Introduction
Puerarin (Pur), which is the major bioactive
ingredient extracted from the root of Pueraria lobata
(Willd.) Ohwi, has been widely used in the treatment of
cardiovascular diseases, cerebrovascular disorders and diabetes in
China (1). Pur has been
demonstrated to exert the following protective effects against
myocardial ischemia/reperfusion (I/R) injury: Amelioration of
oxygen consumption, restriction of the infarct area and improvement
of diastolic function (2,3). Previous studies have further
demonstrated that these protective effects may be associated with
the inhibition of mitochondrial permeability transition pore
opening, activation of the mitochondrial adenosine triphosphate
(ATP)-sensitive potassium channel, opening of the calcium-activated
potassium channel and activation of protein kinase C (4,5).
Although numerous studies have focused on the pharmacological
properties of Pur, the mechanisms underlying the protective effects
of Pur against myocardial I/R injury remain to be fully
elucidated.
The mechanisms underlying myocardial I/R injury have
traditionally been considered to include increased levels of
reactive oxygen species, calcium overload and aberrant ATP
production (6). However, previous
research has also revealed that autophagy is involved in the
mechanisms of myocardial I/R injury (7). Autophagy, by which mammalian cells
degrade and recycle macromolecules and organelles, is vital for
intracellular homeostasis at the basic level. It is activated to
relieve adverse effects under various stress conditions, including
nutritional deprivation, oxidative stress and ischemic injury
(8). Previous studies have
supported the suggestion that enhanced autophagy exerts protective
effects during myocardial ischemia via protein clearance and
restoration of cellular ATP levels (9,10).
However, during the reperfusion phase, increased autophagy in
cardiomyocytes becomes a maladaptive process, which causes further
myocardial damage and cell death (7,11).
Therefore, the manipulation of autophagy may be considered a novel
method to relieve myocardial I/R injury.
The phosphatidylinositol-3-kinase/protein kinase B
(PI3K/Akt) signaling pathway is critical for cell survival under
stress conditions. Akt has previously been reported to be activated
by Pur in various models, including cerebral I/R (12,13).
Furthermore, by activating Akt, autophagy can be negatively
controlled (14). Given the
important role of autophagy in myocardial I/R, the present study
aimed to investigate whether Pur protects against myocardial I/R
injury by inhibiting autophagy via the PI3K/Akt signaling pathway.
The results of the present study provide further understanding of
the protective mechanisms of Pur and the involvement of autophagy
in myocardial I/R injury.
Materials and methods
Reagents
Pur (Fig. 1) was
purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan).
Rapamycin (Ra), 3-methyladenine (3-MA), Akt signaling inhibitor
(API-2) and 5-bromo-2′-deoxyuridine (5-BrdU) were purchased from
Sigma-Aldrich Merck KGaA (Darmstadt, Germany).
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)
−2H-tetrazolium inner salt (MTS) was purchased from Promega
Corporation (Madison, WI, USA). Dulbecco's modified Eagle's medium
(DMEM) and fetal bovine serum (FBS) were purchased from Thermo
Fisher Scientific, Inc. (Beijing, China). All antibodies for
immunoblotting were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA), with the exception of the GAPDH antibody
(KangChen Bio-tech, Inc., Shanghai, China) and goat anti-rabbit
secondary antibody (Boster Systems, Inc., Pleasanton, CA, USA).
Adenovirus-monomeric red fluorescent protein-green fluorescent
protein-light chain 3 (Ad-mRFP-GFP-LC3) was purchased from HanBio
Biotechnology Co., Ltd. (Shanghai, China). Neonatal Sprague Dawley
rats (~220; 1–2 days; 5–6 g) were obtained from the Experimental
Animal Center of Southern Medical University (Guangzhou, China;
license no. scxk-Guangdong-2006-0015) where they had been housed
with female rats under standard conditions with a light/dark cycle
of 12 h and fed by female rats.
Cell isolation and culture
Neonatal rat cardiomyocytes (NRCs) were isolated
from the ventricular heart of 1–2 day old Sprague Dawley rats as
previously described (15),
following which the animals were immediately sacrificed by
decapitation. The isolation procedure was performed in accordance
with the Guide for the Care and Use of Laboratory Animals,
published by the US National Institutes of Health (publication no.
85-23, revised 1996) and was approved by the Southern Medical
University Experimental Animal Ethics Committee (Guangzhou, China).
NRCs were plated at a density of 5×105 cells/60 mm
diameter plate, and cultured in DMEM supplemented with 10% FBS and
1% penicillin/streptomycin (the complete medium) in a 5%
CO2 humidified atmosphere at 37°C. 5-BrdU (0.1 mM) was
added to the culture medium to inhibit fibroblast proliferation
during the first 48 h. The cells were subsequently cultured with
the complete medium for a further 48 h. The culture medium was
changed daily.
Experimental treatment
Following culture for 96 h, NRCs were randomly
divided into the following groups: The control group, in which
cells were incubated under normal conditions; the
hypoxia/reoxygenation (H/R) group, in which cells were exposed to 3
h hypoxia followed by 3 h reoxygenation; the H/R + Pur group, in
which cells were pretreated with Pur and were then exposed to H/R;
H/R + 3-MA/Ra/API-2 group, in which cells were pretreated with
3-MA, Ra or API-2 and were then exposed to H/R; and the H/R + Pur +
3-MA/Ra/API-2 group, in which cells were pretreated with Pur plus
3-MA, Ra or API-2 and were then exposed to H/R. Pur (50, 100 or 200
µM) (16), 3-MA (5 mM), Ra (0.1
µM) and API-2 (10 µM) were added to the culture medium 2 h prior to
H/R.
Model of myocardial H/R
The myocardial H/R model was used to simulate I/R
injury. For experiments under hypoxic conditions, NRCs were
incubated with phosphate-buffered saline (PBS) in a Modular Hypoxic
Chamber (Billups-Rothenberg, Inc., Del Mar, CA, USA) saturated with
<0.1% O2, 95% N2 and 5% CO2 at
37°C for 3 h. Cells were subsequently subjected to reoxygenation by
replacing PBS with the culture medium, and were returned to
normoxic conditions for 3 h (17).
