Introduction
Corneal dystrophies (CDs) are a group of
heterogeneous inherited diseases with non-inflammatory, bilaterally
progressive corneal opacities, which often lead to recurrent
corneal erosion and visual impairment. According to the affected
layer in the cornea, CDs can be categorized as epithelial,
subepithelial, Bowman's layer, stromal or endothelial using
slit-lamp imaging, confocal microscopy or histological staining
However, it has been found that the clinical classifications of CDs
based entirely on ophthalmological and histopathologic examinations
are limited in scope and effectiveness (1).
Advances in molecular genetics have mapped CDs to
eleven chromosomes: 1, 2, 5, 9, 10, 12, 13, 16, 17, 20 and X
(2). Several genes, including the
transforming growth factor β-induced (TGFBI) gene (OMIM
601692), carbohydrate sulfotransferase 6 gene, gelsolin gene,
keratin 3 gene, keratin 12 gene and surface marker 1 gene, have so
far been identified as responsible for CDs (3,4).
Among these, TGFBI-associated CDs have provided the most
reliable evidence. To date, at least five autosomal dominant CDs
have been confirmed to be associated with mutations in the
TFGBI gene: i) Reis-Bücklers CD (RBCD), ii) lattice CD type
1 (LCDI), iii) Thiel-Behnke CD, iv) granular CD type 1 (GCD1) and
v) GCD2 (5,6).
As CD has significant genetic heterogeneity,
different TGFBI gene mutations can lead to the same
phenotype, for example, R124C, V625D, V505D, H626R and other
mutations are associated with LCDI (7–9);
whereas the same TGFBI mutation in a different ethnic
population can exhibit different clinical phenotypes, for example,
the R124C mutation is responsible for GCD2, LCDI and RBCD (10–12).
Due to the complexity of phenotypic and genetic heterogeneity, the
ability of ophthalmologists and geneticists to correctly diagnose
and classify CDs is a challenge, as is understanding their
phenotype-genotype aspects. In the present study, three Chinese
families affected by different types of CD were recruited, and the
gene mutations responsible for the disease were identified. The
results aimed to provide an insight into the clinical-molecular
correlations of the disease.
Materials and methods
Patients and subjects
In the present study, three Chinese pedigrees
comprising individuals diagnosed with LCDI, LCD IIIB and GCD2,
respectively, were recruited from the Tianjin Eye Hospital
(Tianjin, China) between August 2013 and July 2014. The study was
approved by the Ethics Committee of Tianjin Eye Hospital. Informed,
written consent was obtained from each of the participants in
accordance with the Declaration of Helsinki prior to the collection
of their peripheral blood. The participating members underwent
careful ophthalmic examination, including vision tests, slit-lamp
biomicro-examination and anterior segment imaging. In addition, 100
unrelated subjects without CD were recruited from the Tianjin Eye
Hospital as normal controls for the present study.
Genetic analysis
Genomic DNA was extracted from the peripheral blood
samples using a genomic DNA miniprep kit for blood (Roche
Diagnostics, Indianapolis, IN, USA). DNA integrity was evaluated
using 1% agarose gel electrophoresis. The DNA fragments, which
encoded regions of the TGFBI gene, were amplified using
polymerase chain reaction (PCR). The sequences of the primers were
designed according to a previous study by Stix et al
(13) (Table I). Standard PCR reactions were
preformed in a 50 µl reaction mixture including 1 µl of 10X PCR
buffer, 50 ng template DNA, 0.1 µM of each of the forward and
reverse primers, 300 µM dNTP, 5 mM MgCl2 and 0.3U
Hotstart Taq. The PCR program used for DNA amplification was as
follows: 95°C for 3 min; followed by 20 cycles at 95°C for 30 sec,
53–64°C for 30 sec, and 72°C for 45 sec; additional 20 cycles at
95°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec; and a final
extension at 72°C for 7 min, using the Gene Amp PCR 9700 system
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA).
 | Table I.Primers for amplification of
transforming growth factor β1 regions. |
Table I.
