Introduction
Ginseng, the root of Panax ginseng, has been
used worldwide for thousands of years as a herbal drug in oriental
traditional medicine (1).
Ginsenosides (ginseng saponins), the primary active components of
Panax ginseng, have been demonstrated to have anticancer
activities, particularly ginsenoside Rh2 (GRh2) and ginsenoside Rg3
(GRg3) (2,3). GRh2 and GRg3 are protopanaxadiol
(PPD)-type ginsenosides, which have one and two glucose moieties at
the C3 hydroxyl of PPD, respectively (4). Previously, it has been reported that
GRh2 and GRg3 may inhibit growth (5), induce apoptosis (6) and restrict tumor invasion and
metastasis (7,8) in mammalian tumor cells.
Acute lymphoblastic leukemia (ALL), the most common
type of childhood malignancy, comprises a group of hematologic
neoplasms which may be regarded as clonal expansions of B- and
T-lymphocytes arrested at an immature stage of differentiation
(9,10). T-cell (T) immunophenotypes,
associated with poor outcome, have limited prognostic importance in
childhood ALL in the context of contemporary treatment (11,12).
Therefore, novel anticancer agents are required to further improve
survival rates and to avoid serious side effects. GRh2 and GRg3 may
be novel natural products for ALL therapy. However, the underlying
mechanism of GRh2- and GRg3-induced cell death in human T-ALL
Jurkat cells remains unclear.
Apoptosis is a process of genetically programmed
cell death triggered by biological and physical signals, including
chemical reagents (13,14). At present, two major signaling
pathways exist to induce apoptosis: Intrinsic-mitochondrial and
extrinsic-death receptor (15).
The mitochondrial pathway involves the regulation of apoptosis by
mitochondria and is characterized by the release of mitochondrial
intermembrane space proteins (16). Reactive oxygen species (ROS)
primarily generate inside mitochondria, and excess ROS results in
dissipation of the mitochondrial membrane potential (MMP), leading
to the release of cytochrome c and the subsequent engagement of the
Apaf-1-pro-caspase-9 apoptosome complex, which activates downstream
caspases (17,18). In addition, the B-cell lymphoma 2
(Bcl-2) family of proteins regulate permeabilization of the
mitochondrial outer membrane and cytochrome c release (19). Bcl-2 and Bcl-2 X-associated protein
(Bax) have been identified as primary regulators in mitochondrial
control during apoptosis (20).
The present study investigated the anticancer
properties of GRh2 and GRg3 in Jurkat cells. Cell viability,
nuclear morphology and apoptotic levels were examined to evaluate
the cytotoxic effects of GRh2 and GRg3. Mitochondrial ROS
generation, MMP and mitochondria-associated apoptotic proteins were
determined to examine the underlying molecular mechanisms of GRh2-
and GRg3-induced cell death in human acute leukemia Jurkat
cells.
Materials and methods
Materials and cell culture
GRh2 and GRg3 were purchased from Beina Chuanglian
Biotechnology Institute (Beijing, China). A Cell Counting kit-8
(CCK-8) was obtained from Dojindo Molecular Technologies, Inc.
(Kumamoto, Japan). Hoechst 33342 was purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). Annexin V-allophycocyanin (APC)
and 7-amino-actinomycin D (7-AAD) were obtained from BD Pharmingen
(San Diego, CA, USA). MitoSOX™ Red reagent, Roswell Park Memorial
Institute (RPMI) 1640 medium and fetal bovine serum (FBS) were
purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
MitoTEMPO was obtained from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). The primary antibodies against cleaved caspase-9 (9501),
cleaved caspase-3 (9665), Bcl-2 (4223), Bax (5023), β-actin (4970)
and secondary horseradish peroxidase (HRP)-labeled goat-anti-rabbit
antibodies (7074) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA).
The human T-ALL cell line (Jurkat cells) was
purchased from the Cell Bank of Chinese Academic of Science
(Shanghai, China), and cultured in RPMI medium 1640 supplemented
with 10% FBS at 37°C in a 95% air and 5% CO2
incubator.
Cell viability assay
Jurkat cells (5×105 cells/ml) were plated
on a 96-well (100 µl/well) microplate and treated with 15, 30, 45
or 60 µM GRh2 or GRg3. Cell viability was measured by CCK-8
according to the manufacturer's protocol. Following treatment, 10
µl CCK-8 solution was added, and cells were incubated for 4 h at
37°C. The absorbance in each well was measured at a wavelength of
450 nm using an automated ELISA reader (Tecan Austria GmbH,
Salzburg, Austria). IC50 values were calculated using
GraphPad Prism software version 5 (GraphPad Software, Inc., La
Jolla, CA, USA) from CCK-8 assay data after 24 h.
Nuclear staining with hoechst
33342
Apoptotic nuclei were observed by chromatin staining
with Hoechst 33342. Jurkat cells (5×105 cells/ml) were
cultured in a 12-well plate and treated with 35 µM GRh2 or GRg3.
After a 24 h incubation, the cells were washed with PBS three
times, fixed with methanol acetic acid for 10 min and exposed to 1
mg/ml Hoechst 33342 at room temperature in the dark for 3 min. The
nuclear morphology of Jurkat cells was examined under UV
illumination with a fluorescence microscope (Olympus Corporation,
Tokyo, Japan).
