Introduction
Childhood asthma is a group of multifactorial
diseases with similar features, including mast cell and eosinophil
infiltration, causing airway hyper-responsiveness, inflammation and
airway obstruction (1). The
prevalence of childhood asthma is increasing worldwide (2,3).
Airway smooth muscle (ASM) serves an important role in the various
biological processes that are essential for the development of
childhood asthma. Increased ASM mass is associated with increased
asthma severity (4,5). Additionally, studies have indicated
that ASM cell migration towards the airway epithelium, in response
to inflammatory mediators including platelet-derived growth factor
(PDGF), contributes to airway remodeling (6–8).
Therefore, targeting ASM cells may be useful in treating childhood
asthma.
Follistatin-like protein 1 (FSTL1), additionally
referred to as TSC36, is a secreted glycoprotein that belongs to
the follistatin family of proteins. It has been demonstrated to
serve important roles in immunomodulation (9), embryonic development (10) and vascularization (11). In addition, FSTL1 maintains the
proliferation and survival of tumor cells, and overexpression of
FSTL1 significantly suppresses cell proliferation and invasion in
ovarian and endometrial cancers (12). Recently, one study revealed that
FSTL1 is highly expressed by macrophages in the lungs of patients
with severe asthma (13). However,
the role of FSTL1 in ASM cell proliferation and migration remains
unknown. The present study aimed to investigate the role of FSTL1
in cell proliferation and migration mediated by PDGF subunit B
(PDGF-BB) (6), a strong
chemoattractant, in human ASM cells. It was demonstrated that
knockdown of FSTL1 inhibited PDGF-BB-induced ASM cell proliferation
and migration.
Materials and methods
Cell culture
The bronchial-tracheal-derived human ASMC cell line
was purchased from the American Type Culture Collection (Manassas,
VA, USA) and incubated in Dulbecco's Modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere
containing 5% CO2 at 37°C. ASM cells at passage 9 to 20
were used.
FSTL1 suppression by small interfering
RNA (siRNA)
For siRNA transfection, 1×105 cells were
seeded in 6-cm dishes for 24 h in DMEM medium (Gibco; Thermo Fisher
Scientific, Inc.). The cells (1×105 cells/well) were
subsequently transfected with 1 µl (10 nM) FSTL1 siRNA
(5′-UGCAAAUACUUACGGACUUUU-3′) or non-targeting siRNA
(5′-UCUCCGAACGUGUCACGUTT-3′) (Shanghai Sangong Pharmaceutical Co.,
Ltd., Shanghai, China) for 18 h using Lipofectamine®
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). Protein silencing was confirmed by western blot analysis.
ASM cells were subsequently stimulated with 10 ng/ml PDGF-BB
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 24 h.
Cell proliferation assay
Cell proliferation was evaluated using an MTT assay.
Cells were suspended at 1×104 cells/well and 200 µl of
this suspension was plated into each well of a 96-well plate. The
ASMC cells were cultured with DMEM and cells were growth-arrested
prior to every experiment. They were pre-treated with FSTL1 siRNA
for 24 h, followed by PDGF-BB stimulation. Following this, the
medium was removed, and 20 µl 5 mg/ml MTT in DMEMwas added. The
cells were further incubated in 5% CO2 at 37°C for 4 h.
Formazan was solubilized with 100 µl dimethyl sulfoxide for 10 min.
The absorbance of the solution was measured at a wavelength of 570
nm using a microplate reader (Spectra Max 190; Molecular Devices,
LLC, Sunnyvale, CA, USA).
Cell cycle assay
The growth-arrested cells were pre-treated with
FSTL1 siRNA for 24 h, followed by PDGF-BB stimulation. The cells
were harvested and fixed with ice-cold 70% (v/v) ethanol for 24 h.
Following centrifugation at 2,000 × g for 10 min at 37°C,
the cell pellet was washed with PBS and resuspended in PBS
containing 50 µg/ml propidium iodide, 0.1% (v/v) Triton X-100 and 1
µg/ml DNase-free RNase. The cells were subsequently incubated for 1
h in the dark at room temperature. A flow cytometer (FC 500;
Beckman Coulter, Inc., Brea, CA, USA) was used to elucidate cell
cycle progression, and this was analyzed using Modfit software
(version 3.2; Verity Software House, Topsham, ME, USA).
Cell migration assay
ASM cell migration was measured using a Transwell
migration assay. ASM cells at a concentration of 5×104
cells/well, transfected with FSTL1 siRNA, were resuspended gently
in DMEM/F-12 medium containing 2% fetal bovine serum (FBS;
Invitrogen; Thermo Fisher Scientific, Inc.) and added to into the
upper chamber. Following this, 600 µl DMEM/F-12 medium containing
10% FBS with or without PDGF-BB was added into the lower chamber.
Following incubation at 37°C for 24 h, the cells on the upper
membrane surface were removed by washing with PBS, and those that
had migrated to the bottom side of the filter were fixed in
methanol and stained with Harris hematoxylin solution for 5 min.
The mean number of migrated cells from five randomly selected
optical fields under a light microscope (magnification, ×100;
Olympus Corporation, Tokyo, Japan) was determined.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from ASM cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. For mRNA detection,
cDNA was generated using Moloney Murine Leukemia Virus Reverse
Transcriptase (Clontech Laboratories, Inc., Mountainview, CA, USA).
