Introduction
The prevalence of congenital anomalies of the kidney
and urinary tract (CAKUT) is 17.7:1,000 live births (1). CAKUT constitute the major cause of
chronic renal failure in childhood (2) and can occur in isolation
(nonsyndromic CAKUT), or in association with other organ system
malformations (2). While most
cases are sporadic, a familial aggregation has been described in up
to 15% of cases, pointing to a genetic contribution. In a recent
study, 37 different heterozygous mutations (33 novel mutations) in
12 of 17 known CAKUT-causing genes were identified in 6.3% of 650
unrelated families with CAKUT. It thus appears that CAKUT is a
genetically heterogeneous disease with diverse clinical phenotypes
(3).
Submicroscopic chromosomal imbalances (deletions or
duplications), known as copy-number variations (CNVs), have been
used to identify novel genomic regions associated with CAKUT. CNVs
were detected in 10.1% of 178 patients with CAKUT and were found to
be inherited in 90% (9/10) of the families in which they were
identified (4).
Transposable elements (TEs) can sculpt genome
structure, with a profound contribution to genetic variation,
through single nucleotide variations, CNVs (indels) or larger
structural variations (5). Such
TE-mediated rearrangements can be either active, as a direct result
of retrotransposition events, or passive due to their repetitive
nature (6,7), causing diseases in some cases
(7).
The aim of the present study was to investigate CNVs
in a series of children with nonsyndromic upper and lower urinary
tract anomalies, and to search for evidence of a possible causative
role of a transposable element-associated genomic rearrangement.
The novelty of the study is that it focused on first and second
degree relatives with the same or similar CAKUT phenotype.
Materials and methods
Patients
Three unrelated families, each with at least two
members diagnosed with nonsyndromic CAKUT in the Pediatric
Nephrology Department of the University Hospital of Ioannina
(Greece) were invited to participate in the study. The first family
consisted of female monozygotic twins with CAKUT. One twin had a
bilateral completely duplex collecting system, bilateral
vesicoureteral reflux (VUR) grade III–IV in the lower pole, and
right renal hypodysplasia (RHD) (relative function 38%). The second
twin had a left incompletely duplicated system (the two ureters
joined just before entering the bladder), bilateral VUR grade
II–III, and left RHD (relative function 38%). The second family
consisted of two male siblings one of which had bilateral VUR grade
III and left RHD (relative function 26%) and the second bilateral
VUR grade III–IV. In the third family three males, second cousins,
had CAKUT. One had bilateral VUR grade V (normal urethra) and right
RHD (relative function 28%), one had bilateral VUR grade III, and
the third had severe ureterovesical junction obstruction (UVJO),
requiring surgical intervention. The patients of the third family
were members of a cohort of gypsies with a high rate of inbreeding
(first cousin marriages).
All the patients had normal laboratory findings,
apart from the 15-year-old patient with UVJO from the gypsy family
who had persistent hypomagnesemia (serum magnesium levels:
1.02–1.17 mEq/l, normal range 1.3–2.1 mEq/l), hypermagnesuria
[fractional magnesium excretion (FeMg): 5.0–5.5%, and hypocalciuria
(fractional calcium excretion: 0.18–0.2%) [in patients with
hypomagnesemia, hypermagnesuria has been defined as FeMg >4
(8), and hypocalciuria as
fractional calcium excretion <1%) (9)]. Serum creatinine, calcium, phosphate,
albumin, parathyroid hormone, 25-hydroxyvitamin D levels, and
electrolytes were within normal limits.
The CAKUT in these children were investigated and
characterized by urinary tract ultrasound, voiding
cystourethrography, dimercaptosuccinic acid (DMSA) scan, and
technetium-99 m mercaptoacetyltriglycine (MAG3) scan, as indicated.
VUR was classified into grades I–V according to the International
Reflux Classification (10). RHD
was diagnosed in kidneys with reduced renal length and regular
outline, with or without cortical hyperechogenicity and loss of
corticomedullary differentiation on ultrasound, and with persistent
(for ≥6 months), general reduction in 99mTc-DMSA uptake (relative
function <45%).