Cell viability
To assess cell viability, the MTS assay was used
according to the manufacturer's protocol. NRCs were seeded in
96-well plates at a density of 5,000 cells/well. Following the
indicated treatments, 20 µl MTS solution was added to each well,
and plates were subsequently incubated at 37°C for 3 h. The optical
density in each well was measured at 490 nm using a microplate
reader (ELx800; BioTek Instruments, Inc., Winooski, VT, USA).
Western blot analysis
Western blotting was performed to determine
LC3-II/LC3-I, p62, phosphorylated (p-)Akt (Ser473), Akt, P-5′
adenosine monophosphate-activated protein kinase (p-AMPK) (Thr172),
AMPK and GAPDH protein expression levels. Following the indicated
treatments, cells were lysed using a total protein extraction kit
(KangChen Bio-tech, Inc.) and a protease inhibitor cocktail (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Lysates were
subsequently centrifuged at 18,506 × g at 4°C for 15 min.
Protein concentration was determined using an enhanced
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). The loading buffer was added and the protein samples were
boiled in a water bath for 10 min. Western blotting was performed
according to standard procedures. Equal amounts of protein were
resolved via SDS/PAGE (10 or 12%) and transferred to PVDF membranes
(Merck KGaA, Darmstadt, Germany). After blocking in 5% non-fat milk
at room temperature for 2 h, membranes were incubated with the
following primary antibodies overnight at 4°C: anti-LC3B (3868S),
anti-p62 (8025S), anti-p-Akt (4060S), anti-Akt (4691P), anti-p-AMPK
(2531S), anti-AMPK (2532S; all from Cell Signaling Technology,
Inc.) and anti-GAPDH (KC-5G4; KangChen Bio-tech, Inc.). All
dilutions were 1:1,000. After incubation for 2 h at room
temperature with goat anti-rabbit secondary antibodies (1:8,000;
BA1054; Boster Systems, Inc.), the bands were visualized using
enhanced chemiluminescence (Merck KGaA). The blots were quantified
by densitometry using Image-Pro Plus software version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA) and the relative protein
expression was compared with GAPDH.
Transmission electron microscopy
(TEM)
Following the indicated treatments, cells were
washed, scraped, collected in a 1.5 ml tube and centrifuged at 106
× g at 4°C for 5 min. The cell pellets were subsequently
fixed with 2.5% glutaraldehyde at 4°C overnight. Following
fixation, cell pellets were dehydrated with a graded series of
ethanol (50, 70, 90 and 100%), propylene oxide and then infiltrated
with a 1:1 mixture of propylene oxide and EMbed 812. The samples
were sliced into ultrathin sections (75–80 nm), which were stained
and examined using a transmission electron microscope (JEM1200-EX;
JEOL Ltd., Tokyo, Japan). In the samples, vesicles containing
membranous structures (autophagosomes) in each cell were
counted.
Ad-mRFP-GFP-LC3 transfection
Adenoviral transfection was performed according to
the manufacturer's protocol. NRCs were seeded in 15 mm diameter
glass-bottomed dishes at a density of 150 cells/mm2 and
36 h later were incubated in DMEM containing 2% FBS and adenovirus
(multiplicity of infection of 15) at 37°C for 10 h. The
transfection medium was then replaced with the complete medium, and
cells were further cultured at 37°C for 50 h. Following the H/R
treatments, cells were fixed with 4% paraformaldehyde and nuclei
were stained with 2 µg/ml Hoechst 33342 (Sigma-Aldrich; Merck KGaA)
for 5 min. Images of the cells were captured using a confocal
fluorescence microscope (Olympus FV-1,000; Olympus Corporation,
Tokyo, Japan). Autophagic flux was determined by evaluating the
number of mRFP and GFP puncta/cell using Image-Pro Plus software
version 6.0 (Media Cybernetics).
Statistical analysis
All experiments were performed ≥3 times. SPSS 20.0
software (IBM SPSS, Armonk, NY, USA) was used for statistical
analysis. The data were expressed as the mean ± standard deviation.
