs. Cells were maintained under static conditions as controls
or subjected to LSS (1.5 dyn/cm2) for (A) 0, (B) 0.5,
(C) 1, (D) 2 or (E) 3 h. The cells were pretreated with RAPA (5 nM)
(f-j) for 24 h and subjected to LSS (1.5 dyn/cm2) for
(F) 0, (G) 0.5, (H) 1, (I) 2, and (J) 3 h. The cells were
pretreated with CQ (20 µM) for 24 h and subjected to LSS (1.5
dyn/cm2) for (K) 0, (L) 0.5, (M) 1, (N) 2, and (O) 3 h.
Images of the cellular morphology were captured using a microscope.
Scale bar, 1,000 µm. HUVEC, human umbilical vein endothelial cells;
LSS, low shear stress; RAPA, rapamycin; CQ, chloroquine. |
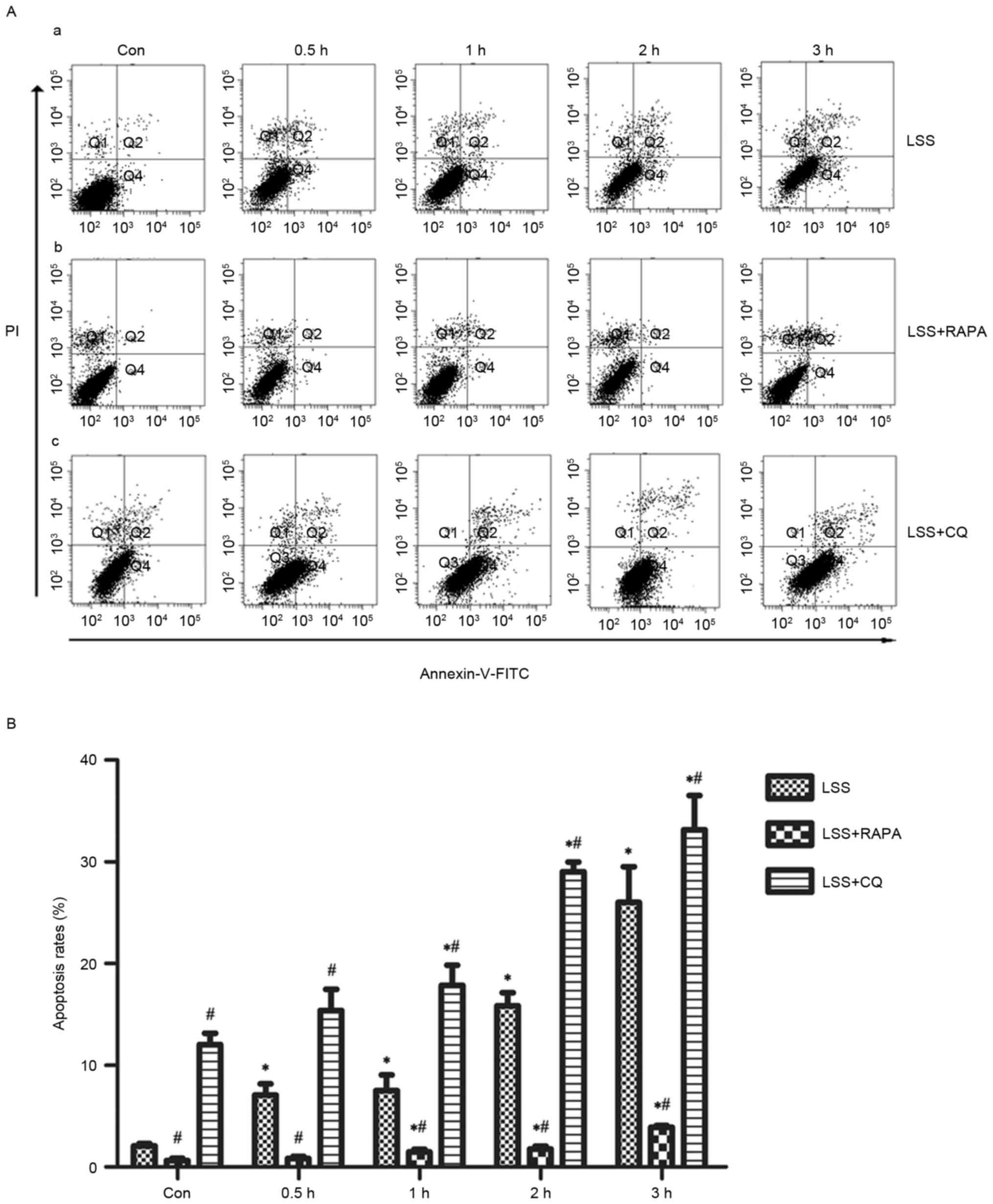
Effects of LSS, LSS+RAPA and LSS+CQ on
cell apoptosis of HUVECs
Flow cytometry analysis demonstrated that LSS-only
treatment resulted in a significant increase in apoptosis in a
time-dependent manner (Fig. 2A-a and
B). Rapamycin treatment reduced the pro-apoptotic effect of LSS
treatment in HUVECs compared with the control and LSS-only group at
the same time points (Fig. 2A-b and
B). In addition, CQ led to a significant increase in the later
apoptotic cell population and an increase of the early apoptotic
cell population compared with the control and LSS-only groups at
the same time points (Fig. 2A-c and
B).
Autophagy and apoptosis-associated
protein levels changes in HUVECs treated with LSS, LSS+RAPA or
LSS+CQ
Beclin-1, LC3 and p62 proteins are reliable markers
for autophagy (Fig. 3). In the
present study, the level of Beclin-1 and conversion of LC3I into
LC3II were markedly increased, whereas the levels of p62 decreased
in the HUVECs treated with LSS for 0.5, 1, 2 and 3 h compared with
control (Fig. 3A and D-F).
Rapamycin treatment significantly increased the level of Beclin-1
and the conversion of LC3I into LC3II (Fig. 3B, D and E); however, the protein
expression of p62 was reduced compared with the control and