CNV detection
For the genetic study, peripheral venous blood was
collected for genomic DNA extraction, according to the
manufacturer's protocol using QIAamp DNA Mini Kit (Qiagen, Hilden,
Germany), at the time of routine blood testing of the patients. DNA
samples were investigated for the presence of CNVs, using
array-comparative genomic hybridization (CGH). Array-CGH was
performed by Cytochip ISCA array (Blue Gnome-version 1.0) with
180,000 oligos, analyzed using buildGRCh37 (hg19).
The following resources were used for the analysis
of the cases: Ensemble (http://www.ensembl.org), Database of Genomic Variants
(http://projects.tcag.ca/variation/),
UCSC Genome Bioinformatics Site (http://genome.ucsc.edu/), and Online Mendelian
Inheritance In Man (http://ncbi.nlm.nih.gov/omim).
CNVs >0.2 Mb were considered significant. CNVs
that did not contain genes, or were <0.2 Mb (unless containing a
gene of known pathogenic significance) were considered of normal
constitution.
Bioinformatics
TE sequences coverage was measured using the in
silico program RepeatMasker (http://www.repeatmasker.org/), and UCSC genome browser
to extract nucleotide sequences from the human genome (GRCh38).
Sequence features were analyzed and visualized in the UCSC browser.
Nucleotide sequence alignments were performed using the Blast
algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Breakpoint
regions (200 bp stretches surrounding the breakpoints) were
analyzed using the Blast algorithm for the identification of
sequence homology.
Results
Of the 7 study children with familial nonsyndromic
CAKUT, 5 had a normal constitution of CNVs (well-established
polymorphism variants).