Statistical significance was analyzed by one-way analysis of
variance followed by Tukey's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Pur and 3-MA protect cardiomyocytes
from H/R-induced reductions in cell viability
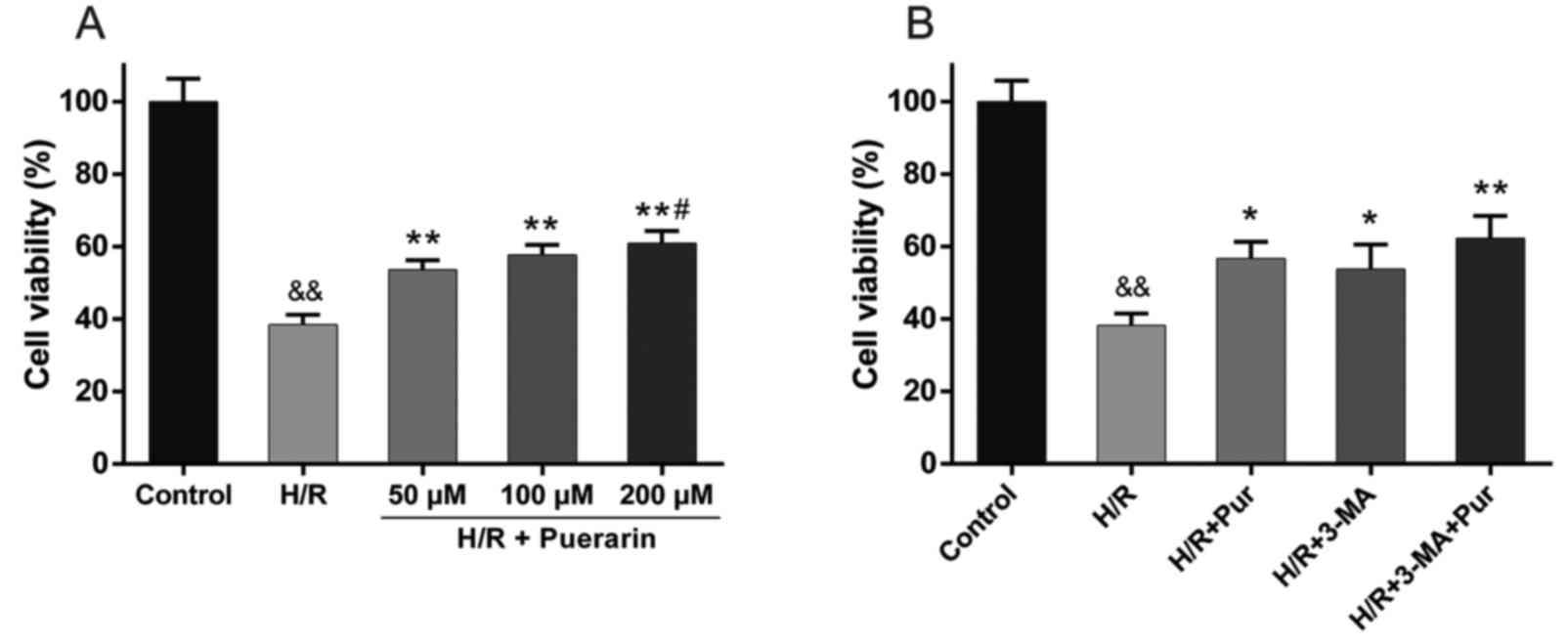
To confirm the protective effect of Pur on
myocardial I/R injury, an MTS assay was performed. Pur (50, 100 and
200 µM) was applied to NRCs 2 h prior to H/R. NRC viability was
markedly decreased to 38.4±2.8% in the H/R group compared with the
control group (Fig. 2A). However,
NRC viability was significantly increased by 15.2–22.5% following
50, 100 and 200 µM Pur pretreatment compared with the H/R group
(Fig. 2A), with the largest
increase observed in the 200 µM group, where NRC viability was
significantly increased compared with the 50 µM Pur pretreatment
group (Fig. 2A). Accordingly, 200
µM was selected as the concentration of Pur used in subsequent
experiments.
3-MA is a classic autophagy inhibitor, which blocks
autophagosome formation via the inhibition of type III PI3K, and
was also tested in the myocardial H/R model. Treatment with 3-MA
significantly increased NRC viability compared with the H/R group
(P<0.05; Fig. 2B), which was
similar to the effect observed following Pur pretreatment. These
results indicated that autophagy during myocardial H/R may
contribute to cell death.
Pur inhibits H/R-induced autophagy in
cardiomyocytes
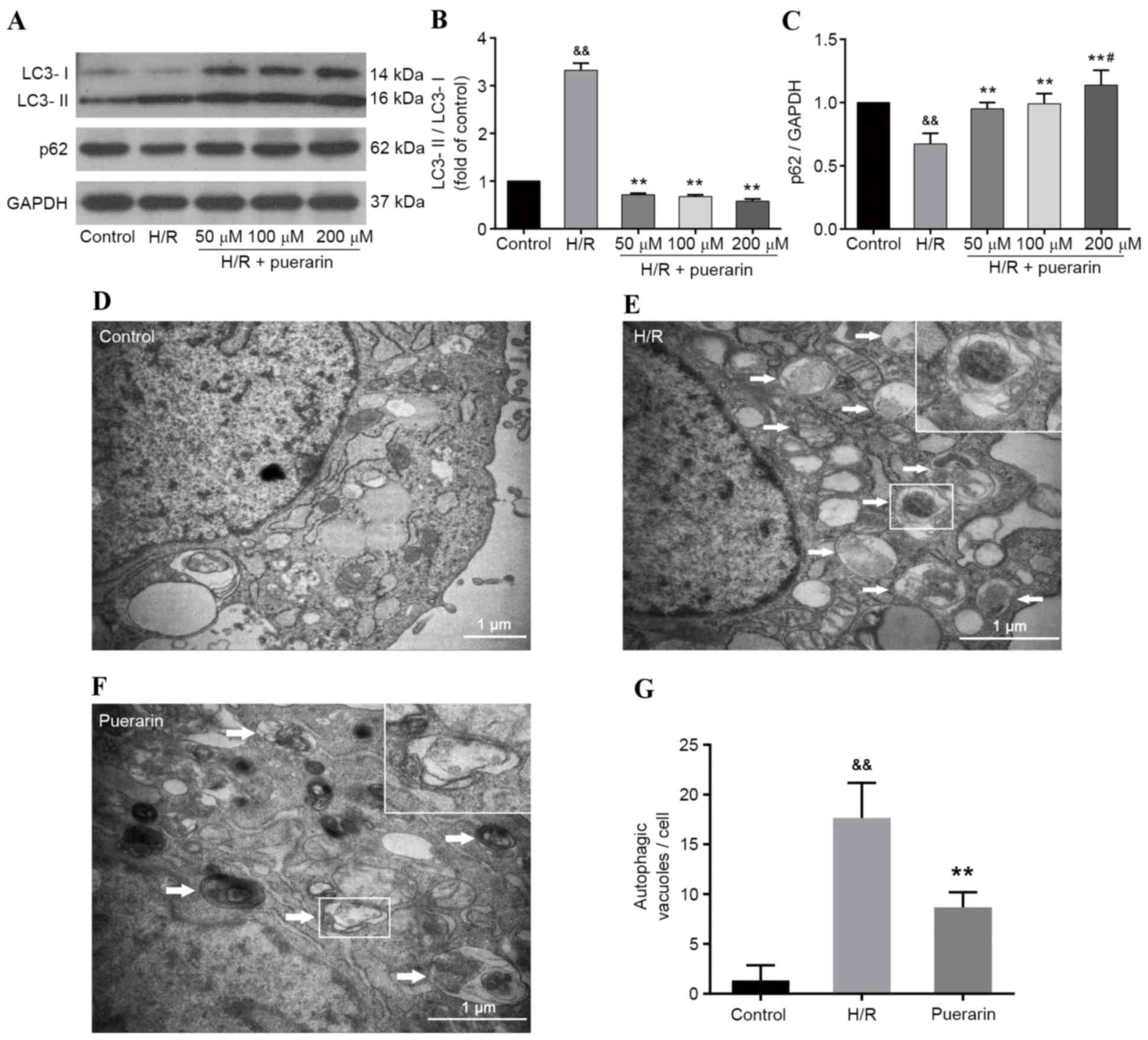
To investigate the effects of Pur on H/R-induced
autophagy in cardiomyocytes, western blot analysis was performed
(Fig. 3A). As presented in
Fig. 3B and C, H/R induced and
increased the ratio of LC3-II/LC3-I, and decreased the expression
levels of p62; however, Pur pretreatment (50, 100 and 200 µM)
decreased LC3-II/LC3-I ratio and increased p62 expression
(P<0.01). These results suggested that Pur may inhibit
H/R-induced autophagy in cardiomyocytes, which was confirmed by the
results of TEM. As shown in Fig.