Primers for amplification of
transforming growth factor β1 regions.
|
| Sequence (5′-3′) |
|---|
|
|
|
|---|
| Exon | Forward | Reverse |
|---|
| 1 | TC TCA CTT CCC TGG
AG | GAC TAC CTG ACC TTC
CGC AG |
| 2 | GGT GGA CGT GCT GAT
CAT CT | AGC CAG CGT GCA TAC
AGC TT |
| 3 | TTC ACC CAC CAT TCC
TCT TC | GGT ACT CCT CTC TCC
CAC CA |
| 4 | ATC CCT CCT TCT GCT
TTC TG | GCA GAC GGA GGT CAT
CTC AC |
| 5 | TTA AAC ACA GAG TCT
GCA GC | TTC ATT ATG CAC CAA
GGG CCA |
| 6 | TGT TGA CTG CTC ATC
CTT GC | CTC TTG GGA GGC AAT
GTG TC |
| 7 | CTT CAG GGA GCA CTC
CAT C | AAT CTA GCT GCA CAA
ATG AGG |
| 8 | CTT GAC CTG AGT CTG
TTT GGA | GGA TGG CAG AAG AGA
TGG TG |
| 9 | CCT GCT GAT GTG TGT
CAT GC | CTG CCT CCA GGG ACA
ATC TA |
| 10 | TCA TTG CAG GAG CAC
ATC TC | CCC AGG AGC ATG ATT
TAG GA |
| 11 | GAG GCC CCT CGT GGA
AGT A | ACA TCC CAC TCC AGC
ATG AC |
| 12 | CTG TTG ACA GGT GAC
ATT TTC | ATG TGC CAA CTG TTT
GCT GCT |
| 13 | GGG ATT AAC TCT ATC
TCC TT | TGT GTA TAA TTC CAT
CCT GG |
| 14 | TCA GTA AAC ACT TGC
TGA GTG AA | ACT GCC ACA TGG AGA
AAA GGA C |
| 15 | CCT CAG TCA CGG TTG
TTA TG | CTC TAT GGC CCA AAC
AGA GG |
| 16 | TTG TCA TAA GCA GTT
GCA GG | GCT TGC TTG GGG GTA
AGG |
| 17 | TCC TAG ACA GAC ATG
GGG AGA | TGA GAG AAA TTG GCG
GAG AG |
Each PCR fragment was purified with a TIANgel Midi
DNA Purification kit (Tiangen Biltech Co., Ltd, Beijing, China),
and the two strands were subsequently sequenced on an ABI 3130
automated DNA sequencer (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The sequence results were compared with the
wild-type TGFBI sequence (GenBank NG_012646.1; https://www.ncbi.nlm.nih.gov/nuccore/NG_012646.1).
The other family members and 100 Chinese control individuals were
also screened for the presence of TGFBI mutations by PCR
amplification and direct sequencing of PCR products.
Histopathological examination
The corneal specimens obtained using penetrating