Annexin V/7-AAD flow cytometry
assay
Jurkat cells were seeded into 12-well plates at a
density of 5×105 cells/ml and treated with 35 µM GRh2 or
GRg3. After a 24 h incubation, cells were washed twice with PBS and
resuspended in 500 µl binding buffer (BD Pharmingen). Annexin V-APC
and 7-AAD were added away from light for 15 min at room
temperature. The cells were analyzed by flow cytometry (FACScan; BD
Biosciences, San Jose, CA, USA) within 1 h. Cells in early
apoptosis are Annexin V-APC-positive and 7-AAD-negative.
Measurement of mitochondrial ROS
generation
Mitochondrial ROS levels were measured using MitoSOX
Red reagent. Jurkat cells (5×105 cells/ml) were seeded
into 12-well plates and treated with GRh2 or GRg3 in the presence
or absence of 50 µM mitoTEMPO, a specific mitochondrial ROS
inhibitor. Following a 24 h incubation, cells were collected and
stained with MitoSOX Red, and incubated at 37°C in the dark for 30
min. MitoSOX Red fluorescence was observed under a fluorescence
microscope and measured by a FACScan™ flow
cytometer.
Measurement of MMP
The JC-1 fluorescent probe (Sigma-Aldrich; Merck
KGaA) was used to detect mitochondrial depolarization during the
early stages of apoptosis. Jurkat cells received either single or a
combination treatment, as previously described, for an incubation
period of 24 h. The cells were stained with JC-1 in the dark for 30
min at 37°C and washed twice with PBS. JC-1 fluorescence was
measured by a FACScan flow cytometer within 1 h.
Western blot analysis
Jurkat cells were cultured in 6-well plates and
treated with 35 µM GRh2 or GRg3 for 24 h. Whole-cell extracts were
lysed using radioimmunoprecipitation assay buffer (Sigma-Aldrich;
Merck KGaA). The supernatant was collected after centrifugation at
15,000 × g for 15 min at 4°C, and heated to 100°C for 5 min and
placed briefly on ice. A total of 20 µl supernatant was separated
by 12% SDS-PAGE. Following this, protein samples were
electrotransferred onto polyvinylidene fluoride membranes. The
membranes were blocked with 5% non-fat dry milk in 1X PBST buffer
(0.1% Tween-20 in PBS) for 1 h at room temperature and incubated in
PBST overnight at 4°C with the appropriate primary antibody. The
membranes were washed with PBST, and incubated in PBST for 1 h at
room temperature with the secondary HRP-conjugated antibody. The
immunoreactive bands were visualized by using an ECL kit (32106;
Thermo Fisher Scientific, Inc.). β-actin served as a loading
control.
Statistical analysis
All experiments were performed in triplicate and
data are expressed as the mean ± standard error of the mean.
Statistical significance was determined by one-way analysis of
variance followed by a multiple comparisons test with a Bonferroni
adjustment. The analysis was performed using GraphPad Prism
software version 5.03. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of GRh2 and GRg3 on cell
proliferation
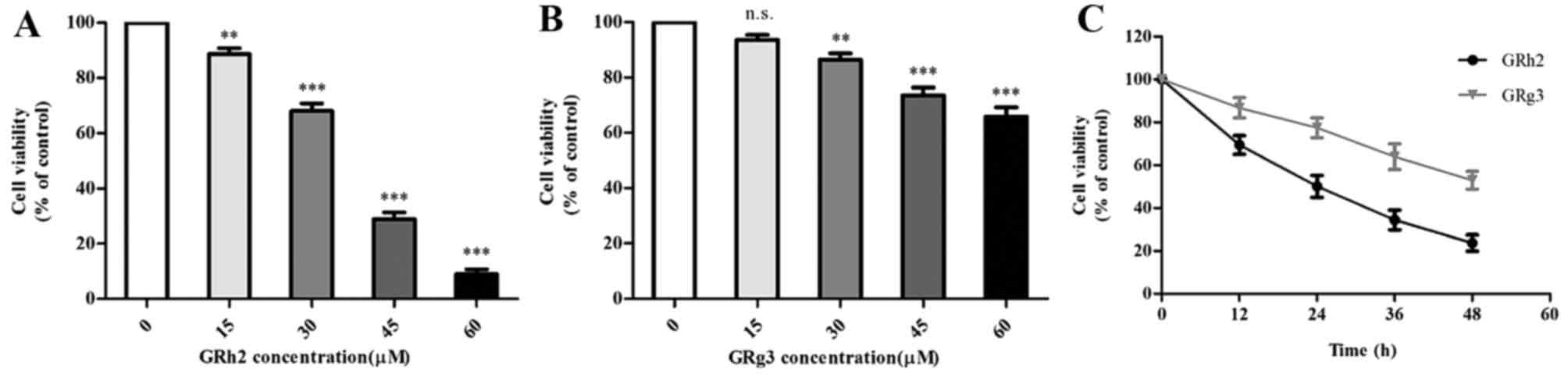
The effect of GRh2 and GRg3 on cell viability in
human ALL cells was assessed by CCK-8 assay. Jurkat cells were
exposed to 0, 15, 30, 45 or 60 µM of GRh2 or GRg3 for 24 h. GRh2
and GRg3 treatment resulted in a dose-dependent decrease in cell
viability with IC50 values of ~35 µM (Fig. 1A) and 90 µM (Fig. 1B), respectively. Jurkat cells were
treated with 35 µM GRh2 or GRg3 for 12, 24, 36 and 48 h. As
presented in Fig. 1C, the survival
of Jurkat cells decreased following GRh2 and GRg3 treatment in a
time-dependent manner. Collectively, these results indicated that
GRh2 and GRg3 may inhibit proliferation of Jurkat cells, and that
GRh2 has a more significant growth-inhibitory effect than GRg3.