3E-F, large vesicles containing membranous structures
(autophagosomes) appeared in the cytoplasm of cells in the H/R
group. Conversely, autophagosomes were scarce in the control group,
which was characterized by normal cytoplasm, mitochondria,
endoplasmic reticulum and nuclei. In addition, the number of
autophagosomes induced by myocardial H/R was significantly
decreased by Pur pretreatment (P<0.01; Fig. 3G).
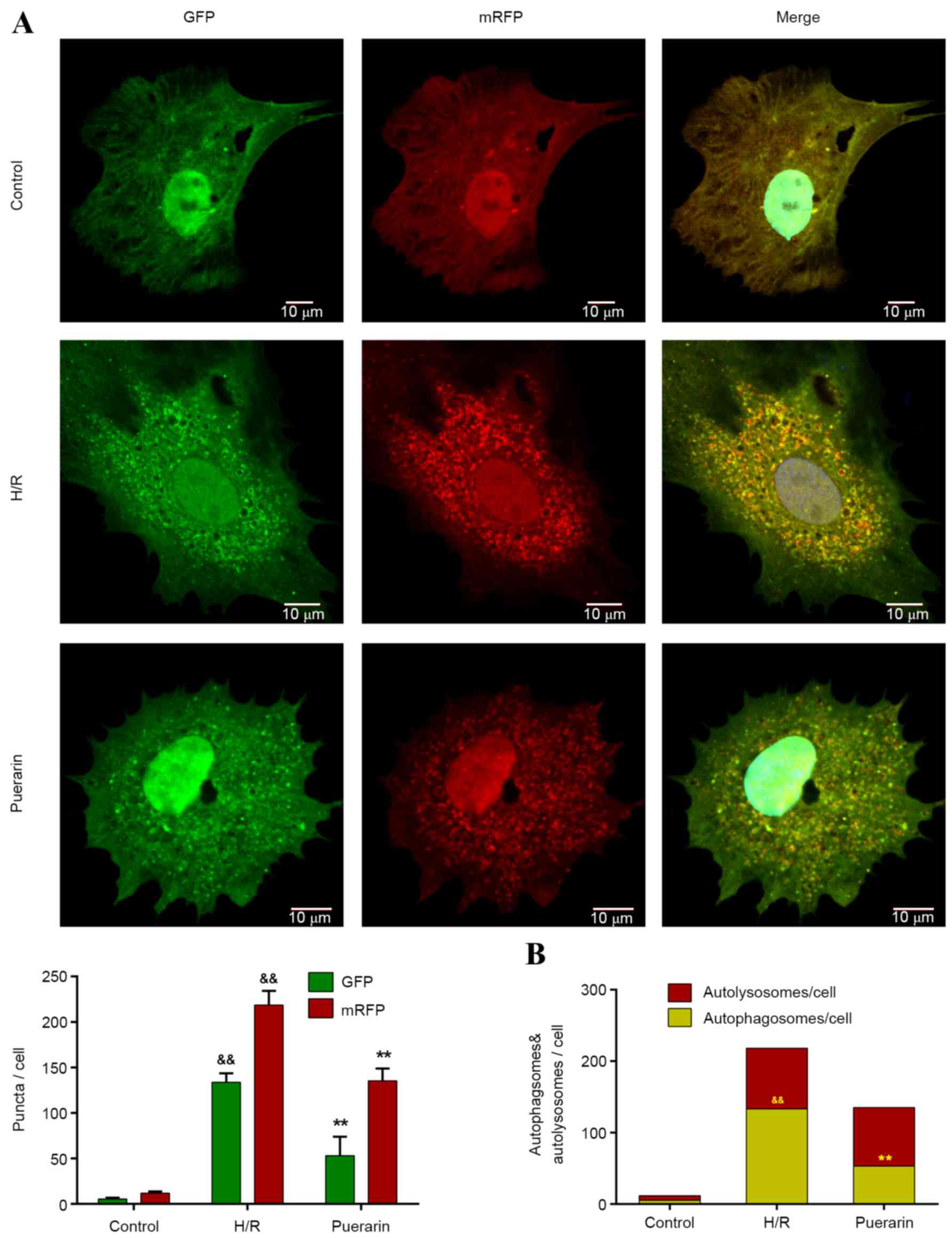
To further investigate the inhibitory effects of Pur
on autophagy, Ad-mRFP-GFP-LC3, a specific marker for autophagosomes
and autolysosomes, was transfected into NSCs. The yellow puncta in
merged pictures, as a result of merged m-RFP and GFP, represent
autophagosomes, whereas the red puncta represent autolysosomes
(Fig. 4A). Only a few yellow
puncta appeared in the control group, whereas the number of yellow
and red puncta visibly increased in the H/R group (Fig. 4A). Following pretreatment with Pur,
the number of yellow puncta (autophagosomes) significantly
decreased by 60.1% compared with the H/R group (P<0.01; Fig. 4B). Conversely, there was no
significant difference in the number of red puncta (autolysosomes)
between the Pur group and H/R group (Fig. 4B).