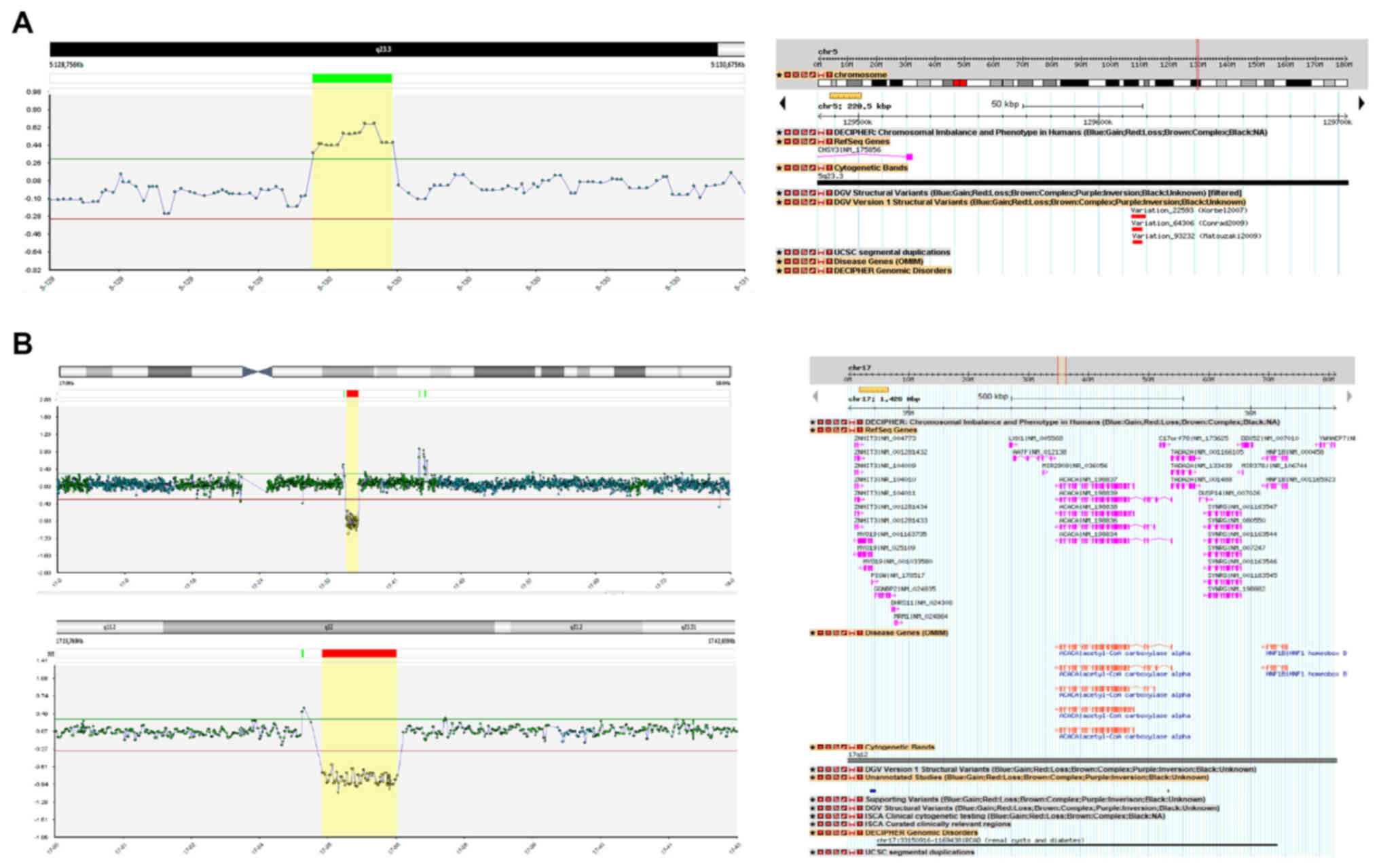
One sibling from the second family, with bilateral
VUR grade III and left RHD had a 0.2 Mb duplication on the long arm
of chromosome 5 (5q-arm) at chromosomal band 5q23.3. The
duplication partially disrupts the Chondroitin Sulfate Sulthase 3
(CHY3) gene (Fig. 1A). His
brother, with a similar CAKUT (bilateral VUR grade III–IV), did not
harbor the same CNV.
One male teenager, from the gypsy family, with UVJO,
was found to have a deletion approximately 1.4 Mb in size on the
long arm of chromosome 17 (17q-arm) at chromosomal band 17q12. This
deletion consists of two OMIM disease genes, the
Acetyl-CoaCarboxylase-Alpha (ACACA, OMIM#200350), and the
Hepatocyte Nuclear Factor-1-Beta (HNF1B, OMIM#189907)
(Fig. 1B).
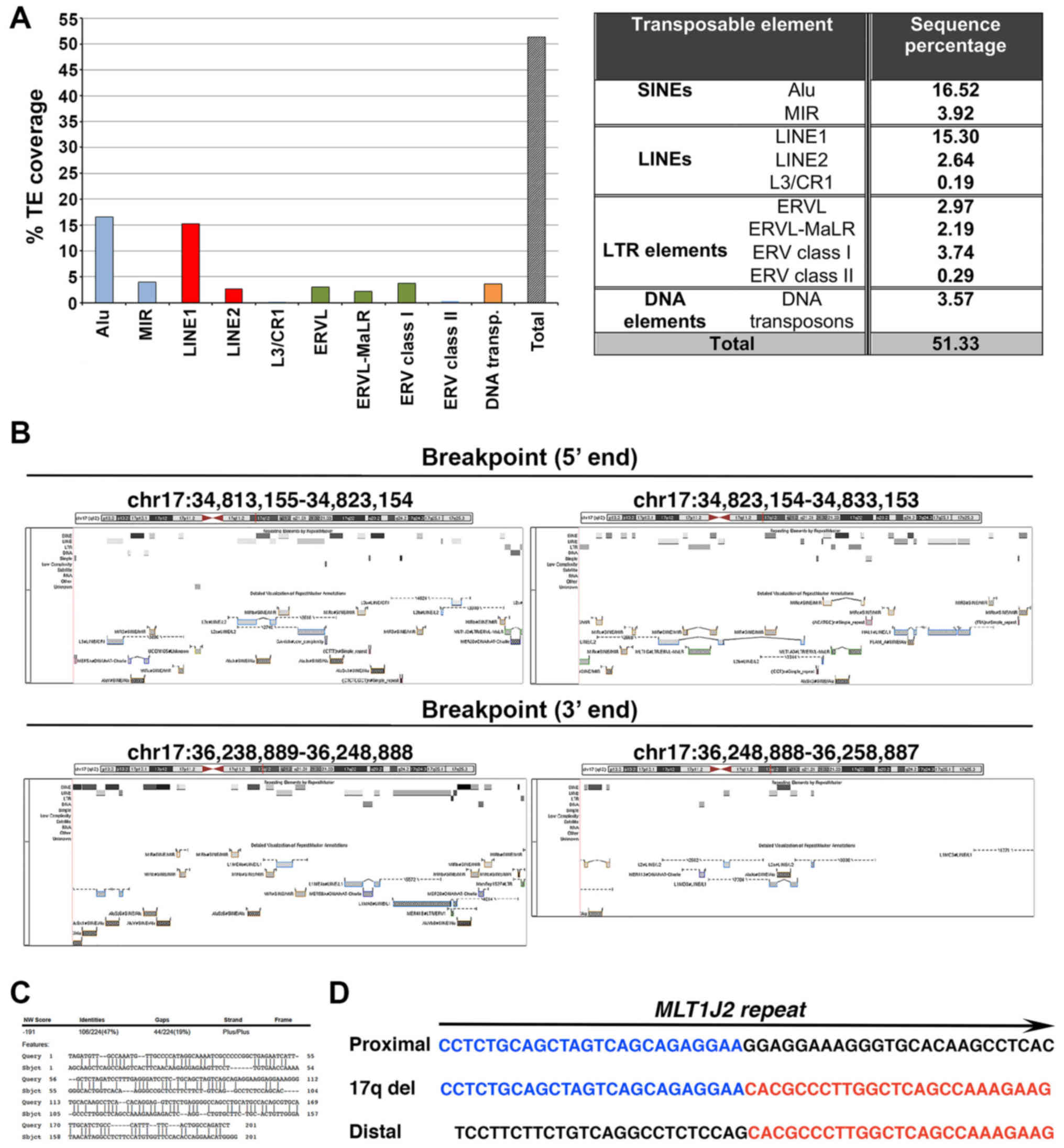
To gain insight into the possible molecular
mechanism underlying the deletion detected, we measured the load in
TE sequences within the deleted region. A higher coverage (51.33%)
was found than expected when inspecting any random genome region
(Fig. 2A). Notably, the 5′
breakpoint was mapped in a solo long terminal repeat (LTR) sequence
(MLT1J2), sharing high similarity (76%) to the solo LTR sequence
MamRep1527 located at the 3′ breakpoint region. Moreover, a
significant load in highly similar (70–89%) mammalian interspersed
repetitive (MIR) element sequences, namely MIRb and MIRc, was found
in the 20Kb-region bilateral to the 5′ and 3′ breakpoints,
amounting to 11.87 and 5.25%, respectively (Fig. 2B). Finally, a 47% nucleotide
sequence homology was identified in a stretch of 200 bp surrounding
the breakpoints, while no microhomology was found in the breakpoint