GRh2 and GRg3 induce apoptosis in
Jurkat cells
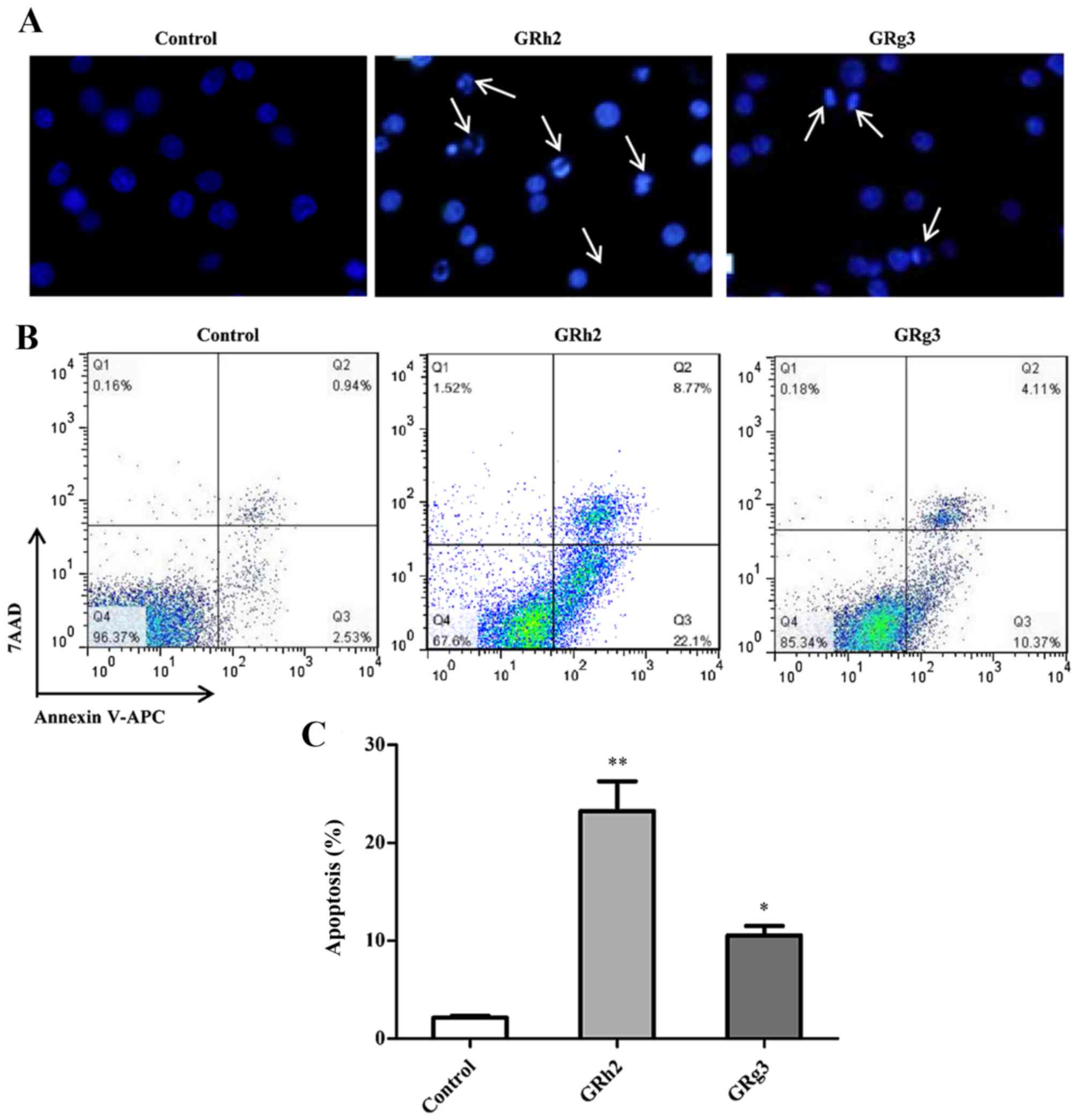
To investigate the cytotoxic effect of GRh2 and
GRg3, the nuclear morphology of dying cells were examined with
Hoechst 33342 staining. As presented in Fig. 2A, following treatment with 35 µM
GRh2 or GRg3 for 24 h, Jurkat cells exhibited condensed and
fragmented nuclei, regarded as a morphological symbol of apoptosis.
Nuclear condensation and apoptotic bodies were increased in
GRh2-treated cells compared with GRg3-treated cells.
Apoptotic cells induced by GRh2 and GRg3 treatment
was assessed using Annexin V-APC and 7-AAD double staining. The
results indicated that the population of Annexin V+ and
7-AAD-apoptotic cells was increased in the GRh2- and GRg3-treated
groups compared with the control group (Fig. 2B). Additionally, the percentage of
early apoptotic cells was 23.23±3.06% in the GRh2-treated group and
10.53±0.98% in the GRg3-treated group (Fig. 2C). These findings suggested that
GRh2 and GRg3 may induce apoptotic cell death in Jurkat cells, and
that GRh2 has greater cytotoxicity than GRg3.