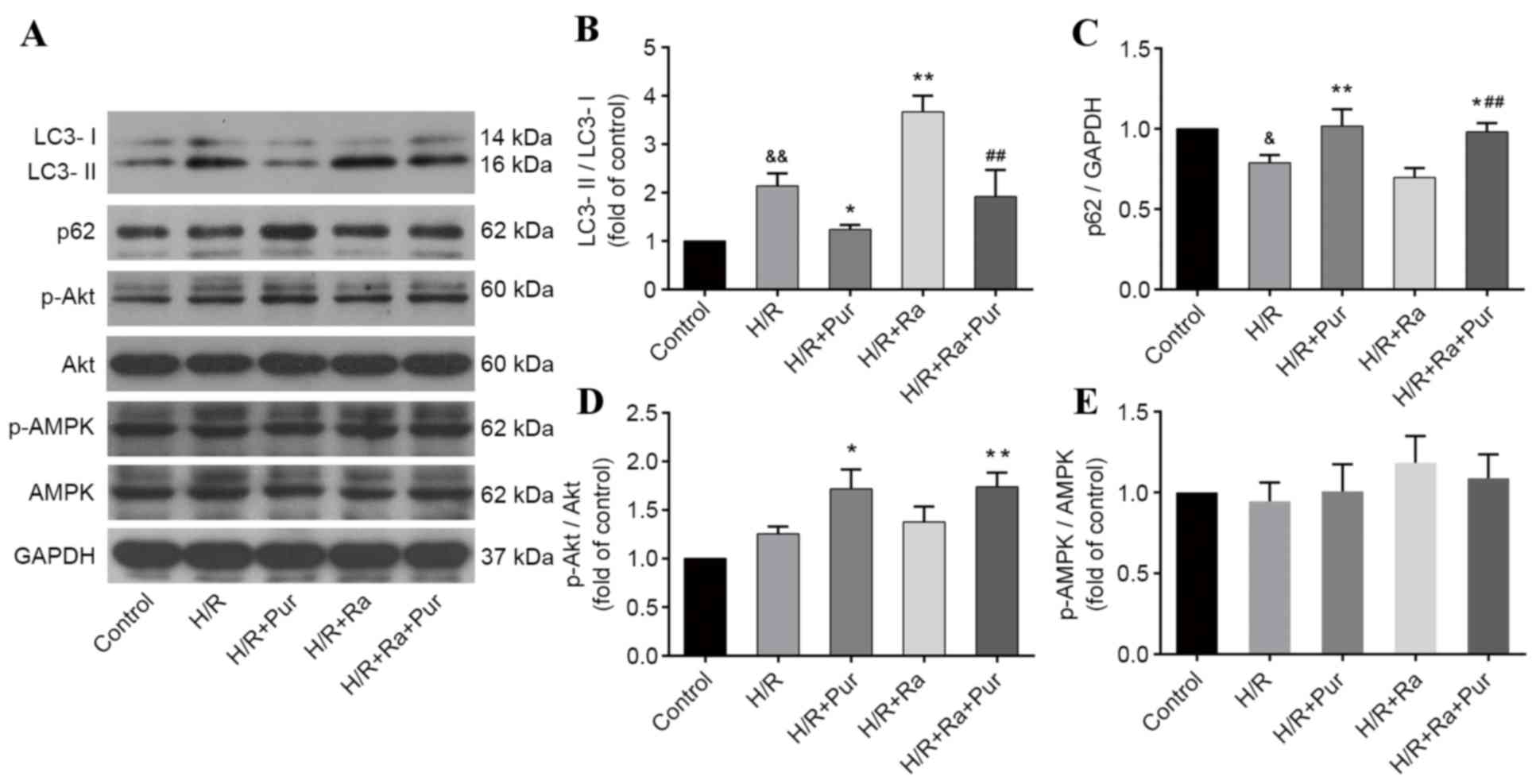
Pur inhibits Ra-induced autophagy
during myocardial H/R
Ra is one of the most commonly used drugs to
stimulate autophagy. As aforementioned, Ra-mediated autophagy
induction was performed in NRCs exposed to H/R, and western
blotting was performed (Fig. 5A)
The LC3-II/LC3-I ratio was significantly increased in the Ra group
compared with the H/R group (P<0.01; Fig. 5B). However, following pretreatment
with Ra + Pur, the LC3-II/LC3-I ratio was significantly decreased
compared with the Ra group (P<0.01; Fig. 5B) and the expression levels of p62
were elevated compared with the Ra group (P<0.01; Fig. 5C). These results suggested that the
increased autophagy induced by Ra during myocardial H/R was
inhibited by Pur pretreatment.
Effects of Pur on the phosphorylation
of Akt and AMPK during myocardial H/R
To investigate the potential molecular mechanisms
involved in the autophagy-inhibiting effect of Pur during
myocardial H/R, the PI3K/Akt and AMPK signaling pathways were
assessed. The expression levels of p-Akt, Akt, p-AMPK and AMPK in
groups were determined by western blotting. There was no
significant difference in the ratios of p-Akt/Akt and p-AMPK/AMPK
between the H/R group and the control group (P>0.05; Fig. 5D and E). Pur pretreatment increased
the phosphorylation of Akt compared with the H/R group (P<0.01;
Fig. 5D), but it did not
significantly affect the phosphorylation of AMPK (Fig. 5E).