keratoplasty were processed for examination using light microscopy.
The tissues were fixed in 10% formalin and embedded in paraffin.
Cross sections of each button were prepared by free-hand sectioning
at a thickness of ~10 mm and were stained with Congo red.
Results
Family with LCDI
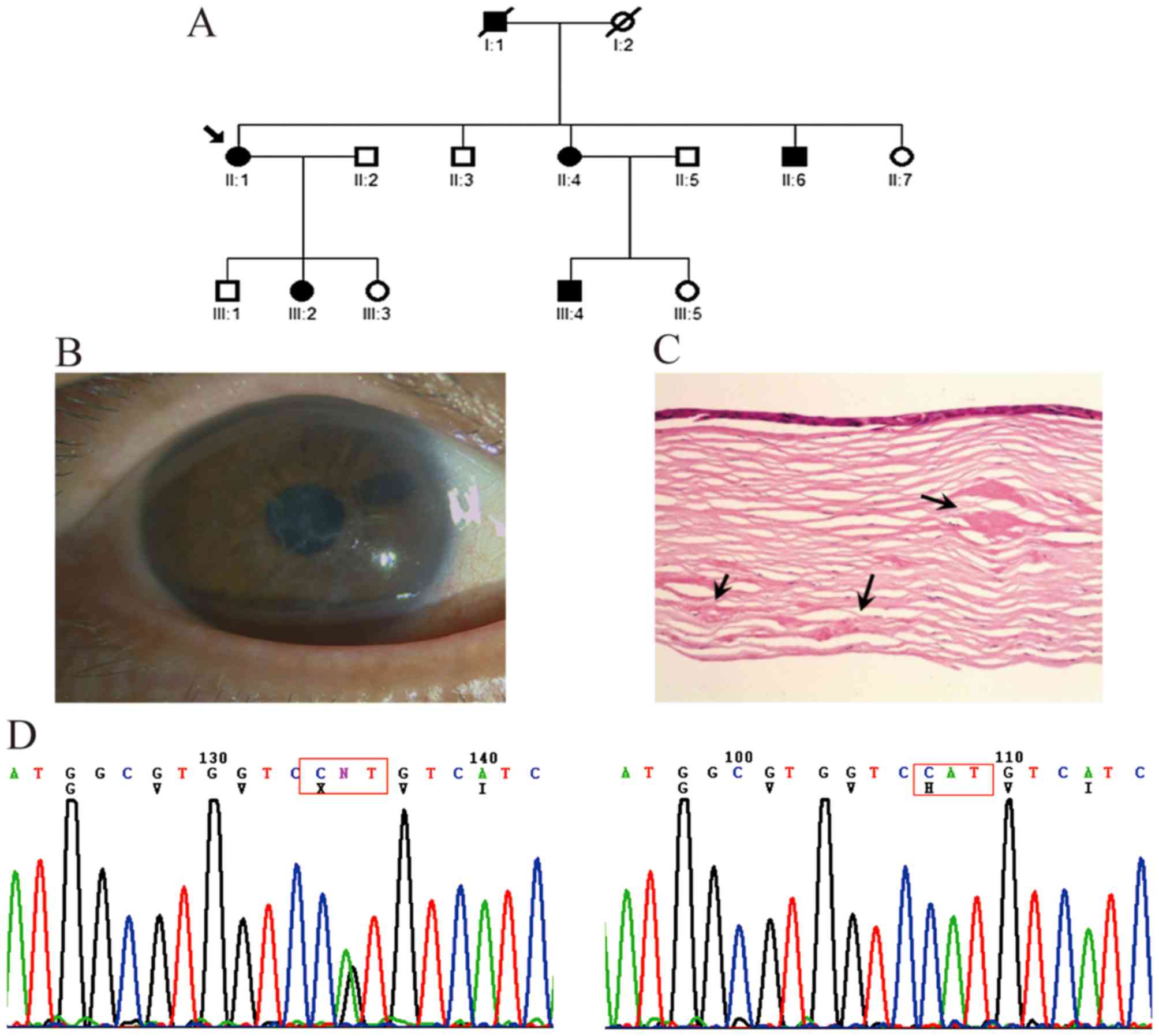
In the first family, comprising three generations,
four affected members and five unaffected members were examined
(Fig. 1A). The symptoms of all
patients began with episodes of acute ocular pain, redness and
photophobia at ~20 years of age. Slit-lamp examination revealed
dots and fine lattice lines in the anterior corneal stroma
(Fig. 1B). The small
lattice-shaped opacity is a typical clinical feature LCDI, which
can be observed using direct and retroillumination. The mutation
analysis showed that a heterozygous c.371 C>T mutation (R124C)
in exon 4 of TGFBI was present in the affected members, but
not in the unaffected members or controls (Fig. 1C).
Family with LCDIIIB
In the second three-generation family, six affected
members and eight unaffected members were examined (Fig. 2A). Thick and ropy lattice lines of
amyloid were visible under direct illumination of the corneas of
the proband patient II-1 (Fig.
2B). This patient was referred to hospital with visual acuity
loss to 0.2 and underwent perforating keratoplasty on her right eye
at 56 years old. The corneal specimens obtained during surgery were
submitted for histologic examination in the present study, which
revealed large patchy amyloid deposits in the subepithelial layer
and deeper corneal stroma (Fig.
2C). The clinical symptoms of the other affected family members
were similar to those of the proband. Mutation analysis showed that
a heterozygous c.1877 A>G mutation (H626R) in exon 14 of
TGFBI was present in the proband and other affected members,
but not in the unaffected members or 100 controls (Fig. 2D).