Mitochondrial ROS is involved in GRh2-
and GRg3-induced cytotoxicity
Previous studies have reported that mitochondria are
the major site of ROS production in mammalian cells, but are major
targets of detrimental effects (17,21).
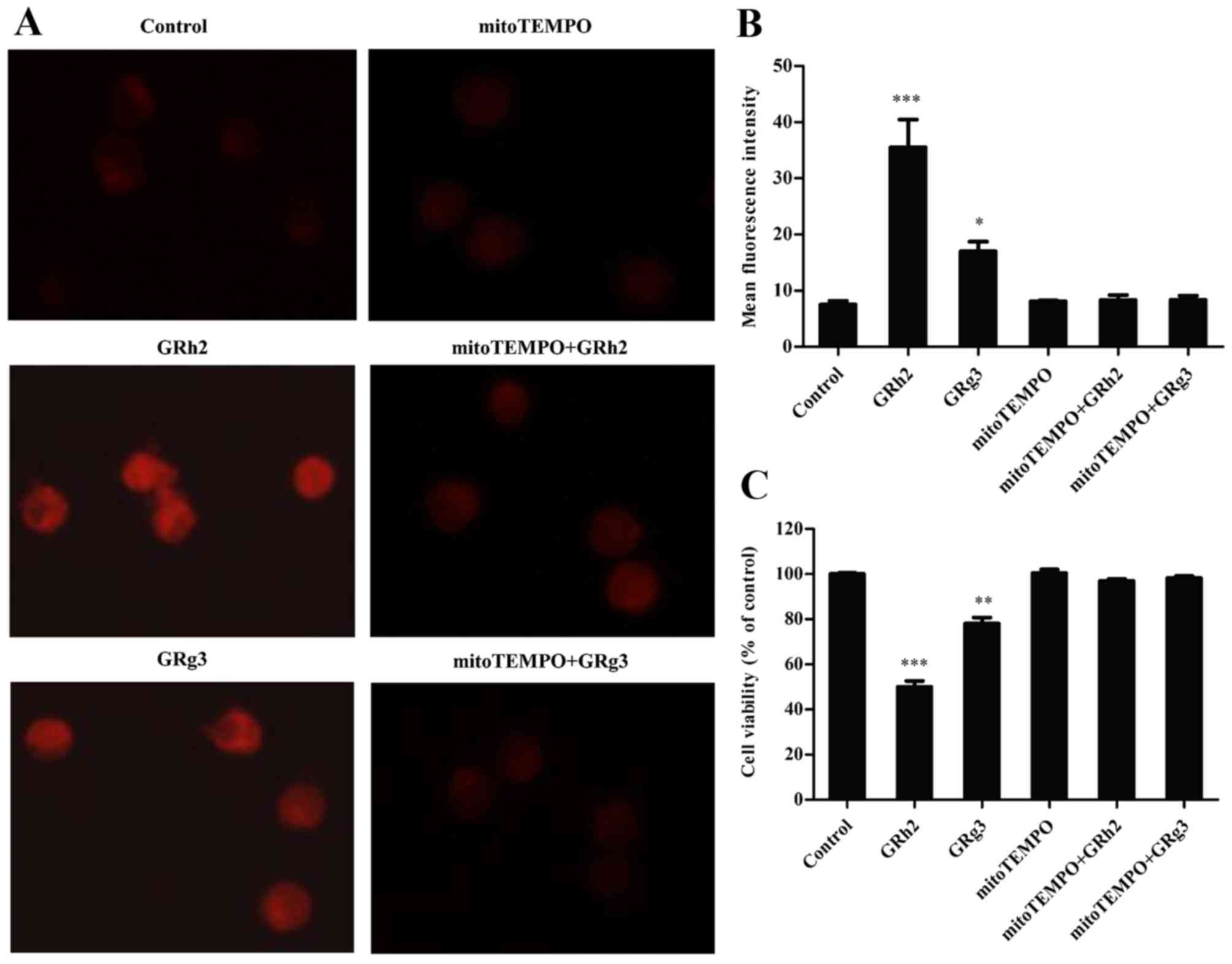
MitoTEMPO, a specific mitochondrial ROS scavenger, was added to
investigate generation of mitochondrial ROS in GRh2- and
GRg3-treated Jurkat cells. The results indicated that the red
fluorescence intensity was clearly increased in the GRh2- and
GRg3-treated groups, and markedly attenuated following concurrent
treatment with mitoTEMPO (Fig.
3A). As presented in Fig. 3B,
GRh2 and GRg3 significantly increased mitochondrial ROS levels, and
mitoTEMPO almost completely blocked GRh2- and GRg3-induced
mitochondrial ROS generation. In addition, GRh2 induced generation
of more mitochondrial ROS than GRg3 in Jurkat cells. Following
this, whether mitochondrial ROS participates in GRh2- and
GRg3-induced cytotoxicity in Jurkat cells was investigated. GRh2
was more effective than GRg3 on decreasing cell viability, whereas
concurrent treatment with mitoTEMPO markedly attenuated GRh2- and
GRg3-induced cell inhibition (Fig.