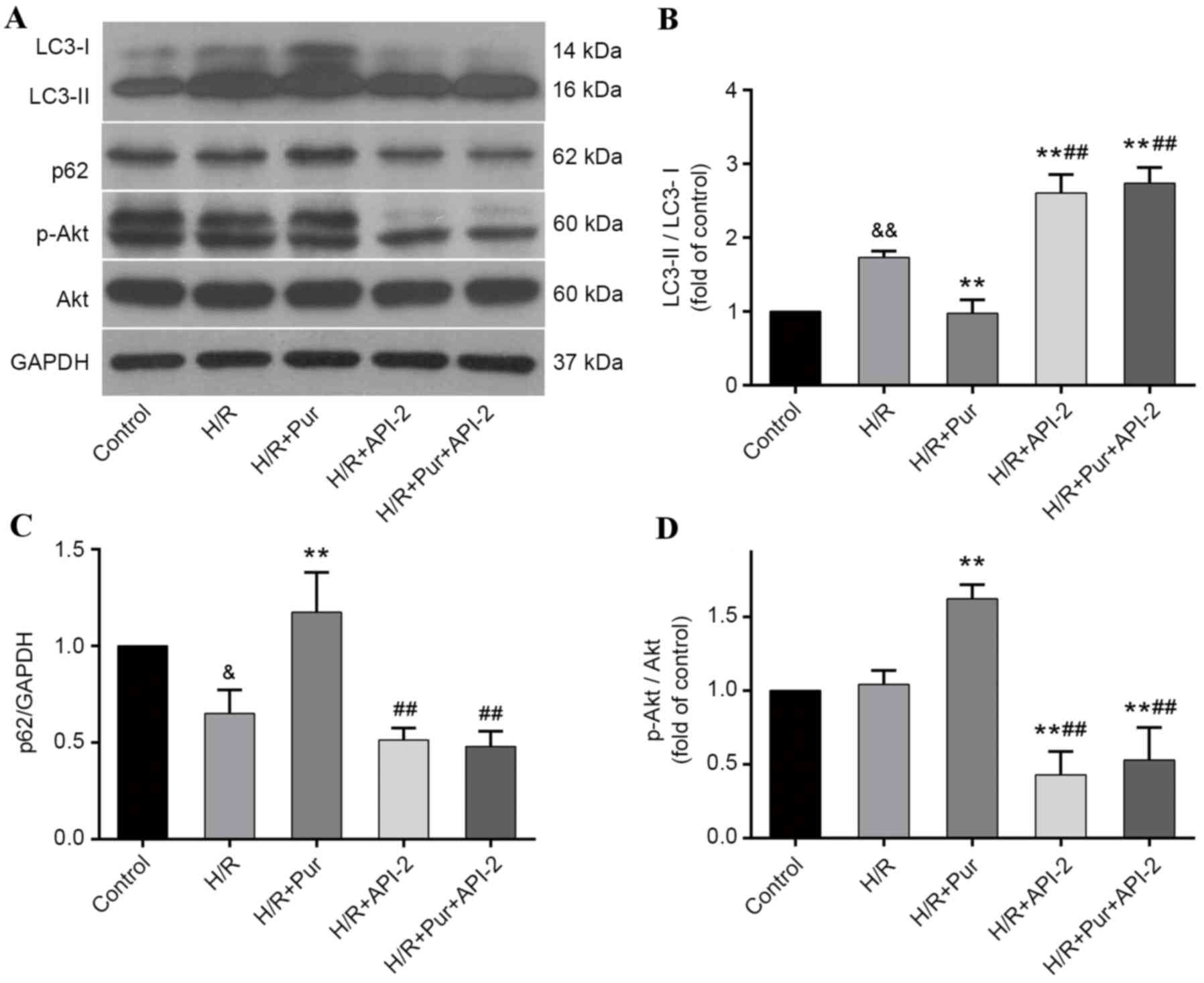
Autophagy-inhibiting effects of Pur
during myocardial H/R were suppressed by API-2
To further elucidate the involvement of the PI3K/Akt
signaling pathway in the autophagy-inhibiting effects of Pur
pretreatment during myocardial H/R, API-2 was applied to cells with
or without Pur pretreatment and the effects were measured by
western blotting (Fig. 6A). The
LC3-II/LC3-I ratio was significantly increased in the H/R +API-2 +
Pur group compared with the H/R + Pur group (P<0.01; Fig. 6B) and p62 protein expression levels
were significantly decreased in the H/R + API-2 + Pur group
compared with the H/R + Pur group (P<0.01; Fig. 6C). Akt phosphorylation was
significantly increased and autophagy was inhibited in the Pur
group compared with the H/R group (P<0.01; Fig. 6B and D); however, following
pretreatment with Pur + API-2, Akt phosphorylation was
significantly decreased and LC3-II/LC3-I ratio was increased
compared with the H/R + Pur group (P<0.01; Fig. 6D). These findings indicated that
the autophagy-inhibiting effects of Pur were abolished by
API-2.
Discussion
Although Pur has been used in clinical practice in
China for decades, the molecular mechanisms and targets underlying
the pharmacological properties of Pur remain unclear, which limits
its further clinical application (1). In the present study, the
cardioprotective effects of Pur against H/R injury in NRCs were
confirmed. Notably, Pur pretreatment inhibited autophagy during
myocardial H/R, and this effect was abolished by inhibition of Akt
signaling, indicating that Akt-dependent autophagy inhibition is
involved in the protective mechanisms of Pur during myocardial
H/R.
Autophagy is a dynamic and complex process wherein
cytoplasm, protein aggregates and organelles are sequestered by
double-membrane vesicles called autophagosomes, and trafficked to
the lysosomes for degradation (18). LC3 and p62 are widely used
autophagy markers. LC3 localizes to all types of autophagic
membranes, and p62 protein is degraded by autophagy. Therefore,
detecting the conversion of LC3-I to LC3-II and the degradation of
p62 by western blotting has been widely used to monitor autophagic
activity (19). In the present
study, Pur was demonstrated to inhibit autophagy during myocardial
H/R, which manifested as decreases in the ratio of LC3-II/LC3-I and
the degradation of p62. As presented in Fig. 3A, the ratio of LC3-II/LC3-I was
decreased in the Pur group due to a significant increase of LC3-I
and a slight decrease of LC3-II protein expression levels, which
suggests that the autophagy-inhibiting effects of Pur during H/R
are mainly due to the inhibition of autophagosome formation, and
thus the conversion of LC3-I to LC3-II, rather than autophagosome
degradation. To further confirm this, Ad-mRFP-GFP-LC3 transfection
was performed. Consistent with previous observations, Pur
pretreatment significantly inhibited autophagosome formation
(Fig. 4C).