junctions (Fig. 2C and D).
CNVs were not studied in the parents of the affected
children, but all the parents were investigated by urinary tract US
and had no CAKUT phenotype.
Discussion
This study investigated the presence of CNVs in a
series of 7 children with familial nonsyndromic CAKUT. Its novelty
is the focus on first and second degree relatives with the same or
similar nonsyndromic CAKUT phenotype, and on evidence for the
causative role of a transposable element-associated genomic
rearrangement.
CNV associated with CAKUT was detected in one of the
7 patients (14%). The number of patients investigated was very
small, but the rate of CNVs was in agreement with previous studies
in which CNVs were identified in 16.6% (11) and 10.1% (4) in large numbers of patients with
nonsyndromic CAKUT (522 and 178 respectively). Sanna-Cherchi and
colleagues (11) studied patients
with congenital solitary kidney and renal hypoplasia, while Caruana
and colleagues (4) included a wide
range of CAKUT, finding a high incidence of CNVs in cases of
multicystic dysplastic kidney (MCDK) (30%), and posterior urethral
valves (24%). Similarly, Westland and colleagues (12), using CNV analysis in 80 patients
with a solitary functioning kidney, found genomic imbalances in 11
of 80 (14%). Weber and colleagues (2) reported a similar CNV frequency (10%)
in 30 children with syndromic CAKUT.
The 0.2 Mb duplication found in one sibling of the
second family is not considered causative for his CAKUT phenotype,
since it does not include any genes known to be associated with
CAKUT (Fig. 1A), and his brother
with the same CAKUT phenotype had a normal constitution of
CNVs.