3C). These results suggested that GRh2 is more potent than GRg3
in inhibiting cell proliferation by mitochondrial ROS
generation.
Mitochondrial ROS contributes to
dissipation of MMP in GRh2- and GRg3-treated Jurkat cells
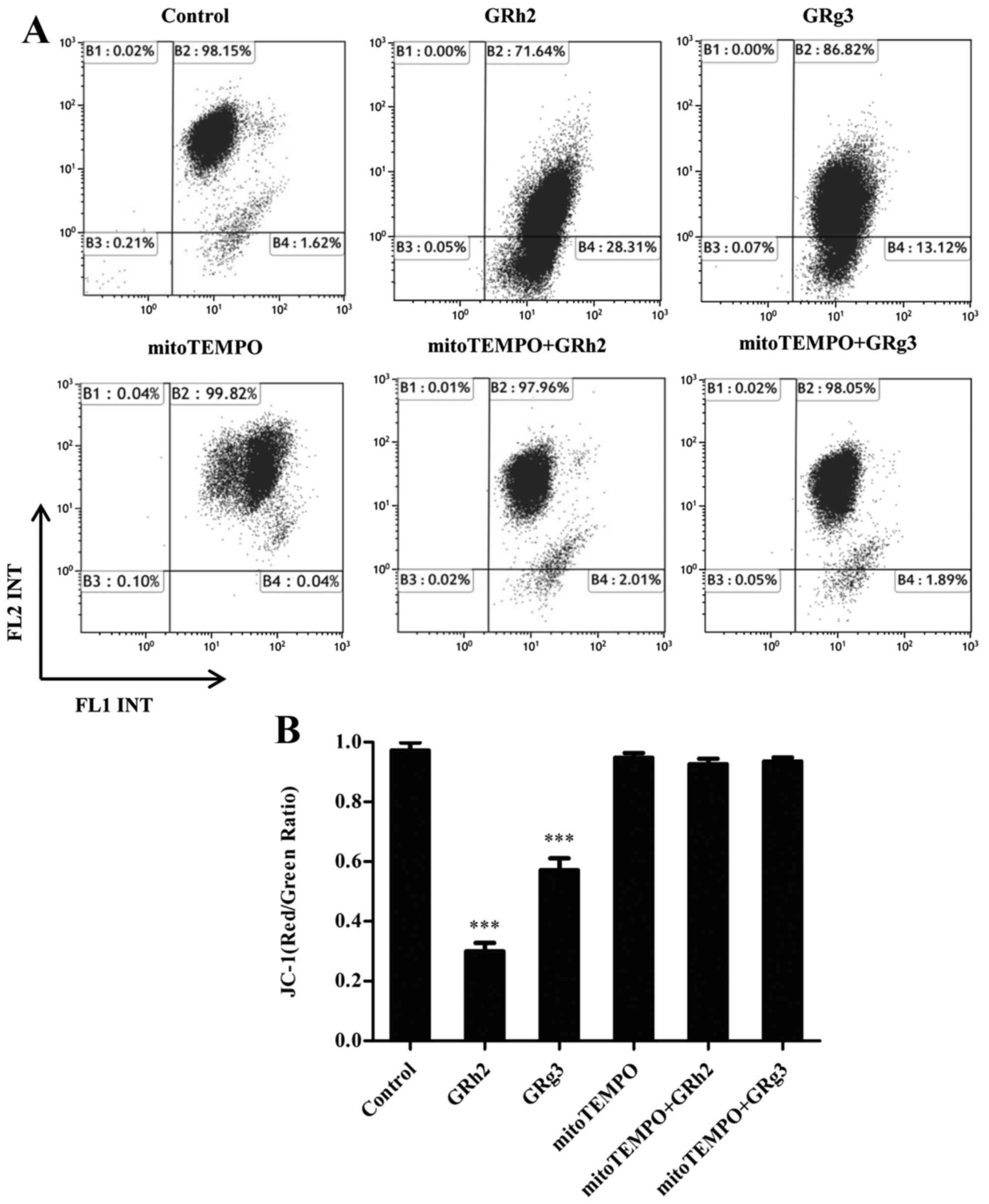
To demonstrate the effect of mitochondrial ROS on
MMP in GRh2- and GRg3-treated Jurkat cells, MMP levels were
examined using a JC-1 sensitive fluorescent probe by flow
cytometry. The results revealed that the ratio of JC-1 (red:green)
was significantly decreased in cells treated with GRh2 compared
with those treated with GRg3. However, concurrent treatment with
mitoTEMPO attenuated the loss of MMP in GRh2- and GRg3-treated
Jurkat cells (Fig. 4A and B).
These results indicated that accumulation of mitochondrial ROS is
more potent than GRg3 in inducing dissipation of MMP in Jurkat
cells.
