Introduction
Sepsis remains a significant health problem among
pediatric patients (1). Worldwide,
it affects a large pediatric population and is the most common
cause of mortality in infants and children (2).· Accumulating evidence has indicated
that myocardial dysfunction is a common complication in adults and
children with severe sepsis (3).
However, the cellular mechanisms underlying sepsis-associated
myocardial dysfunction are not entirely clear. There is increasing
evidence that apoptosis of cardiomyocytes serves an important role
in the pathogenesis of sepsis-induced myocardial dysfunction
(4). Therefore, therapeutic
strategies aimed at preventing the sepsis-induced apoptosis of
cardiomyocytes may be a promising option for the treatment of
sepsis-associated myocardial dysfunction.
Peroxisome proliferator-activated receptors (PPARs)
are ligand-activated nuclear transcriptional activators belonging
to the superfamily of nuclear receptors. A total of three PPAR
subtypes (α, β/δ and γ) have been identified (5). Although the function of the
ubiquitously-expressed PPARδ subtype has received relatively little
attention in comparison with the other two subtypes, it has been
reported to exhibit a potent role in the regulation of cellular
metabolism (6), inflammation
(7), apoptosis (8) and angiogenesis (9). Previous experimental evidence has
indicated that PPARδ activation protects H9c2 cells from oxidative
stress-induced apoptosis (10).
However, the possible anti-apoptotic effect of PPARδ activation in
lipopolysaccharide (LPS)-stimulated H9c2 cells has not been tested.
Furthermore, the precise mechanism underlying this response remains
unclear.
Molecular and pharmacological strategies that
prevent nuclear factor-κB (NF-κB) activation have been demonstrated
to provide considerable myocardial protection in models of
endotoxin challenge, thus confirming the vital role of this
transcription factor in mediating sepsis-associated myocardial
dysfunction (11). Heme
oxygenase-1 (HO-1), which is the rate-limiting enzyme responsible
for the degradation of heme into free ferrous iron, CO and
bilirubin, exerts cytoprotective effects in various diseases
(12). It is reported that HO-1
induction serves an important role in the cytoprotective actions of
PPARδ in vascular endothelia (13). Recent experimental evidence
indicates that heme oxygenase-1 overexpression protects human
cardiac endothelial cells from IL-18-induced apoptosis by
inhibiting NF-κB activation (14).
Therefore, the present study was designed to investigate the effect
of the PPARδ-selective agonist GW501516 (GW) on LPS-induced
apoptosis in the rat cardiomyoblast cell line H9c2, and to analyze
the role of NF-κB and HO-1 in this effect.
Materials and methods
Materials
GW was obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Zinc protoporphyrin-IX (Znpp-IX, an HO-1
inhibitor), dimethyl sulfoxide (DMSO), trypsin and MTT were from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). All culture
reagents were from Invitrogen (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). LPS, phenylmethanesulfonyl fluoride (PMSF),
radioimmunoprecipitation assay (RIPA) buffer, the enhanced
bicinchoninic acid (BCA) protein assay kit, all SDS-PAGE reagents,
the nuclear and cytoplasmic protein extraction kit, the enhanced
chemiluminescence (ECL) substrate reagent kit and the caspase-3
activity kit were obtained from Beyotime Institute of Biotechnology
(Jiangsu, China). The Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) detection kit was from KeyGen Biology
(Nanjing, China). Rabbit polyclonal antibodies specific for cleaved
caspase-3 (CC3; cat. no. 9661), apoptosis regulator Bcl-2 (bcl-2;
cat. no. 2876), apoptosis regulator BAX (bax; cat. no. 2772), NF-κB
p65 (cat. no. 3034) and anti-GAPDH (cat. no. 2118; 1:1,000) were
from Cell Signaling Technology, Inc. (Danvers, MA, USA). Rabbit
polyclonal antibodies specific for HO-1 (cat. no. SPA-896) and
histone 3 (H3; cat. no. ab1791) were obtained from Stressgen
Bioreagents Corporation (Victoria, BC, Canada) and Abcam
(Cambridge, UK), respectively.
Cell culture and treatment
protocol
H9c2 cardiomyoblasts (CRL-1446; American Type
Culture Collection, Manassas, VA, USA) were maintained in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and
100 µg/ml streptomycin in a humidified atmosphere of 5%
CO2 and 95% air at 37°C. Cells were passaged regularly
and subcultured to 90% confluence prior to experimental
procedures.