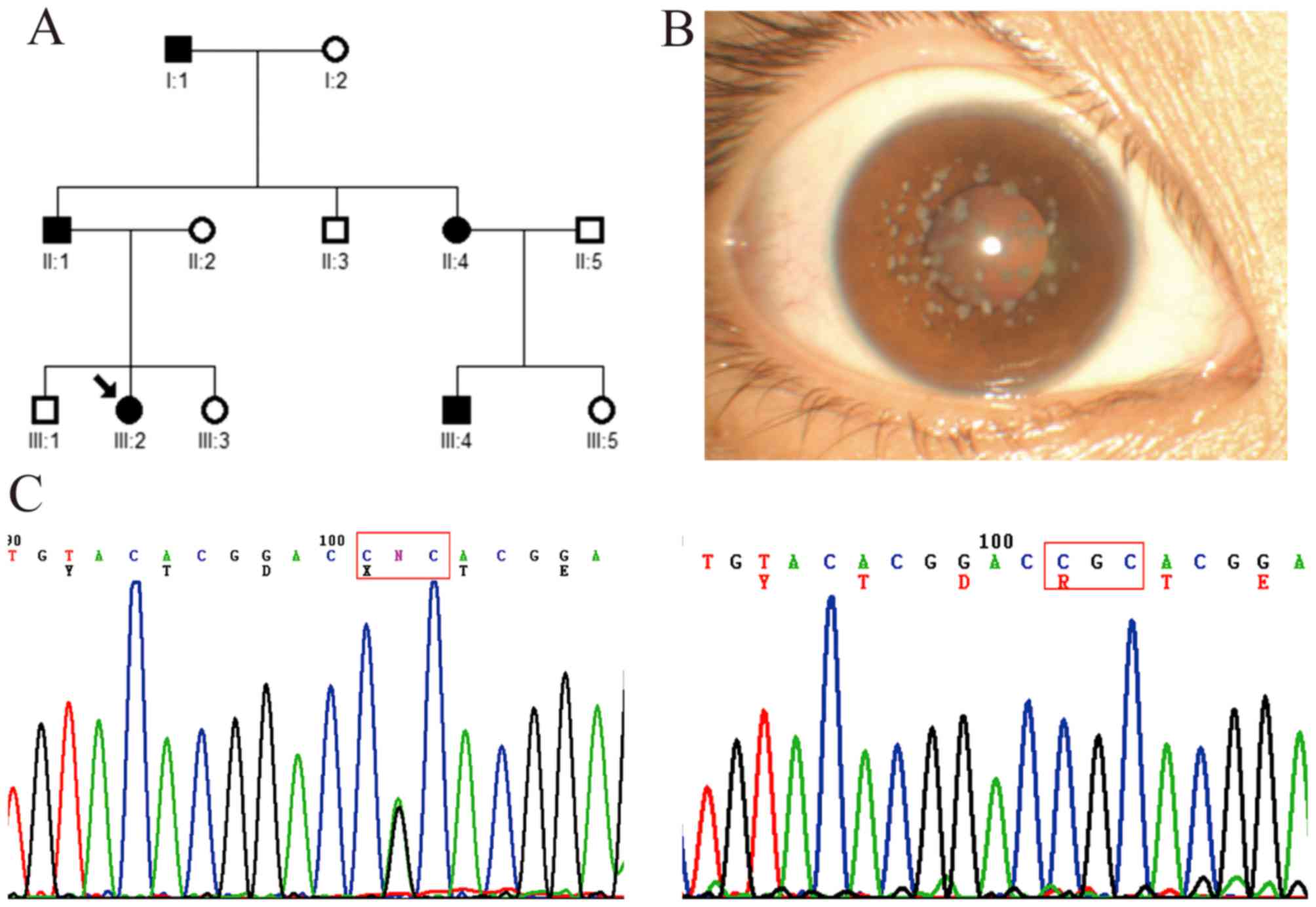
Family with GCD2
In this three-generation family, five affected
patients and seven unaffected members were examined (Fig. 3A). The proband (a 22-year-old
female patient; III:2) had recurrent corneal epithelial erosion
with visual impairment, and symptoms of redness, photophobia,
lacrimation and foreign-body sensation in both eyes. The visual
acuity recovered to 1.0 when corneal epithelial erosion
disappeared. Slit-lamp examination showed bilateral scattered
grayish small dot, annular and snow flake-like opacities in the
superficial mid-stroma of the central cornea (Fig. 3B). Mutation analysis showed that a
heterozygous c.371 G>A mutation (R124H) in exon 4 of
TGFBI was identified in the proband and other affected
members, but not in the unaffected members and healthy controls
(Fig. 3C).
Discussion
The TGFBI gene (also known as BIGH3)
was first identified by Skonier et al in 1992 (14), and has been closely associated with
inherited CDs. In the present study, mutations of TGFBI were
identified in three unrelated Chinese families affected with LCDI,
LCDIIIB and GCD2, respectively. Mutations were identified in all of
the families, and there were perfect correlations between LCDI and
R124C, between LCDIIIB and H626R, and between GCD2 and R124H.