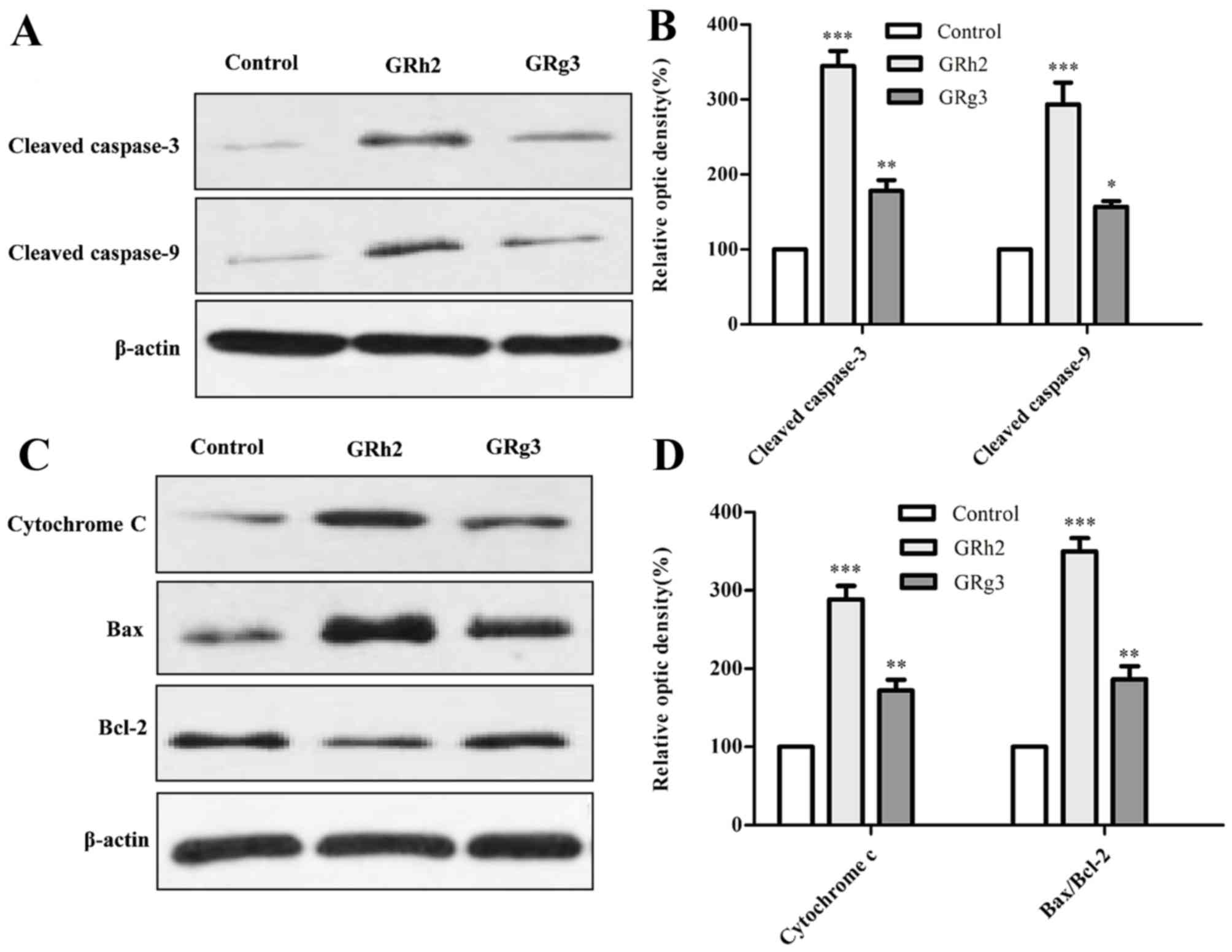
GRh2 and GRg3 induce expression of
apoptosis-related proteins in Jurkat cells
To investigate the involvement of the
mitochondrial-related pathway in GRh2- and GRg3-induced apoptosis,
the expression levels of numerous mitochondrial-associated
apoptosis proteins were examined by western blot analysis. It is
understood that caspase and Bcl-2 family members serve critical
roles in mitochondrial-associated apoptosis (22). The present study demonstrated that
protein expression levels of cleaved-caspase-3 and −9 were
significantly increased in cells treated with GRh2 compared with
cells treated with GRg3 (Fig. 5A and
B). As presented in Fig. 5C and
D, the ratio of Bax to Bcl-2 and cytochrome c was significantly
increased in the GRh2-treated group compared with the GRg3-treated
group.
Discussion
ALL is the most prevalent type of childhood
malignancy, and T-ALL is associated with poor prognosis (11). To increase survival rates and
improve quality of life, novel natural medicines are required to
treat T-ALL. GRh2 and GRg3, extracted from the root of the Panax
ginseng, are recognized as major active anticancer saponins in
ginsenosides (23). GRg3 may be
metabolized to GRh2 by human fecal microflora (24). It has been reported that GRh2 and
GRg3 have anticancer effects on numerous strains of human tumor
cells (6,25,26),
and GRh2 has a more potent anticancer activity than GRg3 (6,27,28).
The present study investigated the underlying mechanisms of GRh2-
and GRg3-induced toxicity, and examined whether mitochondrial ROS
contributes to apoptosis via mitochondrial damage in Jurkat cells.
These findings provide a potential strategy for T-ALL therapy.
Previous studies have demonstrated that GRh2 and
GRg3 are extracted from ginsenosides and have anticancer activities
(5,7,29).
The present study demonstrated that GRh2 and GRg3 inhibited cell
growth in a dose- and time-dependent manner, and GRh2 was
significantly more potent at inhibiting Jurkat proliferation than
GRg3. Therefore, GRh2 and GRg3 may have apoptosis-inducing
activities in Jurkat cells. This hypothesis was supported by the
generation of nuclear condensation and apoptotic bodies induced by
GRh2 and GRg3 treatment. Furthermore, GRh2 and GRg3 treatment
significantly increased the percentage of apoptotic cells; GRh2 to
a greater extent. Collectively, these findings revealed that GRh2
and GRg3 induce apoptosis in the T-ALL cell line, and that GRh2 has
greater cytotoxicity than GRg3.