One child in the gypsy family, with UVJO, had a
deletion approximately 1.4 Mb in size on the long arm of chromosome
17q12. The deletion consists of two OMIM disease genes, the
Acetyl-CoaCarboxylase-Alpha (ACACA, OMIM#200350), and the
Hepatocyte Nuclear Factor-1beta (HNF1B, OMIM#189907)
(Fig. 1B). The same CNV including
the HNF1B gene has also been reported recently by Caruana
and colleagues (4) in a female
infant with MCDK. The HNF1B gene has been associated with
renal cyst, diabetes mellitus and genital malformation (RCAD
syndrome: OMIM 137920), where a 75 bp deletion in exon 2 causes a
loss-of-function mutation (13).
HNF1B mutations are well described in patients with upper
urinary tract malformations. Nakayama and colleagues (14) identified HNF1B mutations in
5 of 50 patients (10%) with CAKUT (3 with MCDK and 2 with RHD),
where de novo heterozygous complete deletions of
HNF1B were found in patients with MCDK. CNV analysis showed
1.4 Mb deletion of chromosome 7, involving the whole HNF1B
gene with breakpoints in flanking segmental duplications. Thomas
and colleagues (15) reported that
among 50 North American Caucasian children with RHD, 4 (8%) carried
mutations in HNF1B gene. In our study, a CNV involving the
whole HNF1B gene was found in one patient with a lower
urinary tract malformation (UVJO). Considering that the HNF1B is
expressed in the ureteric bud derivatives (16,17),
this finding is not surprising. The association of HNF1B
alteration with the UVJO phenotype has not been reported as often
as its association with upper urinary tract malformations. Adalat
et al (18) reported a
mutation in the HNF1B gene in one patient with
hydronephrosis/hydroureter. Specifically, they found heterozygous
mutations in HNF1B gene in 23% of 91 cases with renal
malformation. The range of phenotypes included large bright
kidneys, MCDK, and hydronephrosis/hydroureter. One patient has been
reported with complete prune-belly syndrome associated with a
complete HNF1B gene deletion (19).
Our patient had also persistent hypomagnesemia,
hypermagnesuria and hypocalciuria. Adalat et al (18) found that 44% of the 18 HNF1B
mutation carriers had hypomagnesemia. They detected highly
conserved HNF1-recognition sites in FXYD2, and demonstrated
HNF1B-mediated transactivation of FXYD2, a gene that can
cause autosomal dominant hypomagnesemia, hypermagnesuria and
hypocalciuria when mutated (20).
Our findings, even though not novel, depict a phenotype of
HNF1B gene alternation, which includes both lower urinary
tract malformations and renal magnesium wasting. Therefore, our
patient's phenotypic features both overlap and expand on reported
cases of nonsyndromic CAKUT. Given, however, that the two other
family members (cousins) with CAKUT did not have the 17q12
deletion, we suggest that either the CAKUT in this family is
unrelated or that the UVJO and the VUR in the cousins are due to a
separate undetected genetic defect. Considering that these patients
had a highly consanguineous background, a possibly recessive cause
of CAKUT cannot be excluded. It should be pointed out that the
etiology of the majority of CAKUT cases remains unknown. The
genomic imbalance, such as copy number variants, genomic, or de
novo mutations, can explain up to one-third of all CAKUT cases.
Moreover, findings from several studies suggest a contribution of
epigenetic and environmental factors on the pathogenesis of CAKUT,
supporting the theory of its multifactorial character (21).
Congenital diseases, such as CAKUT, have
developmental origin (22), and
TEs and their control mechanisms regulate development (23). It is known that TEs are involved in
recombination events, representing a major source of structural
variation in the genomic landscape (24). Even if deletions of chromosome
17q12 spanning HNF1B gene are one of the most commonly
identified mutations associated with CAKUT (21), their molecular characterization,
and, especially, the coverage in TEs sequences within or at the
boundaries of such deletions has not been previously reported. The
novelty of our study was the bioinformatic analysis of a
CAKUT-causative 1.4 Mb deletion on the 17q-arm based on the in
silico program RepeatMasker, which effectively identifies TEs
sequences within any given genomic region. An additional advantage
of such approach comes from the use of the alignment heuristic
program cross_match for the performance of sequence comparisons,
providing a high sensitivity. Our analysis revealed: (a) a high
coverage in TEs within the 17q12-deleted region (Fig. 2A), (b) a MLT1J2 solo LTR sequence
at the 5′ breakpoint (Fig. 2B),
(c) the presence of 4.6- and 2-fold higher load of MIR sequences on
either side of the 5′ and 3′ breakpoint regions respectively,
compared to their expected genomic percentages (25) (Fig.