LSS-only groups at the same time points (Fig. 3B and F). LSS+CQ treatment resulted
in a significant increase of Beclin-1 expression and the LC3I to
LC3II ratio compared with control and LSS group at the same time
point (Fig. 3C-E). It also
increased p62 expression in HUVECs compared with LSS group at the
same time point; however, the p62 expression following LSS+CQ
treatment was lower compared with CQ only treatment (Fig. 3C and F). Bcl-2 and Bax are
important members of the Bcl-2 family which have an important role
the regulation of apoptosis. In the present study, the expression
of Bcl-2 was downregulated and the expression of Bax was
upregulated in the LSS treatment group compared with the control
group (Fig. 3A, G and H). Compared
with RAPA treatment alone, LSS+RAPA treatment for 0.5, 1, 2 and 3 h
significantly decreased the Bcl-2 levels (Fig. 3B and G) and increased the Bax
levels (Fig. 3B and H). However,
compared with LSS groups at the same time points, LSS+RAPA
treatment significantly increased the Bcl-2 levels (Fig. 3B and G) and reduced Bax levels
(Fig. 3B and H). CQ treatment
increased the Bax levels and reduced the Bcl-2 levels (Fig. 3C, G and H) compared with control
and LSS group at the same time point.
 | Figure 3.LSS induces autophagy and apoptosis in
HUVECs. The expression levels of the Beclin-1, LC3I, LC3II, p62,
Bcl-2 and Bax proteins were determined by western blotting,
following treatment with (A) LSS, (B) LSS+RAPA and (C) LSS+CQ. (D)
Beclin-1 intensity, (E) LC3II/LC3I intensity, (F) p62 intensity,
(G) Bcl-2 intensity and (H) Bax intensity were expressed as the
fold change between LSS+RAPA, LSS+CQ and LSS. The bar graphs
represent the mean ± standard error (n=3). *P<0.01 vs. the
control. †P<0.05 and #P<0.01 vs. the
cells pretreated with the various modulators at the same point.