LCD is generally divided into three subtypes, and
classification has been assigned according to clinical and
pathological findings. LCDI is an autosomal dominantly inherited
corneal amyloidosis, which is characterized by a network of
delicate linear opacities within the superficial corneal stroma.
The disease usually begins in the first decade of life with
symptoms of recurrent painful epithelial erosions. Lattice lines
and diffuse opacification of the central cornea develop gradually
subsequent to the formation of amyloid accumulations or erosions.
Several studies from different ethnic groups have demonstrated an
R124C mutation of the TGFBI gene as the most common cause of
LCDI (11,15,16).
In the present study, the clinical features of the affected members
of the family with LCDI were similar, however, Liu et al
(17) reported that the clinical
features of Chinese patients with the same R124C mutation were
variable, even within the same family. Therefore, the mechanism
underlying phenotypic variability with the same mutation and within
the same family remains unclear and requires further
investigation.
The LCDIIIB subtype is characterized by bilateral
progressive visual impairment and has an intermediate age of onset,
compared with LCDI and LCDIIIA. The LCDIIIB subtype was first
described in England (16), and
was named by Chau et al (18). In 2010, Yang et al
identified this mutation in a population in North China, which had
clinical features of an intermediate subtype between LCDI and IIIA
(19). In 2013, Wang et al
first reported the TGFBI p.H626R mutation in a pedigree from
South Chinese with LCDIIIB (20).
All these findings suggested that TGFBI p.H626R may be a mutation
hotspot across different populations in LCDIIIB. Similar to the
previous studies, the present study found the p.H626R mutation in
the family affected by LCDIIIB.
The onset of GCD2, previously termed avellino
dystrophy, often occurs in the second decade and demonstrates fewer
opacities, compared with GCD1, exhibiting granular, branching
deposits in a clear superficial mid-stroma, with lattice lines
sometimes in deeper corneas (21).
This disease has clinical and histologic features of granular and
lattice dystrophy. To date, the molecular genetic techniques used
for the majority of cases of GCD2 have revealed the R124H mutation
in TGFBI, however, the phenotype of the affected individuals
vary markedly in severity between families (22). Direct sequencing in the present
study revealed that the c.371 G>A mutation of TGFBI
resulting in Arg124His was responsible for GCD2 in the pedigree,
which was consistent with the results of a previous study (21). These results further support the
importance of TGFBI in maintaining corneal transparency.
In conclusion, although no novel mutations were
found in the three pedigrees with CD in the present study, the
previously reported phenotype-genotype correlations were confirmed
in all patients with TGFBI-linked CDs. As the mutation at
TGFBI can be readily, rapidly and cost-effectively detected
via PCR sequencing or using PCR-RFLP, the identification of these
mutations enables Chinese patients to benefit from the timely and
accurate molecular diagnosis of CDs.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 30940081) and the
Xuzhou Science and Technology Foundation (grant no. XF10C050).
References
|
1
|
Weiss JS: Molecular genetics and the
classification of the corneal dystrophies: What is next? Am J
Ophthalmol. 148:477–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weiss JS, Møller HU, Lisch W, Kinoshita S,
Aldave AJ, Belin MW, Kivelä T, Busin M, Munier FL, Seitz B, et al:
The IC3D classification of the corneal dystrophies. Cornea.
27:(Suppl 2). S1–S83. 2008.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klintworth GK: Corneal dystrophies.
Orphanet J Rare Dis. 4:72009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klintworth GK: The molecular genetics of
the corneal dystrophies-current status. Front Biosci. 8:d687–d713.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aldave AJ and Sonmez B: Elucidating the
molecular genetic basis of the corneal dystrophies: Are we there
yet? Arch Ophthalmol. 125:177–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Munier FL, Korvatska E, Djemaï A, Le
Paslier D, Zografos L, Pescia G and Schorderet DF: Kerato-epithelin
mutations in four 5q31-linked corneal dystrophies. Nat Genet.
15:247–251. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho KJ, Mok JW, Na KS, Rho CR, Byun YS,
Hwang HS, Hwang KY and Joo CK: TGFBI gene mutations in a Korean
population with corneal dystrophy. Mol Vis. 18:2012–2021.