Mitochondria are key regulators of apoptotic cell
death. The mitochondrial pathway of apoptosis is activated in
response to a number of stress conditions including DNA damage and
oxidative stress, which is a common cause of tumor cell death
induced by chemotherapeutic agents (30,31).
ROS is primarily generated within the mitochondrial electron
transport chain during typical cellular metabolism. However,
various stimuli, including tumor necrosis factor-α, Fas ligand and
growth factors, rapidly provoke ROS accumulation in target cells
(32,33). In addition, excess ROS induces
mitochondrial membrane permeabilization, which, in turn, results in
the loss of MMP by activating mitochondrial permeability transition
(34). In the present study,
mitoTEMPO, a specific mitochondrial ROS inhibitor, was used to
assess the role of mitochondrial ROS in GRh2- and GRg3-treated
Jurkat cells. The results revealed that GRh2 induced increased
generation of mitochondrial ROS compared with GRg3 in Jurkat cells;
however, this effect was ameliorated by subsequent treatment with
mitoTEMPO. Furthermore, excess mitochondrial ROS induced by GRh2
was more potent than GRg3 in inhibiting cell proliferation and
reducing MMP.
It has been reported that numerous anticancer agents
may trigger the release of mitochondrial-associated apoptotic
proteins and induce cell death by promoting the intrinsic apoptotic
signaling pathway (18,35). It is understood that caspases,
another family of kinases, serve an important role in the
regulation of cell apoptosis. Activation of caspase-3 and −9
stimulates mitochondrial cell death signals (36). In addition, gene members of the
Bcl-2 family, particularly Bax (an pro-apoptotic gene) and Bcl-2
(an anti-apoptotic gene), are key mediators in regulating the
mitochondrial cell death signaling pathway (37). The present study demonstrated that
expression levels of apoptosis-associated proteins were
significantly increased in Jurkat cells treated with GRh2 compared
with GRg3. These findings supported that GRh2 and GRg3 induce
apoptosis via mitochondria-dependent signaling pathways, and that
GRh2 is more potent than GRg3 in promoting apoptosis of Jurkat
cells.
In conclusion, the current study revealed the
underlying mechanisms of GRh2- and GRg3-induced cell death in
Jurkat cells. GRh2 and GRg3 may inhibit growth and induce
apoptosis, and GRh2 has greater cytotoxicity than GRg3.
Furthermore, GRh2 inhibits proliferation and induces apoptosis more
effectively than GRg3 by stimulating the generation of
mitochondrial ROS and promoting the loss of MMP in Jurkat cells.
Collectively, these results suggested that GRh2 and GRg3 may be
used as potential chemopreventive agents for the treatment of
ALL.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81600126, 81570140,
31471722 and 31671851), the National High Technology Research and
Development Program of China (grant no. 2013AA102106) and the
Program for Changjiang Scholars and Innovative Research Team in
University (grant no. IRT15R49) and National Ministry of Science
and Technology (grant no. 2016YFD0400505).
References
|
1
|
Attele AS, Wu JA and Yuan CS: Ginseng
pharmacology: Multiple constituents and multiple actions. Biochem
Pharmacol. 58:1685–1693. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bak MJ, Jeong WS and Kim KB: Detoxifying
effect of fermented black ginseng on
H2O2-induced oxidative stress in HepG2 cells.
Int J Mol Med. 34:1516–1522. 2014.PubMed/NCBI
|
|
3
|
Wang JH, Nao JF, Zhang M and He P:
20(s)-ginsenoside Rg3 promotes apoptosis in human ovarian cancer
HO-8910 cells through PI3K/Akt and XIAP pathways. Tumour Biol.
35:11985–11994. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang P, Wei Y, Fan Y, Liu Q, Wei W, Yang
C, Zhang L, Zhao G, Yue J, Yan X and Zhou Z: Production of
bioactive ginsenosides Rh2 and Rg3 by metabolically engineered
yeasts. Metab Eng. 29:97–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim HS, Lee EH, Ko SR, Choi KJ, Park JH
and Im DS: Effects of ginsenosides Rg3 and Rh2 on the proliferation
of prostate cancer cells. Arch Pharm Res. 27:429–435. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park HM, Kim SJ, Kim JS and Kang HS:
Reactive oxygen species mediated ginsenoside Rg3- and Rh2-induced
apoptosis in hepatoma cells through mitochondrial signaling
pathways. Food Chem Toxicol. 50:2736–2741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang XP, Tang GD, Fang CY, Liang ZH and
Zhang LY: Effects of ginsenoside Rh2 on growth and migration of
pancreatic cancer cells. World J Gastroenterol. 19:1582–1592. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim JW, Jung SY, Kwon YH, Lee JH, Lee YM,
Lee BY and Kwon SM: Ginsenoside Rg3 attenuates tumor angiogenesis
via inhibiting bioactivities of endothelial progenitor cells.
Cancer Biol Ther. 13:504–515. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Richardson RB: Promotional etiology for
common childhood acute lymphoblastic leukemia: The infective
lymphoid recovery hypothesis. Leuk Res. 35:1425–1431. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vicente C and Cools J: The origin of
relapse In Pediatric T-Cell acute lymphoblastic leukemia.