The expression levels of gene mRNA transcripts were analyzed using
specific primers, SYBR®-Green I reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) and the Bio-Rad iQ5 qPCR system
(Takara Biotechnology Co., Ltd., Dalian, China), according to the
manufacturer's protocol. The specific primers for FSTL1 were as
follows: Sense, 5′-CGAGGTGGAGTTGACGAGAAAC-3′ and antisense,
5′-AGGACTGGATCATCATGACGTTCT-3′. GAPDH were
5′-GGCAAATTCAACGGCACAGTC-3′ (sense), 5′-GCTGACAATCTTGAGTGAGTT-3′
(antisense). The PCR procedure was as follows: An initial
denaturation step at 94°C for 4 min, followed by 40 cycles of
denaturation at 94°C for 20 sec, annealing at 55°C for 30 sec and
extension at 72°C for 20 sec, and a melting curve from 65 to 95°C.
GAPDH served as a control for normalizing the gene expression
levels and the results were analyzed using the
2−ΔΔCq method (14). The sequences for GAPDH are as
follows: 5′-GGCAAATTCAACGGCACAGTC-3′ (sense) and
5′-GCTGACAATCTTGAGTGAGTT-3′ (antisense).
Western blot analysis
Cells were lysed using RIPA Cell lysis buffer
(Takara Biotechnology, Co., Ltd., Dalian, China) and protein
concentrations were determined using the Bicinchoninic Acid assay
kit (Pierce; Thermo Fisher Scientific, Inc.). Equal amounts of
protein (30 µg) were separated by 10% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (Whatman; GE Healthcare Life
Sciences, Chalfont, UK). The membranes were blocked in 5% nonfat
milk in TBS with Tween-20 (TBST; 5 mM Tris-HCl at pH 7.4, 136 mM