2012.PubMed/NCBI
|
|
8
|
Tian X, Fujiki K, Zhang Y, Murakami A, Li
Q, Kanai A, Wang W, Hao Y and Ma Z: A novel variant lattice corneal
dystrophy caused by association of mutation (V625D) in TGFBI gene.
Am J Ophthalmol. 144:473–475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tian X, Fujiki K, Wang W, Murakami A, Xie
P, Kanai A and Liu Z: Novel mutation (V505D) of the TGFBI gene
found in a Chinese family with lattice corneal dystrophy, type I.
Jpn J Ophthalmol. 49:84–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang QN, Zhao YW, Guo LH, Yan NH, Liu XY
and Cai SP: Arg124Cys mutation of the TGFBI gene in a Chinese
pedigree of Reis-Bücklers corneal dystrophy. Int J Ophthalmol.
4:235–238. 2011.PubMed/NCBI
|
|
11
|
Patel DA, Chang SH, Harocopos GJ, Vora SC,
Thang DH and Huang AJ: Granular and lattice deposits in corneal
dystrophy caused by R124C mutation of TGFBIp. Cornea. 29:1215–1222.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Munier FL, Frueh BE, Othenin-Girard P,
Uffer S, Cousin P, Wang MX, Héon E, Black GC, Blasi MA, Balestrazzi
E, et al: BIGH3 mutation spectrum in corneal dystrophies. Invest
Ophthalmol Vis Sci. 43:949–954. 2002.PubMed/NCBI
|
|
13
|
Stix B, Leber M, Bingemer P, Gross C,
Rüschoff J, Fändrich M, Schorderet DF, Vorwerk CK, Zacharias M,
Roessner A and Röcken C: Hereditary lattice corneal dystrophy is
associated with corneal amyloid deposits enclosing C-terminal
fragments of keratoepithelin. Invest Ophthalmol Vis Sci.
46:1133–1139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Skonier J, Neubauer M, Madisen L, Bennett
K, Plowman GD and Purchio AF: cDNA cloning and sequence analysis of
beta ig-h3, a novel gene induced in a human adenocarcinoma cell
line after treatment with transforming growth factor-beta. DNA Cell
Biol. 11:511–522. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim HS, Yoon SK, Cho BJ, Kim EK and Joo
CK: BIGH3 gene mutations and rapid detection in Korean patients
with corneal dystrophy. Cornea. 20:844–849. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stewart H, Black GC, Donnai D, Bonshek RE,
McCarthy J, Morgan S, Dixon MJ and Ridgway AA: A mutation within
exon 14 of the TGFBI (BIGH3) gene on chromosome 5q31 causes an
asymmetric, late-onset form of lattice corneal dystrophy.
Ophthalmology. 106:964–970. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Z, Wang YQ, Gong QH and Xie LX: An
R124C mutation in TGFBI caused lattice corneal dystrophy type I
with a variable phenotype in three Chinese families. Mol Vis.
14:1234–1239. 2008.PubMed/NCBI
|
|
18
|
Chau HM, Ha NT, Cung LX, Thanh TK, Fujiki
K, Murakami A and Kanai A: H626R and R124C mutations of the TGFBI
(BIGH3) gene caused lattice corneal dystrophy in Vietnamese people.
Br J Ophthalmol. 87:686–689. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang J, Han X, Huang D, Yu L, Zhu Y, Tong
Y, Zhu B, Li C, Weng M and Ma X: Analysis of TGFBI gene mutations
in Chinese patients with corneal dystrophies and review of the
literature. Mol Vis. 16:1186–1193. 2010.PubMed/NCBI
|
|
20
|
Wang D, Yao Y, Zhang M and Chen J: Genetic
and phenotypic investigation of a Chinese pedigree with lattice
corneal dystrophy IIIB subtype. Eye Sci. 28:144–147.
2013.PubMed/NCBI
|
|
21
|
Gu Z, Zhao P, He G, Wan C, Ma G, Yu L,
Zhang J, Feng G, He L and Gao L: An Arg124His mutation in TGFBI
associated to Avellino corneal dystrophy in a Chinese pedigree. Mol
Vis. 17:3200–3207. 2011.PubMed/NCBI
|
|
22
|
Abazi Z, Magarasevic L, Grubisa I and
Risovic D: Individual phenotypic variances in a family with
Avellino corneal dystrophy. BMC Ophthalmol. 13:302013. View Article : Google Scholar : PubMed/NCBI
|