2B), and (d) 47% nucleotide sequence microhomology in a 200 bp
region surrounding the breakpoints (Fig. 2C). Based on these findings, it is
tempting to suggest that TEs may have served as a substrate for the
generation of the deletion. We believe that the deletion detected
may have originated from the mispairing of TE sequences, possibly
via non-allelic homologous recombination. Given that TEs can form
non B-DNA structures promoting the formation of DNA nicks, double
strand breaks or stall replication forks (26), we cannot exclude the involvement of
microhomology-mediated replication-dependent recombination
mechanisms, as previously reported in human Xq isochromosome
formation (27).
In conclusion, among 7 children with familial
nonsyndromic CAKUT, one with UVJO was found to have a significant
CNV, suggesting this genomic imbalance as causative of the anomaly,
since it includes a known CAKUT gene, HNF1B. The phenotype
of the HNF1B deletion was extended, and included both lower
urinary tract malformation and renal magnesium wasting. Moreover,
we determined the topological features, in terms of nucleotide
sequence identity, of the deleted genomic region. Overall, our
findings provide evidence of a correlation between a TE-associated
genomic rearrangement and CAKUT. Future studies will shed more
light for the role of TEs as causative factors of pathogenic
variants. Finally, CNV analysis could reveal novel causative
genomic regions in patients with CAKUT as well as in other
multifactorial diseases, and further studies in larger cohorts are
needed.
References
|
1
|
Melo BF, Aguiar MB, Bouzada MC, Aguiar RL,
Pereira AK, Paixão GM, Linhares MC, Valerio FC, Simões E Silva AC
and Oliveira EA: Early risk factors for neonatal mortality in
CAKUT: Analysis of 524 affected newborns. Pediatr Nephrol.
27:965–972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weber S, Landwehr C, Renkert M, Hoischen
A, Wühl E, Denecke J, Radlwimmer B, Haffner D, Schaefer F and Weber
RG: Mapping candidate regions and genes for congenital anomalies of
the kidneys and urinary tract (CAKUT) by array-based comparative
genomic hybridization. Nephrol Dial Transplant. 26:136–143. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hwang DY, Dworschak GC, Kohl S, Saisawat
P, Vivante A, Hilger AC, Reutter HM, Soliman NA, Bogdanovic R,
Kehinde EO, et al: Mutations in 12 known dominant disease-causing
genes clarify many congenital anomalies of the kidney and urinary
tract. Kidney Int. 85:1429–1433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caruana G, Wong MN, Walker A, Heloury Y,
Webb N, Johnstone L, James PA, Burgess T and Bertram JF:
Copy-number variation associated with congenital anomalies of the
kidney and urinary tract. Pediatr Nephrol. 30:487–495. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hancks DC and Kazazian HH Jr: Active human
retrotransposons: Variation and disease. Curr Opin Genet Dev.
22:191–203. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kidd JM, Graves T, Newman TL, Fulton R,
Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK and Eichler EE: A
human genome structural variation sequencing resource reveals
insights into mutational mechanisms. Cell. 143:837–847. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hancks DC and Kazazian HH Jr: Roles for
retrotransposon insertions in human disease. Mob DNA. 7:92016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elisaf M, Panteli K, Theodorou J and
Siamopoulos KC: Fractional excretion of magnesium in normal
subjects and in patients with hypomagnesemia. Magnes Res.