NaCl, 0.1% Tween-20) for 1 h at room temperature prior to
hybridization with the following primary antibodies overnight at
4°C. Detection of rabbit anti-FSTL1 (dilution, 1:1,500; Abcam,
Cambridge, UK; cat. no. 111969), mouse anti-phospho-extracellular
signal-regulated kinase (ERK; dilution, 1:2,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; cat. no. sc-81492), rabbit
anti-ERK (dilution, 1:2,000; Santa Cruz Biotechnology, Inc.; cat.
no. sc-292838), mouse anti-AKT (dilution, 1:2,000; Santa Cruz
Biotechnology, Inc.; sc-5298), rabbit anti-phospho-AKT (dilution,
1:2,000; Santa Cruz Biotechnology, Inc.; cat. no. sc-135650) was
performed. The membranes were subsequently washed three times for 5
min with TBST and incubated with bovine anti-rabbit horseradish
peroxidase-conjugated secondary antibodies (dilution, 1:3,000; cat.
no. sc-2385) for 1 h at room temperature. The resulting protein
bands were visualized using an Enhanced Chemiluminescence reagent
according to the manufacturer's protocol. The relative expression
levels of each protein compared with GAPDH were analyzed. BandScan
software (version 5.0; Glyko, Novato, CA, USA) was used for the
quantification of all proteins following western blot analysis.
Statistical analysis
All experiments were performed in at least
triplicate and results are expressed as the mean ± standard
deviation. Statistical comparisons were performed using one-way