Con, control; LSS, low shear stress; HUVEC, human umbilical vein
endothelial cells; RAPA, rapamycin; CQ, chloroquine; LC3, MAP1
light chain 3-like protein; Bcl-2, apoptosis regulator Bcl-2; Bax,
apoptosis regulator BAX. |
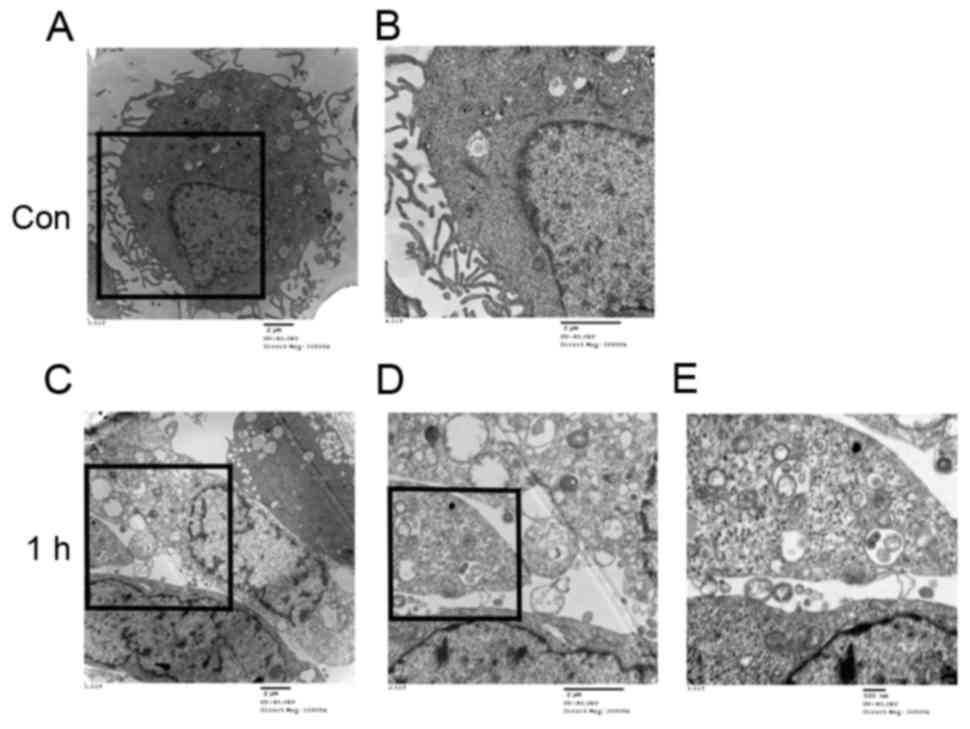
Observation of autophagosomes and
lysosomes in HUVECs
A normal cytoplasm, mitochondria and nuclei and a
small number of autophagosomes and lysosomes were observed in the
TEM images of the control group (Fig.
4A and B). HUVECs treated with LSS-only for 1 h exhibited
numerous autophagosomes at various stages of development (Fig. 4C-E).
In addition, the treatment with LSS induced
extensive formation of LC3 puncta compared with static cells, as
determined by LC3 immunofluorescence staining (Fig. 5A-a, -b and B). Pretreatment with
rapamycin significantly increased the formation of the LC3 puncta
in HUVECs (Fig. 5A-c, -d and B)
compared with control and LSS-only group. Pretreatment with CQ
resulted in a significant accumulation of LC3 puncta (Fig. 5A-e, -f and B) in HUVECs compared
with control and LSS group.
Discussion
In the present study, it was demonstrated that
atheroprone LSS conditions were able to induce cell autophagy and
apoptosis by regulating the balance of Bcl-2/Beclin-1 and
Bcl-2/Bax. The induction of autophagy, by pretreatment with
rapamycin, protected the HUVECs against LSS-induced apoptotic cell
death. Autophagy inhibition by pretreatment with CQ resulted in
elevated apoptotic cell death. With these results, it was concluded
that autophagy served an important role in protecting against
LSS-induced apoptosis.