Haematologica. 100:1373–1375. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pui CH, Robison LL and Look AT: Acute
lymphoblastic leukemia. Lancet. 371:1030–1043. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meleshko AN, Belevtsev MV, Savitskaja TV
and Potapnev MP: The incidence of T-cell receptor gene
rearrangements in childhood B-lineage acute lymphoblastic leukemia
is related to immunophenotype and fusion oncogene expression. Leuk
Res. 30:795–800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reed JC: Apoptosis-regulating proteins as
targets for drug discovery. Trends Mol Med. 7:314–319. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chakraborty JB, Oakley F and Walsh MJ:
Mechanisms and biomarkers of apoptosis in liver disease and
fibrosis. Int J Hepatol. 2012:6489152012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marchi S, Giorgi C, Suski JM, Agnoletto C,
Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti
F, et al: Mitochondria-ros crosstalk in the control of cell death
and aging. J Signal Transduct. 2012:3296352012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Indran IR, Hande MP and Pervaiz S: hTERT
overexpression alleviates intracellular ROS production, improves
mitochondrial function, and inhibits ROS-mediated apoptosis in
cancer cells. Cancer Res. 71:266–276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng EH, Wei MC, Weiler S, Flavell RA,
Mak TW, Lindsten T and Korsmeyer SJ: Bcl-2, Bcl-X(L) sequester BH3
domain-only molecule spreventing BAX-and BAK-mediated mitochondrial
apoptosis. Mol Cell. 8:705–711. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kroemer G: The proto-oncogene Bcl-2 and
its role in regulating apoptosis. Nat Med. 3:614–620. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi JS, Chun KS, Kundu J and Kundu JK:
Biochemical basis of cancer chemoprevention and/or chemotherapy
with ginsenosides (Review). Int J Mol Med. 32:1227–1238.
2013.PubMed/NCBI
|
|
24
|
Bae EA, Han MJ, Kim EJ and Kim DH:
Transformation of ginseng saponins to ginsenoside Rh2 by acids and
human intestinal bacteria and biological activities of their
transformants. Arch Pharm Res. 27:61–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Liu T, Zhao L, Chen W, Hou H, Ye Z
and Li X: Ginsenoside 20(S) Rg3 inhibits the Warburg effect through
STAT3 pathways in ovarian cancer cells. Int J Oncol. 46:775–781.
2015.PubMed/NCBI
|
|
26
|
Guo XX, Guo Q, Li Y, Lee SK, Wei XN and
Jin YH: Ginsenoside Rh2 induces human hepatoma cell apoptosis via
bax/bak triggered cytochrome c release and caspase-9/caspase-8
activation. Int J Mol Sci. 13:15523–15535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang W, Wang H, Rayburn ER, Zhao Y, Hill
DL and Zhang R: 20(S)-25-methoxyldammarane-3beta, 12beta, 20-triol,
a novel natural product for prostate cancer therapy: Activity in
vitro and in vivo and mechanisms of action. Br J Cancer.
98:792–802. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cheong JH, Kim H, Hong MJ, Yang MH, Kim
JW, Yoo H, Yang H, Park JH, Sung SH, Kim HP and Kim J:
Stereoisomer-specific anticancer activities of ginsenoside Rg3 and
Rh2 in HepG2 Cells: Disparity in cytotoxicity and
autophagy-inducing effects due to 20(S)-epimers. Biol Pharm Bull.
38:102–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu N, Wu GC, Hu R, Li M and Feng H:
Ginsenoside Rh2 inhibits glioma cell proliferation by targeting
microRNA-128. Acta Pharmacol Sin. 32:345–353. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Favaloro B, Allocati N, Graziano V, Di
Ilio C and De Laurenzi V: Role of apoptosis in disease. Aging
(Albany NY). 4:330–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Green DR and Kroemer G: Pharmacological
manipulation of cell death: Clinical applications in sight? J Clin
Invest. 115:2610–2617. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Apel K and Hirt H: Reactive oxygen
species: Metabolism, oxidative stress, and signal transduction.
Annu Rev Plant Biol. 55:373–399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fujisawa S, Atsumi T, Ishihara M and
Kadoma Y: Cytotoxicity, ROS-generation activity and
radical-scavenging activity of curcumin and related compounds.
Anticancer Res. 24:563–569. 2004.PubMed/NCBI
|
|
34
|
Gogvadze V, Norberg E, Orrenius S and
Zhivotovsky B: Involvement of Ca2+ and ROS in alpha-tocopheryl
succinate-induced mitochondrial permeabilization. Int J Cancer.
127:1823–1832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yadav N, Kumar S, Marlowe T, Chaudhary AK,
Kumar R, Wang J, O'Malley J, Boland PM, Jayanthi S, Kumar TK, et
al: Oxidative phosphorylation-dependent regulation of cancer cell
apoptosis in response to anticancer agents. Cell Death Dis.
6:e19692015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ola MS, Nawaz M and Ahsan H: Role of bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou J, Zhang S, Ong CN and Shen HM:
Critical role of pro-apoptotic bcl-2 family members in
andrographolide-induced apoptosis in human cancer cells. Biochem
Pharmacol. 72:132–144. 2006. View Article : Google Scholar : PubMed/NCBI
|