analysis of variance followed by the Student's t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
FSTL1 is upregulated in
PDGF-BB-treated ASM cells
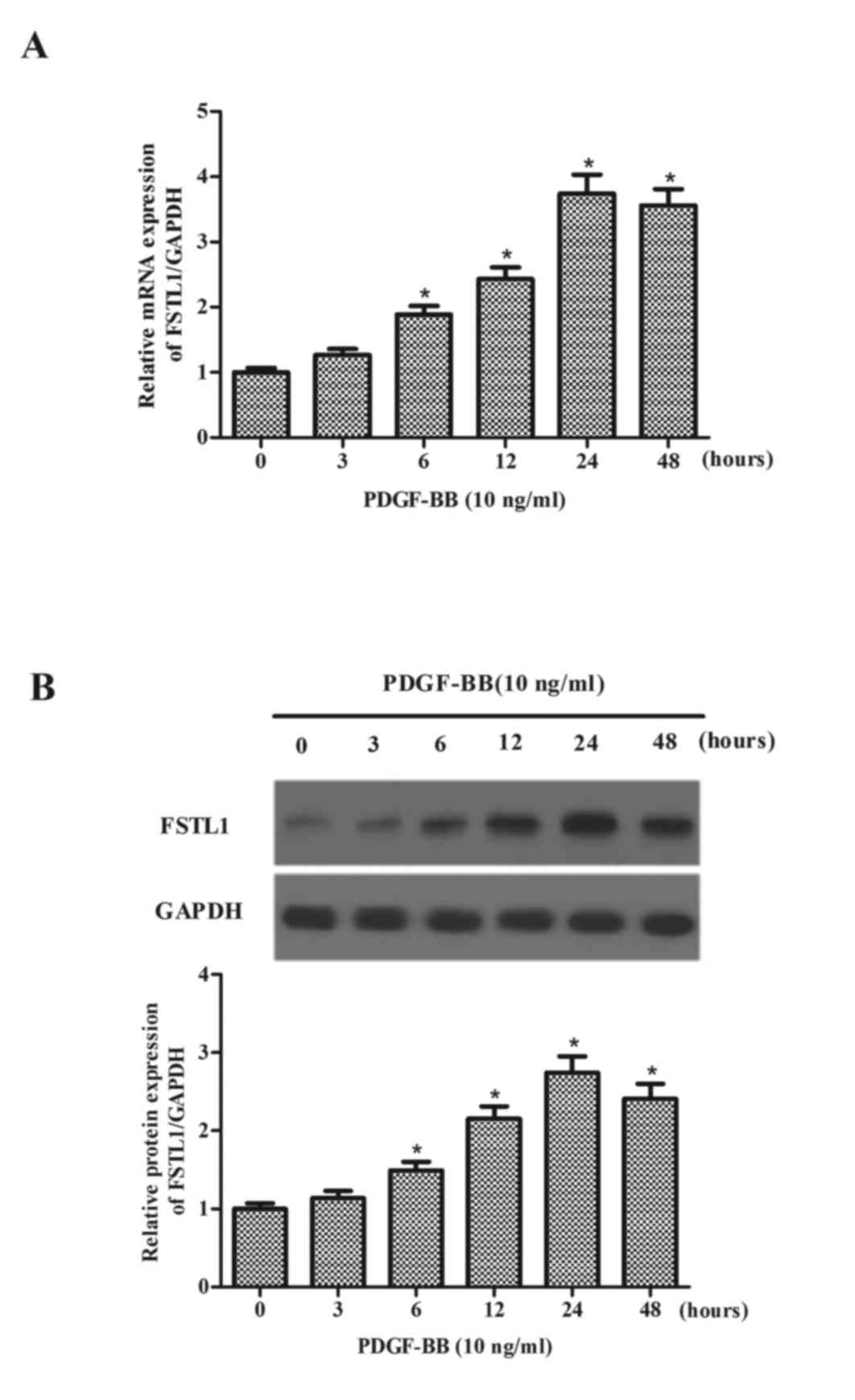
The present study initially measured the expression
levels of FSTL1 in PDGF-BB-treated ASM cells. As presented in
Fig. 1A, after 6 h PDGF-BB
markedly increased the mRNA expression levels of FSTL1, reaching a
peak at 24 h. Additionally, the expression levels of the FSTL1
protein were increased by PDGF-BB (Fig. 1B) in a time-dependent manner.
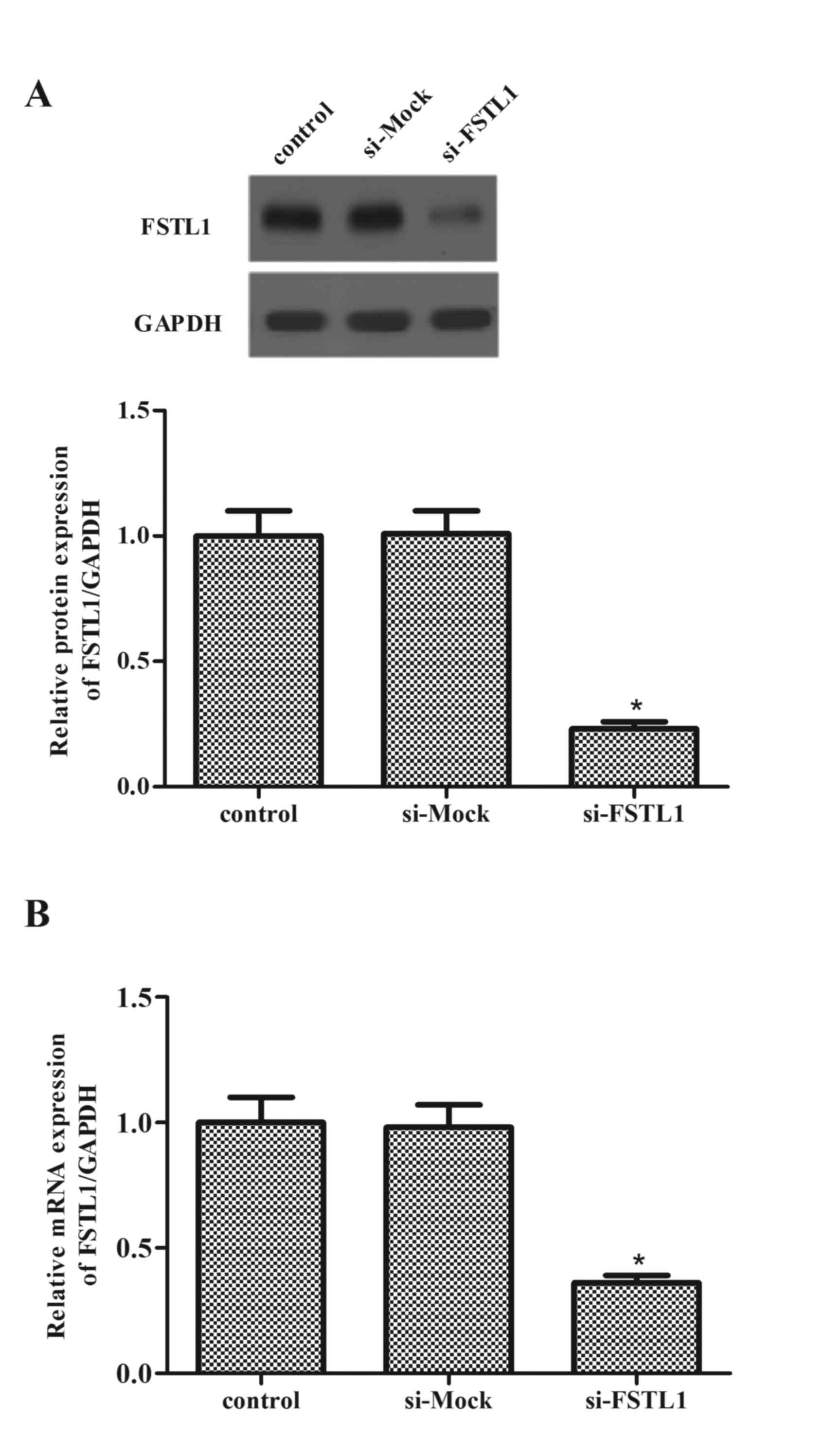
Knockdown of FSTL1 expression levels
in ASM cells
Following this, ASM cells were transfected with
FSTL1 siRNA, and the efficiency of transfection was evaluated by
western blot and RT-qPCR analysis. Western blot data revealed that
transfection of FSTL1 siRNA markedly decreased the expression
levels of FSTL1 protein (P=0.016; Fig.
2A). Consistent with the results of western blot, western blot
analysis demonstrated that FSTL1 siRNA inhibited the mRNA
expression levels of FSTL1 (P=0.029; Fig. 2B).
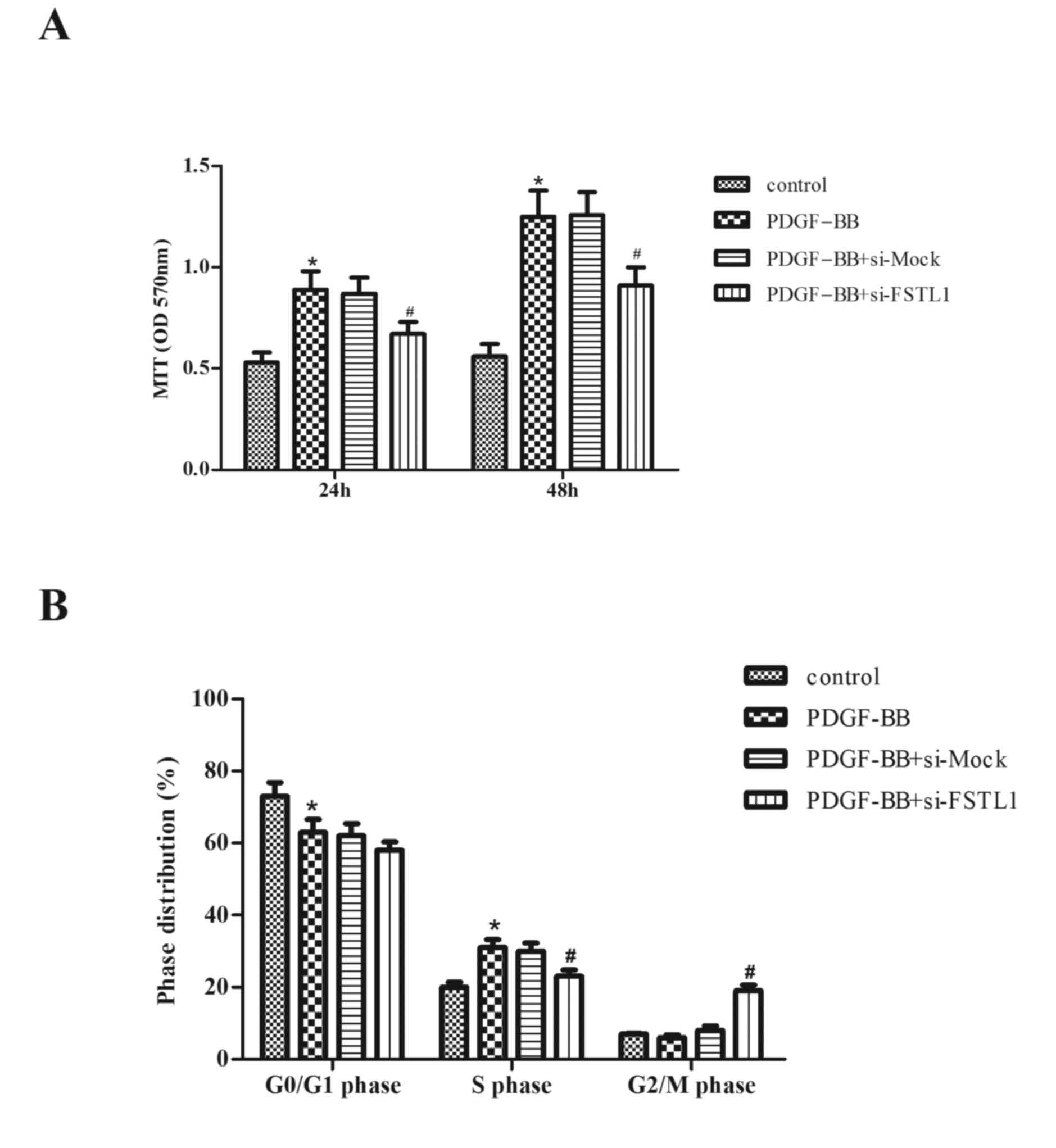
Knockdown of FSTL1 inhibits
PDGF-BB-induced ASM cell proliferation
The role of FSTL1 in PDGF-BB-stimulated
proliferation of ASM cells was subsequently investigated. As
presented in Fig. 3A, compared
with the control group, PDGF-BB treatment markedly increased cell
proliferation (P=0.034; P=0.026). However, knockdown of FSTL1
significantly inhibited PDGF-BB-induced ASM cell proliferation
(P=0.043; P=0.036).