Apoptosis is a highly regulated cell death process
characterized by cell shrinkage, membrane blebbing, DNA
fragmentation and chromatin condensation (11). It constitutes an initial step in
endothelial cell dysfunction, which is an important feature in
atherosclerosis (12). LSS is a
well-established risk factor resulting in atherosclerosis, and it
serves a critical role in modulating endothelial cell function.
Recently, studies indicate that LSS is able to induce endothelial
cell apoptosis (13). Consistent
with these previous results, the data from flow cytometry with
Annexin-V-FITC/PI dual staining, demonstrated that LSS induced the
apoptosis of HUVECs. It is well-known that the Bcl-2 family serves
a key role in the process of apoptosis. This family includes
anti-apoptotic proteins, including Bcl-2, Bcl-2-like protein 1 and
induced myeloid leukemia cell differentiation protein Mcl-1, and
pro-apoptotic proteins, including Bax, Bcl-2-associated agonist of
cell death and Bcl-2 homologous antagonist/killer (14). A previous study demonstrated that
Bcl-2/Bax ratio is a rheostat which determines the incidence of
apoptosis (15). In the present
study, a marked decrease in Bcl-2 and an increase in Bax in HUVECs
treated with LSS was observed. This observation suggests that LSS
increases apoptosis by regulating the balance between Bcl-2 and
Bax.
Autophagy, another type of programmed cell death,
serves an important role in a number of physiological and
pathological processes, including aging and cardiac ischemia
(16,17). Studies have demonstrated that
endothelial cells exhibit characteristics of autophagy when the
cells are exposed to pro-atherogenic factors (18), which indicates that autophagy may
serve a crucial role in regulating the formation and progression of
atherosclerosis (7). Beclin-1, LC3
and p62 have been reported as reliable markers of autophagy
(19). Beclin-1 was originally
identified as a Bcl-2-interacting protein, and was shown to be
essential to autophagy. Beclin-1 induces autophagy by interacting
with certain cofactors to activate the
phosphatidylinositol-3-kinase Vps34 (20). LC3 are essential proteins that
regulate the autophagosomal membrane. Under normal conditions, the
majority of LC3 proteins present in the cytosol are in the LC3I
form. Upon autophagy induction, the cytosolic LC3I form is
conjugated with phosphatidylethanolamine and becomes LC3II, which
forms a stable association with the autophagosomal membrane
(21). p62 (also known as
sequestosome 1) serves as an association between LC3 and
ubiquitinated substrates to facilitate autophagic clearance. p62
decreases when autophagy is induced, and accumulates when autophagy
is inhibited. Therefore, p62 is used as a readout of autophagic
degradation and a marker of autophagy flux (22,23).
In the present study, an increase in double-membrane autophagosomes
by TEM and LC3 puncta was observed by fluorescence microscopy in
HUVECs treated with LSS. Monitoring the levels of autophagic
proteins revealed an increase in Beclin-1 and LC3II, but an
opposite trend in p62 in the LSS-treated HUVECs. Additionally, the
level of p62 was downregulated by rapamycin by upregulating the
protein level of LC3II. CQ, an inhibitor of autophagy, was able to
disrupt the fusion of autophagosomes with lysosomes and suppress
the activity of lysosomal acid hydrolases as a weak base, thereby
blocking the degradation of autolysosome and accumulating LC3II at
a late stage, exhibiting anti-autophagic characteristics (24). Therefore, CQ up-regulated the level
of LC3II and p62 by blocking autophagy. This observation indicated
that LSS induced autophagy flux, which in turn proved that an
autophagy process was activated by pro-atherogenic LSS.