10:315–320. 1997.PubMed/NCBI
|
|
9
|
Bianchetti MG, Edefonti A and Bettinelli
A: The biochemical diagnosis of Gitelman disease and the definition
of ‘hypocalciuria’. Pediatr Nephrol. 18:409–411. 2003.PubMed/NCBI
|
|
10
|
Medical versus surgical treatment of
primary vesicoureteral reflux: Report of the International Reflux
Study Committee. Pediatrics. 67:392–400. 1981.PubMed/NCBI
|
|
11
|
Sanna-Cherchi S, Kiryluk K, Burgess KE,
Bodria M, Sampson MG, Hadley D, Nees SN, Verbitsky M, Perry BJ,
Sterken R, et al: Copy-number disorders are a common cause of
congenital kidney malformations. Am J Hum Genet. 91:987–997. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Westland R, Verbitsky M, Vukojevic K,
Perry BJ, Fasel DA, Zwijnenburg PJ, Bökenkamp A, Gille JJ,
Saraga-Babic M, Ghiggeri GM, et al: Copy number variation analysis
identifies novel CAKUT candidate genes in children with a solitary
functioning kidney. Kidney Int. 88:1402–1410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lindner TH, Njolstad PR, Horikawa Y,
Bostad L, Bell GI and Sovik O: A novel syndrome of diabetes
mellitus, renal dysfunction and genital malformation associated
with a partial deletion of the pseudo-POU domain of hepatocyte
nuclear factor-1beta. Hum Mol Genet. 8:2001–2008. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakayama M, Nozu K, Goto Y, Kamei K, Ito
S, Sato H, Emi M, Nakanishi K, Tsuchiya S and Iijima K: HNF1B
alterations associated with congenital anomalies of the kidney and
urinary tract. Pediatr Nephrol. 25:1073–1079. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thomas R, Sanna-Cherchi S, Warady BA,
Furth SL, Kaskel FJ and Gharavi AG: HNF1B and PAX2 mutations are a
common cause of renal hypodysplasia in the CKiD cohort. Pediatr
Nephrol. 26:897–903. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Coffinier C, Barra J, Babinet C and Yaniv
M: Expression of the vHNF1/HNF1beta homeoprotein gene during mouse
organogenesis. Mech Dev. 89:211–213. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kolatsi-Joannou M, Bingham C, Ellard S,
Bulman MP, Allen LI, Hattersley AT and Woolf AS: Hepatocyte nuclear
factor-1beta: A new kindred with renal cysts and diabetes and gene
expression in normal human development. J Am Soc Nephrol.
12:2175–2180. 2001.PubMed/NCBI
|
|
18
|
Adalat S, Woolf AS, Johnstone KA, Wirsing
A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van't
Hoff W, et al: HNF1B mutations associate with hypomagnesemia and
renal magnesium wasting. J Am Soc Nephrol. 20:1123–1131. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Murray PJ, Thomas K, Mulgrew CJ, Ellard S,
Edghill EL and Bingham C: Whole gene deletion of the hepatocyte
nuclear factor-1beta gene in a patient with the prune-belly
syndrome. Nephrol Dial Transplant. 23:2412–2415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meij IC, Koenderink JB, Van Bokhoven H,
Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van
den Heuvel LP and Knoers NV: Dominant isolated renal magnesium loss
is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat
Genet. 26:265–266. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nicolaou N, Renkema KY, Bongers EM, Giles
RH and Knoers NV: Genetic, environmental, and epigenetic factors
involved in CAKUT. Nat Rev Nephrol. 11:720–731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schedl A: Renal abnormalities and their
developmental origin. Nat Rev Genet. 8:791–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Friedli M and Trono D: The developmental
control of transposable elements and the evolution of higher
species. Annu Rev Cell Dev Biol. 31:429–451. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Babatz TD and Burns KH: Functional impact
of the human mobilome. Curr Opin Genet Dev. 23:264–270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mandal PK and Kazazian HH Jr: SnapShot:
Vertebrate transposons. Cell. 135:192.e12008. View Article : Google Scholar
|
|
26
|
Pearson CE, Edamura K Nichol and Cleary
JD: Repeat instability: Mechanisms of dynamic mutations. Nat Rev
Genet. 6:729–742. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koumbaris G, Hatzisevastou-Loukidou H,
Alexandrou A, Ioannides M, Christodoulou C, Fitzgerald T, Rajan D,
Clayton S, Kitsiou-Tzeli S, Vermeesch JR, et al: FoSTeS, MMBIR and
NAHR at the human proximal Xp region and the mechanisms of human Xq
isochromosome formation. Hum Mol Genet. 20:1925–1936. 2011.
View Article : Google Scholar : PubMed/NCBI
|