To further examine the effect of FSTL1 on
PDGF-BB-induced ASM cell proliferation, cell cycle progression was
evaluated by flow cytometry. As presented in Fig. 3B, PDGF-BB induced the entry of ASM
cells to the synthesis phase (S-phase) to undergo proliferation,
with 62.5±1.6% cells in G0/G1 phase (compared with 73.1±2.5% in the
control; P=0.041) and 31.2±1.9% in S-phase (compared with 19.4±1.3%
in the control; P=0.031), whereas the knockdown of FSTL1
significantly arrested the cell cycle at the G2/M phase (18.6±1.7
vs. 5.8±1.1% in the PDGF-BB group; P=0.028).
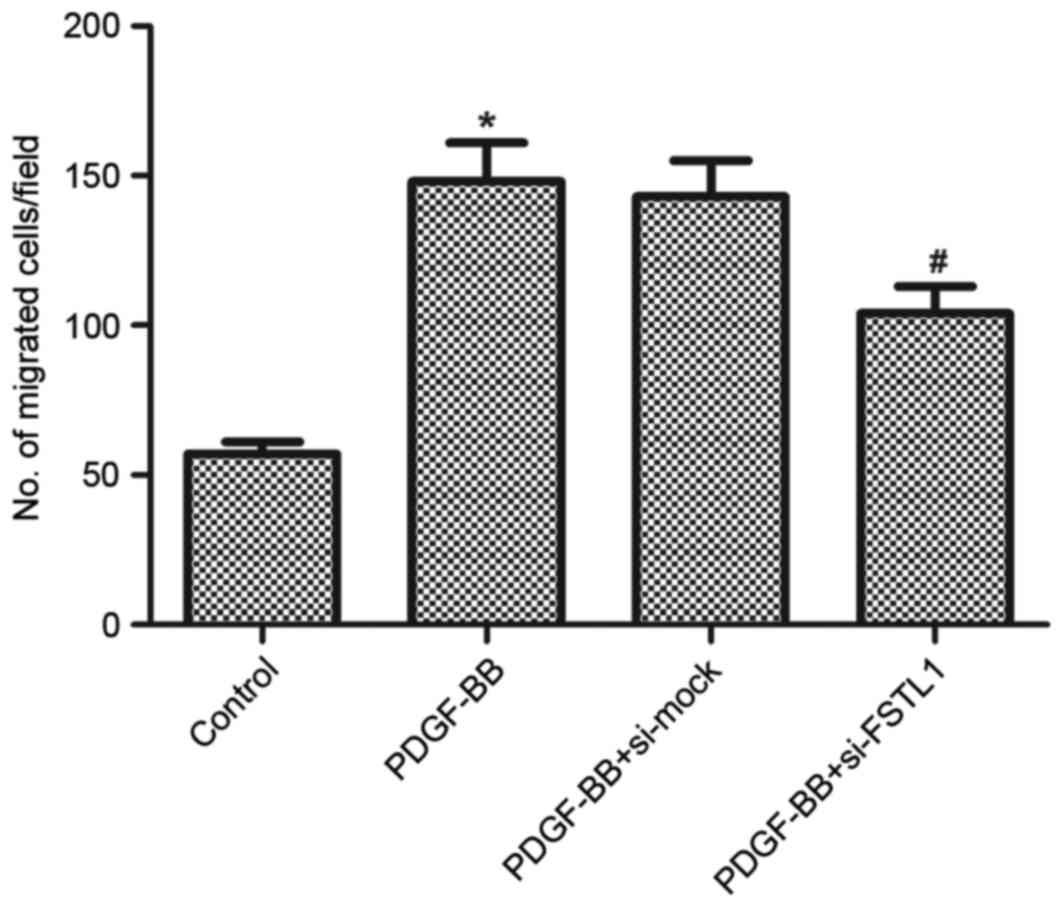
Knockdown of FSTL1 inhibits
PDGF-BB-induced ASM cell migration
Following this, the effect of FSTL1 on ASM cell
migration induced by PDGF-BB was examined using a Transwell
migration assay. As presented in Fig.