Autophagy can be either protective or detrimental,
depending on the specific cellular context. It is activated as an
adaptive response to stress conditions, including nutritional
deprivation and ischemic injury (8). However, excessively activated
autophagy during reperfusion may result in the loss of necessary
proteins or organelles, leading to cellular dysfunction and
autophagic cell death, which is detrimental for cell survival
(7,11,20).
Therefore, inhibiting excessive autophagy may be a potential
therapeutic strategy to attenuate myocardial I/R injury.
Consistently, Huang et al (21) previously demonstrated that
berberine exerts protective effects during myocardial I/R by
suppressing autophagy (21). The
results of the present study demonstrated that inhibition of
autophagy by 3-MA reduced cell viability loss (Fig. 2B), which indicated that the
increased autophagy response to H/R is detrimental to
cardiomyocytes. Therefore, by inhibiting autophagosome formation,
Pur may maintain autophagic activity within a moderate range, thus
exerting a protective effect against myocardial H/R injury.
Once the autophagy-inhibiting effects of Pur during
myocardial H/R were determined, the underlying mechanisms were
explored further. It has previously been reported that Pur
activates the PI3K/Akt signaling pathway in various models,
including oxidative stress and cerebral I/R (12,22).
Consistent with these observations, the results of the present
study demonstrated that Pur pretreatment increased Akt
phosphorylation in cardiomyocytes exposed to H/R. Furthermore, the
autophagy-inhibiting effects of Pur were abolished following
treatment with an Akt signaling inhibitor, thus suggesting that Akt
phosphorylation may be critical for the effects of Pur on autophagy
inhibition. Combined with the inhibition of autophagosome formation
by Pur, these findings indicated that Pur inhibits autophagy during
myocardial H/R at the stage of induction via the Akt signaling
pathway.
The PI3K/Akt signaling pathway is one of the most
extensively studied pathways regulating autophagy (23). Previous studies have demonstrated
that Akt activation inhibits autophagy by activating mammalian
target of rapamycin complex 1 (mTORC1) (14), blocking the activation of forkhead
box O3 (FOXO3) (24), or
regulating the phosphorylation of Beclin1 (25). Ra inhibits mTORC1, which is one of
the downstream targets of Akt, thereby activating autophagy
(26). However, the
autophagy-inhibiting effects of Pur were not suppressed by Ra in
the present study (Fig. 5B), thus
suggesting that the Akt-mTORC1-Atg1 is not the main pathway that
mediates these effects. FOXO3 controls the transcription of
autophagy-related genes, including LC3. Akt activation has been
demonstrated to suppress FOXO3 activation, downregulate LC3
transcription and inhibit autophagy in skeletal muscle (24). However, the increased LC3-I levels
observed in the Pur group do not support this hypothesis. Wang
et al (25) demonstrated
that Akt inhibits autophagy by mediating the phosphorylation of
Beclin1 in tumor cells. Beclin1 is upregulated during the
reperfusion phase (11), and it is
essential for the PI3K complex, which is involved in the conversion
of LC3-I to LC3-II (27). In
combination with the findings of the present study and the
aforementioned previous studies, there is a possibility that Pur
inhibits autophagy by affecting the formation of the PtdIns3K
complex, via Akt-mediated Beclin1 phosphorylation. Further
investigation is required to test this hypothesis.
Pur has also been demonstrated to restore defective
autophagy in the cardiac hypertrophy model and the ethanol-treated
hepatocyte model via AMPK activation (28,29).
However, AMPK phosphorylation was not increased following
pretreatment with Pur during myocardial H/R (Fig. 5E). During hypoxia and ischemia,
AMPK acts as a sensor for energy deprivation and is activated by
decreased ATP concentration. The energy crisis is resolved at the
time of reperfusion, which in turn causes the inactivation of AMPK
signaling (11). So the difference
between models may be one explanation as to why AMPK
phosphorylation was not increased following Pur pretreatment in the
present study. The different intracellular environment may also
enable Pur to act on the Akt signaling pathway and exert an
inhibitory effect on autophagy.
In conclusion, the present study demonstrated that
Pur pretreatment inhibited autophagy during myocardial H/R, and
Akt-dependent autophagy inhibition may be one of the mechanisms
underlying the protective effects of Pur against myocardial H/R
injury. The results also suggested that excessive autophagy during
the reperfusion phase is detrimental to cell survival. Taken
together, the autophagy-inhibiting mechanism may be a basis for
further clinical applications of Pur to relieve myocardial I/R
injury.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 81270218,
to Dr Chen), the National Natural Science Foundation of China
(grant no. 81400190, to Dr Wang) and the Scientific and
Technological Research Project of Guangzhou City (grant no.
2014A020212191, to Dr Yang).