Although Bcl-2 family proteins were initially
characterized as cell death regulators, it has recently become
clear that they also control autophagy. A study indicated that
autophagy induction correlated with the dissociation of Bcelin-1
from Bcl-2 (25). In normal,
Beclin-1 is bound to Bcl-2 through interaction involving Bcl-2
homology 3 (BH3) domain in Beclin-1 and the BH3 binding groove of
Bcl-2 (20). Phosphorylation of
Bcl-2 can lead to Bcl-2 separating from Beclin-1, thereby
alleviating the inhibitory effect on Beclin-1 (25). In the present study, we observed
that the LSS treatment induced HUVEC autophagy with decreased Bcl-2
levels and increased Beclin-1 levels, indicating that LSS is able
to alter the balance between Bcl-2 and Beclin-1 which may be the
mechanism of autophagy induced by LSS.
Although the upregulation of autophagy has been
observed in HUVECs treated with LSS in the present study, it is
unclear whether the autophagy is protective or detrimental. The
cross talk between the autophagic and apoptotic cell death pathways
is complex (26). The theory that
autophagy is initiated as a protective response has become accepted
(27). To investigate the effect
of autophagy on the LSS-induced apoptosis in HUVECs, rapamycin, a
mammalian target of rapamycin (mTOR) inhibitor, was used to induce
autophagy. The results demonstrated that rapamycin upregulated the
level of autophagy and downregulated the apoptosis rate. Similarly,
previous studies demonstrated that rapamycin was able to reduce
tert-butyl hydroperoxide-induced apoptosis (28) and mechanical stress-induced
endothelial apoptosis (29).
Furthermore, rapamycin upregulated the level of Bcl-2, but failed
to inhibit the expression of Beclin-1. This may be due to the fact
that inhibition of mTOR promotes Beclin-1 expression and prevents
the decreasing of Beclin-1 (30).
To further investigate the association between autophagy and
apoptosis induced by LSS, CQ was used to inhibit autophagy and
investigate changes in apoptotic cell death. In the present study,
with the decreased level of autophagy induced in CQ, the rate of
apoptosis in the HUVECs treated with LSS increased significantly.
These results are consistent with the data demonstrating that an
increased level of autophagy protects HUVECs from LSS-induced
apoptosis, whereas a deceased level of autophagy led to increased
apoptosis in HUVECs, which suggests that LSS-induced apoptosis is
regulated by autophagy. Autophagy serves a protective role in
LSS-induced apoptosis.
The results of the present study suggest that LSS
was able to induce autophagy through the modulation of
Bcl-2/Beclin-1 in HUVECs. Furthermore, it was observed that the
cross talk between autophagy and apoptosis contributes to the
autophagic protection of HUVECs from LSS-induced apoptosis.
Although these results represent an advancement in the
understanding of the association between LSS-induced autophagy and
apoptosis, additional work is necessary to further characterize the
protective effect of autophagy in the progression of
atherosclerosis induced by LSS.
Acknowledgments
The authors would like to thank Dr Chunlai Wang
(Harbin Veterinary Research Institute, Harbin, China) and Professor
Zuyan Liu (Harbin Institute of Technology, Harbin, China) for their
technical assistance. The present study was supported by the
Natural Science Foundation of Heilongjiang Province Project (grant
no. H201345) and by the Postdoctoral Foundation of Heilongjiang
Province Project (grant no. LBH-Z11062).
References
|
1
|
Farmakis TM, Soulis JV, Giannoglou GD,
Zioupos GJ and Louridas GE: Wall shear stress gradient topography
in the normal left coronary arterial tree: Possible implications
for atherogenesis. Curr Med Res Opin. 20:587–596. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davies PF, Polacek DC, Shi C and Helmke
BP: The convergence of haemodynamics, genomics, and endothelial
structure in studies of the focal origin of atherosclerosis.