4, PDGF-BB resulted in a 2.59-fold increase in the number of
cells that migrated through the membrane compared with the
untreated control (P=0.023), whereas knockdown of FSTL1 markedly
inhibited PDGF-BB-induced ASM cell migration (P=0.036).
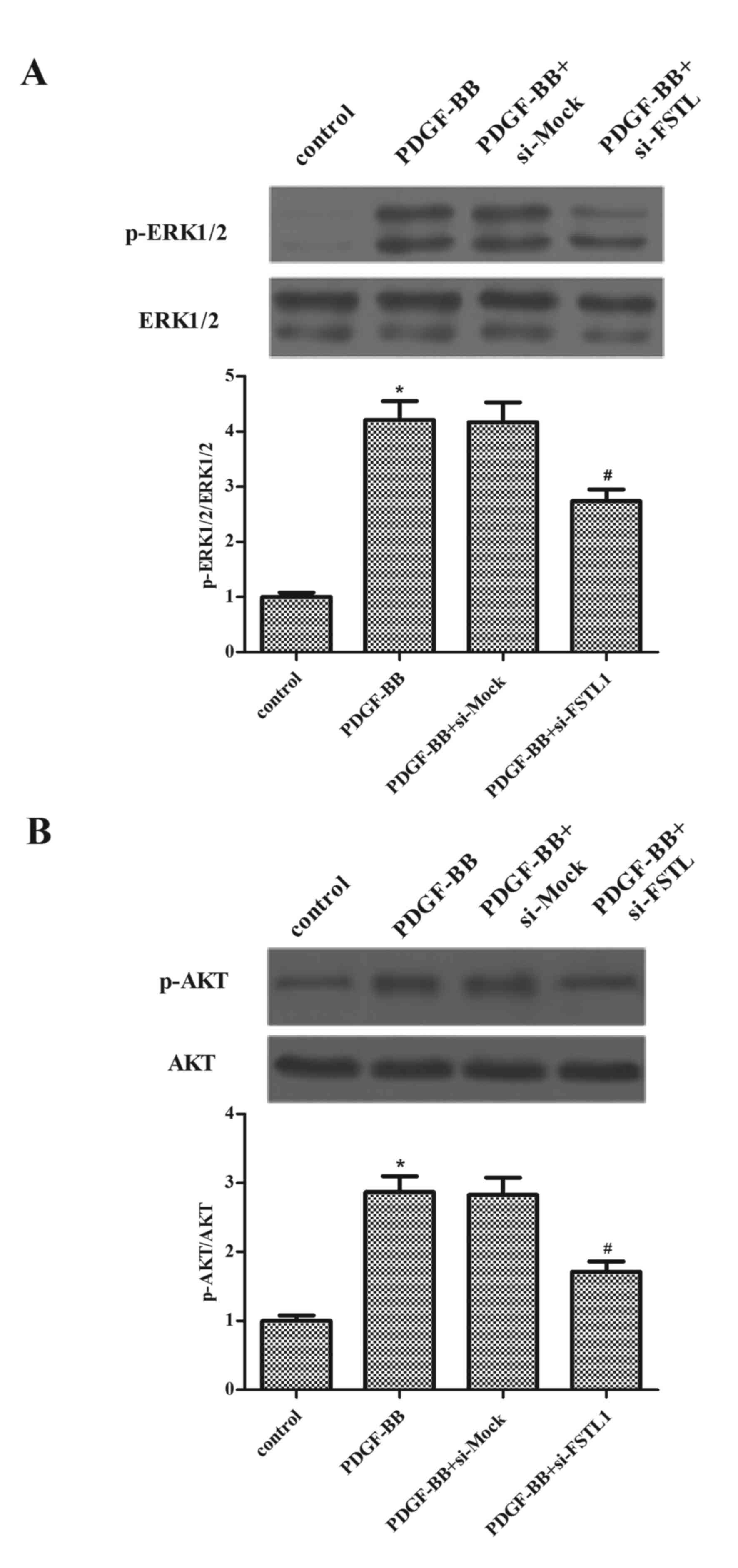
Knockdown of FSTL1 inhibits
PDGF-BB-induced activation of ERK and AKT in ASM cells
To investigate the underlying mechanisms by which
FSTL1 affected PDGF-BB-induced ASM cell proliferation and
migration, the effect of FSTL1 on the activation of ERK and AKT in
PDGF-BB-stimulated ASM cells was examined. As presented in Fig. 5, the protein expressions levels of
p-ERK1/2 and p-AKT increased following PDGF-BB stimulation, whereas
the knockdown of FSTL1 markedly inhibited PDGF-induced expression
levels of p-ERK1/2 and p-AKT.
Discussion
Abnormal proliferation and migration of ASM cells
serve roles in airway remodeling and contribute to airway
hyper-responsiveness. The present study demonstrated that PDGF-BB
stimulation upregulated FSTL1 expression levels in ASM cells in
vitro. Knockdown of FSTL1 inhibited cell proliferation and
arrested the cell cycle in the G2/M phase in PDGF-BB-stimulated ASM
cells. Additionally, FSTL1 knockdown inhibited PDGF-BB-induced ASM
cell migration. Furthermore, knockdown of FSTL1 caused
downregulation of p-ERK and p-AKT protein expression levels induced
by PDGF-BB in ASM cells.