Glossary
Abbreviations
Abbreviations:
|
Pur
|
puerarin
|
|
3-MA
|
3-methyladenine
|
|
Ra
|
rapamycin
|
|
API-2
|
Akt signaling inhibitor
|
|
5-BrdU
|
5-bromo-2′- deoxyuridine
|
|
I/R
|
ischemia/reperfusion
|
|
H/R
|
hypoxia/reoxygenation
|
|
PI3K/Akt
|
phosphatidylinositol-3-kinase/protein
kinase B
|
|
AMPK
|
5′ adenosine monophosphate-activated
protein kinase
|
|
TEM
|
transmission electron microscope
|
References
|
1
|
Zhou YX, Zhang H and Peng C: Puerarin: A
review of pharmacological effects. Phytother Res. 28:961–975. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan LL, Sun LH, Li J, Yue XH, Yu HX, Wang
SY and Dong SQ: Protective effect of puerarin against myocardial
reperfusion injury. Myocardial metabolism and ultrastructure. Chin
Med J (Engl). 105:451–456. 1992.PubMed/NCBI
|
|
3
|
Pan HY, Gao Q, Yao H and Xia Q: The
protective role and the mechanisms of puerarin on isolated rat
heart during ischemia/reperfusion. Zhongguo Ying Yong Sheng Li Xue
Za Zhi. 22:455–459. 2006.(In Chinese). PubMed/NCBI
|
|
4
|
Gao Q, Pan HY, Qiu S, Lu Y, Bruce IC, Luo
JH and Xia Q: Atractyloside and 5-hydroxydecanoate block the
protective effect of puerarin in isolated rat heart. Life Sci.
79:217–224. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao Q, Yang B, Ye ZG, Wang J, Bruce IC and
Xia Q: Opening the calcium-activated potassium channel participates
in the cardioprotective effect of puerarin. Eur J Pharmacol.
574:179–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murphy E and Steenbergen C: Mechanisms
underlying acute protection from cardiac ischemia-reperfusion
injury. Physiol Rev. 88:581–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma S, Wang Y, Chen Y and Cao F: The role
of the autophagy in myocardial ischemia/reperfusion injury. Biochim
Biophys Acta. 1852:271–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yan L, Vatner DE, Kim SJ, et al: Autophagy
in chronically ischemic myocardium. Proc Natl Acad Sci USA. 102:pp.
13807–13812. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vilahur G, Juan-Babot O, Peña E, Oñate B,
Casani L and Badimon L: Molecular and cellular mechanisms involved
in cardiac remodeling after acute myocardial infarction. J Mol Cell
Cardiol. 50:522–533. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han JQ, Yu KY and He M: Effects of
puerarin on the neurocyte apoptosis and p-Akt (Ser473) expressions
in rats with cerebral ischemia/reperfusion injury. Zhongguo Zhong
Xi Yi Jie He Za Zhi. 32:1069–1072. 2012.(In Chinese). PubMed/NCBI
|
|
13
|
Wei SY, Chen Y and Xu XY: Progress on the
pharmacological research of puerarin: A review. Chin J Nat Med.
12:407–414. 2014.PubMed/NCBI
|
|
14
|
Arico S, Petiot A, Bauvy C, Dubbelhuis PF,
Meijer AJ, Codogno P and Ogier-Denis E: The tumor suppressor PTEN
positively regulates macroautophagy by inhibiting the
phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol
Chem. 276:35243–35246. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dong M, Ding W, Liao Y, Liu Y, Yan D,
Zhang Y, Wang R, Zheng N, Liu S and Liu J: Polydatin prevents
hypertrophy in phenylephrine induced neonatal mouse cardiomyocytes
and pressure-overload mouse models. Eur J Pharmacol. 746:186–197.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang L, Liu D, Yi X, Xu T, Liu Y, Luo Y,
Yin D and He M: The protective effects of puerarin in
cardiomyocytes from anoxia/reoxygenation injury are mediated by
PKCε. Cell Biochem Funct. 32:378–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ngoh GA, Facundo HT, Hamid T, Dillmann W,
Zachara NE and Jones SP: Unique hexosaminidase reduces metabolic
survival signal and sensitizes cardiac myocytes to
hypoxia/reoxygenation injury. Circ Res. 104:41–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang P and Mizushima N: LC3- and
p62-based biochemical methods for the analysis of autophagy
progression in mammalian cells. Methods. 75:13–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Valentim L, Laurence KM, Townsend PA,
Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS and
Stephanou A: Urocortin inhibits Beclin1-mediated autophagic cell
death in cardiac myocytes exposed to ischaemia/reperfusion injury.
J Mol Cell Cardiol. 40:846–852. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Z, Han Z, Ye B, Dai Z, Shan P, Lu Z,
Dai K, Wang C and Huang W: Berberine alleviates cardiac
ischemia/reperfusion injury by inhibiting excessive autophagy in
cardiomyocytes. Eur J Pharmacol. 762:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hwang YP and Jeong HG: Mechanism of
phytoestrogen puerarin-mediated cytoprotection following oxidative
injury: Estrogen receptor-dependent up-regulation of PI3K/Akt and
HO-1. Toxicol Appl Pharmacol. 233:371–381. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shanware NP, Bray K and Abraham RT: The
PI3K, metabolic, and autophagy networks: Interactive partners in
cellular health and disease. Annu Rev Pharmacol Toxicol. 53:89–106.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mammucari C, Milan G, Romanello V, Masiero
E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J,
et al: FoxO3 controls autophagy in skeletal muscle in vivo. Cell
Metab. 6:458–471. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang RC, Wei Y, An Z, Zou Z, Xiao G,
Bhagat G, White M, Reichelt J and Levine B: Akt-mediated regulation
of autophagy and tumorigenesis through Beclin 1 phosphorylation.
Science. 338:956–959. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yue Z and Zhong Y: From a global view to
focused examination: Understanding cellular function of lipid
kinase VPS34-Beclin 1 complex in autophagy. J Mol Cell Biol.
2:305–307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu B, Wu Z, Li Y, Ou C, Huang Z, Zhang J,
Liu P, Luo C and Chen M: Puerarin prevents cardiac hypertrophy
induced by pressure overload through activation of autophagy.
Biochem Biophys Res Commun. 464:908–915. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Noh BK, Lee JK, Jun HJ, Lee JH, Jia Y,
Hoang MH, Kim JW, Park KH and Lee SJ: Restoration of autophagy by
puerarin in ethanol-treated hepatocytes via the activation of
AMP-activated protein kinase. Biochem Biophys Res Commun.
414:361–366. 2011. View Article : Google Scholar : PubMed/NCBI
|