Biorheology. 39:299–306. 2002.PubMed/NCBI
|
|
3
|
Kinlay S and Ganz P: Role of endothelial
dysfunction in coronary artery disease and implications for
therapy. Am J Cardiol. 80:11I–16I. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stone PH, Saito S, Takahashi S, Makita Y,
Nakamura S, Kawasaki T, Takahashi A, Katsuki T, Nakamura S, Namiki
A, et al: Prediction of progression of coronary artery disease and
clinical outcomes using vascular profiling of endothelial shear
stress and arterial plaque characteristics: The PREDICTION study.
Circulation. 126:172–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cunningham KS and Gotlieb AI: The role of
shear stress in the pathogenesis of atherosclerosis. Lab Invest.
85:9–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis: A cell survival and death phenomenon with
therapeutic potential. Circ Res. 104:304–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schrijvers DM, De Meyer GR and Martinet W:
Autophagy in atherosclerosis: A potential drug target for plaque
stabilization. Arterioscler Thromb Vasc Biol. 31:2787–2791. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levesque MJ and Nerem RM: The elongation
and orientation of cultured endothelial cells in response to shear
stress. J Biomech Eng. 107:341–347. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu W, Gu K, Yu Z, Yuan D, He M, Ma N, Lai
S, Zhao J, Ren Z, Zhang X, et al: Sorafenib potentiates irradiation
effect in hepatocellular carcinoma in vitro and in vivo. Cancer
Lett. 329:109–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gimbrone MA Jr, Topper JN, Nagel T,
Anderson KR and Garcia-Cardeña G: Endothelial dysfunction,
hemodynamic forces, and atherogenesis. Ann N Y Acad Sci.
902:230–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, Wang Z, Zhang J, Zuo G, Li B, Mao
W and Chen S: Rapamycin attenuates endothelial apoptosis induced by
low shear stress via mTOR and sestrin1 related redox regulation.
Mediators Inflamm. 2014:7696082014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin HC and Lai IR: Isolated mitochondria
infusion mitigates ischemia-reperfusion injury of the liver in
rats: Reply. Shock. 39:5432013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Korsmeyer SJ: BCL-2 gene family and the
regulation of programmed cell death. Cancer Res. 59:(7 Suppl).
1693s–1700s. 1999.PubMed/NCBI
|
|
16
|
Nowicki M, Zabirnyk O, Duerrschmidt N,
Borlak J and Spanel-Borowski K: No upregulation of lectin-like
oxidized low-density lipoprotein receptor-1 in serum-deprived
EA.hy926 endothelial cells under oxLDL exposure, but increase in
autophagy. Eur J Cell Biol. 86:605–616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Khan MJ, Alam Rizwan M, Waldeck-Weiermair
M, Karsten F, Groschner L, Riederer M, Hallström S, Rockenfeller P,
Konya V, Heinemann A, et al: Inhibition of autophagy rescues
palmitic acid-induced necroptosis of endothelial cells. J Biol
Chem. 287:21110–21120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie Y, You SJ, Zhang YL, Han Q, Cao YJ, Xu
XS, Yang YP, Li J and Liu CF: Protective role of autophagy in
AGE-induced early injury of human vascular endothelial cells. Mol
Med Rep. 4:459–464. 2011.PubMed/NCBI
|
|
19
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar :
|
|
21
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan WJ, Dong HL and Xiong LZ: The
protective roles of autophagy in ischemic preconditioning. Acta
Pharmacol Sin. 34:636–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shin YJ, Cho DY, Chung TY, Han SB, Hyon JY
and Wee WR: Rapamycin reduces reactive oxygen species in cultured
human corneal endothelial cells. Curr Eye Res. 36:1116–1122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Raaz U, Kuhn H, Wirtz H and Hammerschmidt
S: Rapamycin reduces high-amplitude, mechanical stretch-induced
apoptosis in pulmonary microvascular endothelial cells. Microvasc
Res. 77:297–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang T, Li D, Liu F, Qi L, Yan G and Wang
M: Regulation on Beclin-1 expression by mTOR in CoCl2-induced HT22
cell ischemia-reperfusion injury. Brain Res. 1614:60–66. 2015.
View Article : Google Scholar : PubMed/NCBI
|