A previous study reported that FSTL1 acted as a bone
morphogenetic protein 4 antagonist during lung development
(15). FSTL1 expression is induced
in response to lung injury and mediates the accumulation of
myofibroblasts and subsequent fibrosis; inhibition of FSTL1 with a
neutralizing antibody in mice reduced bleomycin-induced fibrosis
in vivo (16). The present
study demonstrated that PDGF-BB stimulation upregulated FSTL1
expression levels in ASM cells in vitro. These data
suggested that FSTL1 may serve a critical role in in asthmatic ASM
remodeling.
FSTL1 has been reported to inhibit proliferation and
invasion, and to induce apoptosis of cancer cells in vitro
(17,18). In addition, increasing evidence
indicates that PDGF is involved in the pathophysiology of asthma,
and that PDGF is a potent mitogen that may mediate ASM cell
proliferation and migration (19–21).
The present study revealed that knockdown of FSTL1 inhibited cell
proliferation and arrested the cell cycle in the G2/M phase in
PDGF-BB-stimulated ASM cells. In addition, it was observed that the
knockdown of FSTL1 inhibited PDGF-BB-induced ASM cell
migration.
It has been reported that the dual phosphoinositide
3-kinase (PI3K)/AKT and ERK pathways may regulate ASM cell
proliferation and migration (22–24).
AKT is a primary downstream target of PI3K, activated in response
to various stimuli, growth factors and hormones. ERK1/2 belongs to
the mitogen-activated protein kinase family and is located
downstream of the Raf/Ras/mitogen-activated protein kinase kinase
cascade. A previous study demonstrated that ERK and AKT activation
was significantly greater in ASM cells from asthmatic, compared
with non-asthmatic, subjects (25), and that the inhibition of the
ERK1/2 or PI3K/AKT signaling pathway significantly inhibited ASM
proliferation (26). Additionally,
one study reported that the overexpression of FSTL1 protected
cultured neonatal rat ventricular myocytes from
hypoxia/reoxygenation-induced apoptosis, and that this protective
effect was dependent on the upregulation of AKT and ERK activities.
The knockdown of FSTL1 in cardiac myocytes decreased basal AKT
signaling and increased cell apoptosis (27). The present study revealed that
PDGF-BB-induced ASM cell migration was partially mediated by
inhibition of the activation of ERK and AKT. These data are
consistent with those of previous studies in which the activation
of ERK and AKT were demonstrated to mediate ASM proliferation and
migration induced by various growth factors, including PDGF-BB
(20,28,29).
Furthermore, the present study revealed that the knockdown of FSTL1
markedly inhibited PDGF-BB-induced protein expression levels of
p-ERK and p-AKT.
In conclusion, the results of the present study
demonstrated that knockdown of FSTL1 inhibited ASM cell
proliferation and migration induced by PDGF-BB at least partially
via inhibiting the activation of ERK and AKT. These results provide
novel insight into the pathogenesis of airway remodeling in
childhood asthma and FSTL1 may be a possible therapeutic strategy
for the treatment of asthma.
References
|
1
|
Noutsios GT and Floros J: Childhood
asthma: Causes, risks, and protective factors; a role of innate
immunity. Swiss Med Wkly. 144:w140362014.PubMed/NCBI
|
|
2
|
Asher MI, Montefort S, Björkstén B, Lai
CK, Strachan DP, Weiland SK and Williams H; ISAAC Phase Three Study
Group, : Worldwide time trends in the prevalence of symptoms of
asthma, allergic rhinoconjunctivitis, and eczema in childhood:
ISAAC Phases One and Three repeat multicountry cross-sectional
surveys. Lancet. 368:733–743. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
De Marco R, Cappa V, Accordini S, Rava M,
Antonicelli L, Bortolami O, Braggion M, Bugiani M, Casali L,
Cazzoletti L, et al: Trends in the prevalence of asthma and
allergic rhinitis in Italy between 1991 and 2010. Eur Respir J.
39:883–892. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zuyderduyn S, Sukkar M, Fust A, Dhaliwal S
and Burgess JK: Treating asthma means treating airway smooth muscle
cells. Eur Respir J. 32:265–274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirst SJ, Martin JG, Bonacci JV, Chan V,
Fixman ED, Hamid QA, Herszberg B, Lavoie JP, McVicker CG, Moir LM,
et al: Proliferative aspects of airway smooth muscle. J Allergy
Clin Immunol. 114(2 Suppl): S2–S17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Carlin SM, Roth M and Black JL: Urokinase
potentiates PDGF-induced chemotaxis of human airway smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol. 284:L1020–L1026. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Madison JM: Migration of airway smooth
muscle cells. Am J Respir Cell Mol Biol. 29:8–11. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parameswaran K, Radford K, Zuo J, Janssen
L, O'byrne P and Cox P: Extracellular matrix regulates human airway
smooth muscle cell migration. Eur Respir J. 24:545–551. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Le Luduec JB, Condamine T, Louvet C,
Thebault P, Heslan JM, Heslan M, Chiffoleau E and Cuturi MC: An
immunomodulatory role for follistatin-like 1 in heart allograft
transplantation. Am J Transplant. 8:2297–2306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Esterberg R, Delalande JM and Fritz A:
Tailbud-derived Bmp4 drives proliferation and inhibits maturation
of zebrafish chordamesoderm. Development. 135:3891–3901. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ouchi N, Oshima Y, Ohashi K, Higuchi A,
Ikegami C, Izumiya Y and Walsh K: Follistatin-like 1, a secreted
muscle protein, promotes endothelial cell function and
revascularization in ischemic tissue through a nitric-oxide
synthase-dependent mechanism. J Biol Chem. 283:32802–32811. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan QK, Ngan HY, Ip PP, Liu VW, Xue WC
and Cheung AN: Tumor suppressor effect of follistatin-like 1 in
ovarian and endometrial carcinogenesis: A differential expression
and functional analysis. Carcinogenesis. 30:114–121. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miller M, Beppu A, Rosenthal P, Pham A,
Das S, Karta M, Song DJ, Vuong C, Doherty T, Croft M, et al: Fstl1
promotes asthmatic airway remodeling by inducing oncostatin M. J
Immunol. 195:3546–3556. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Geng Y, Dong Y, Yu M, Zhang L, Yan X, Sun
J, Qiao L, Geng H, Nakajima M, Furuichi T, et al: Follistatin-like
1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling
antagonist in controlling mouse lung development. Proc Natl Acad
Sci USA. 108:pp. 7058–7063. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dong Y, Geng Y, Li L, Li X, Yan X, Fang Y,
Li X, Dong S, Liu X, Li X, et al: Blocking follistatin-like 1
attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med.
212:235–252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnston I, Spence HJ, Winnie JN, McGarry
L, Vass JK, Meagher L, Stapleton G and Ozanne BW: Regulation of a
multigenic invasion programme by the transcription factor, AP-1:
Re-expression of a down-regulated gene, TSC-36, inhibits invasion.
Oncogene. 19:5348–5358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sumitomo K, Kurisaki A, Yamakawa N,
Tsuchida K, Shimizu E, Sone S and Sugino H: Expression of a
TGF-beta1 inducible gene, TSC-36, causes growth inhibition in human
lung cancer cell lines. Cancer Lett. 155:37–46. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei Y, Xu YD, Yin LM, Wang Y, Ran J, Liu
Q, Ma ZF, Liu YY and Yang YQ: Recombinant rat CC10 protein inhibits
PDGF-induced airway smooth muscle cells proliferation and
migration. Biomed Res Int. 2013:6909372013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu W, Kong H, Zeng X, Wang J, Wang Z, Yan
X, Wang Y, Xie W and Wang H: Iptakalim inhibits PDGF-BB-induced
human airway smooth muscle cells proliferation and migration. Exp
Cell Res. 336:204–210. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ning Y, Huang H, Dong Y, Sun Q, Zhang W,
Xu W and Li Q: 5-Aza-2-deoxycytidine inhibited PDGF-induced rat
airway smooth muscle cell phenotypic switching. Arch Toxicol.
87:871–881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Day RM, Lee YH, Park AM and Suzuki YJ:
Retinoic acid inhibits airway smooth muscle cell migration. Am J
Respir Cell Mol Biol. 34:695–703. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JH, Johnson PR, Roth M, Hunt NH and
Black JL: ERK activation and mitogenesis in human airway smooth
muscle cells. Am J Physiol Lung Cell Mol Physiol. 280:L1019–L1029.
2001.PubMed/NCBI
|
|
24
|
Xie S, Sukkar MB, Issa R, Oltmanns U,
Nicholson AG and Chung KF: Regulation of TGF-beta 1-induced
connective tissue growth factor expression in airway smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol. 288:L68–L76. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Burgess JK, Lee JH, Ge Q, Ramsay EE,
Poniris MH, Parmentier J, Roth M, Johnson PR, Hunt NH, Black JL and
Ammit AJ: Dual ERK and phosphatidylinositol 3-kinase pathways
control airway smooth muscle proliferation: Differences in asthma.
J Cell Physiol. 216:673–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stewart AG, Xia YC, Harris T, Royce S,
Hamilton JA and Schuliga M: Plasminogen-stimulated airway smooth
muscle cell proliferation is mediated by urokinase and annexin A2,
involving plasmin-activated cell signalling. Br J Pharmacol.
170:1421–1435. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oshima Y, Ouchi N, Sato K, Izumiya Y,
Pimentel DR and Walsh K: Follistatin-like 1 is an Akt-regulated
cardioprotective factor that is secreted by the heart. Circulation.
117:3099–3108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Movassagh H, Shan L, Halayko AJ, Roth M,
Tamm M, Chakir J and Gounni AS: Neuronal chemorepellent Semaphorin
3E inhibits human airway smooth muscle cell proliferation and
migration. J Allergy Clin Immunol. 133:560–567. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Walker TR, Moore SM, Lawson MF, Panettieri
RA Jr and Chilvers ER: Platelet-derived growth factor-BB and
thrombin activate phosphoinositide 3-kinase and protein kinase B:
Role in mediating airway smooth muscle proliferation. Mol
Pharmacol. 54:1007–1015. 1998.PubMed/NCBI
|