GW dissolved in DMSO was diluted with low-serum
medium (1% FBS/DMEM) to the final concentrations (25, 50 and 100
nmol/l) before use. The final concentration of DMSO in the
incubation mixture was not more than 0.1% (v/v). Prior to
experimental intervention, confluent cultured cells were
serum-starved for 24 h in low-serum medium. Cells were pretreated
with different doses of GW for 24 hat 37°C, and then incubated with
1 µg/ml of LPS for 24 h. For the inhibitor experiment, cells were
pretreated with 100 nM of GW for 24 h at 37°C in the absence or
presence of 10 µmol/l of ZnPP-IX, which was added 1 h before GW,
and then the cells were incubated for 24 h with 1 µg/ml LPS.
Cell viability assay
Cell viability was assessed by MTT assay. Following
incubation with the test reagents, 5 mg/ml MTT was added to the
culture media and the cells were incubated for an additional 4 h.
Subsequent to this incubation, the resulting formazan was
solubilized by adding DMSO, and the optical density of the
solubilized cell extract was measured at 490 nm using a microplate
reader.
Evaluation of cell apoptosis by
Annexin V-FITC/PI staining
Apoptosis was detected using an Annexin V-FITC/PI
detection kit according to the manufacturer's protocol. The cells
were digested with 0.25% trypsin, washed with ice-cold PBS and
resuspended in binding buffer (5×105 cells/ml). The
cells were centrifuged at 1,000 × g for 5 min at 4°C. Subsequent to
the supernatant being discarded, 500 µl binding buffer, 5 µl
Annexin V-FITC and 5 µl PI were added to the cell suspension.
Following gentle mixing, the suspensions were incubated for 15 min
at room temperature without light. The cells were analyzed by flow
cytometry (BD LSRII; BD Biosciences, Franklin Lakes, NJ, USA). Data
analysis was performed using CellQuest version 3.3 (BD
Biosciences).
Caspase-3 activity assay
Caspase-3 activity was determined using a caspase-3
activity kit, which detects the production of the chromophore
p-nitroanilide following its cleavage from the peptide substrate
DEVD-p-nitroanilide. According to the manufacturer's protocol,
following washing with cold PBS, cells were lysed with lysis buffer
(100 µl/2×106 cells) for 15 min on ice. The mixture,
composed of 10 µl cell lysate, 80 µl reaction buffer and 10 µl 2
mmol/l caspase-3 substrate, was incubated in 96-well plates at 37°C
for 4 h, and caspase-3 activity was quantified in the samples using
a microplate reader at an absorbance of 405 nm.
Western blot analysis
Cells from each group were washed twice with
ice-cold PBS and lysed in RIPA buffer (10 mmol/l Tris, pH 7.4; 150
mmol/l NaCl; 1 mmol/l EDTA; 0.1% SDS; 1% Triton X-100; and 1%
sodium deoxycholate) with protease inhibitors (1 mmol/l PMSF, 1
µg/ml aprotinin, 1 µg/ml pepstatin, and 1 µg/ml leupeptin) at 4°C.
The lysate was centrifuged at 12,000 × g at 4°C for 20 min to
remove the insoluble material. Supernatants were collected. The
protein concentration was determined using the BCA assay. Equal
quantities of protein (30 µg) were separated by 10% SDS-PAGE, and
subsequently transferred onto a polyvinylidene difluoride (PVDF)
membrane. The membranes were blocked in TBS with Tween-20 (TBST)
with 5% (w/v) skimmed milk at room temperature for 2 h, followed by
overnight incubation at 4°C with primary antibodies diluted in 0.1%
TBST. Following washing in TBST, the membranes were incubated at
room temperature for 1 h with goat anti-rabbit horseradish
peroxidase-conjugated secondary antibody (cat. no. 7074; 1:3,000;
Cell Signaling Technology, Inc.) diluted in 0.1% TBST. Following
washing once more in TBST, the immunoreactive bands were visualized
by using an ECL kit and exposed to X-ray film. The band intensity
was quantified using Quantity One software (Bio-Rad Laboratories
Inc. Hercules, CA, USA).
Nuclear protein extraction
The extraction and isolation of nuclear proteins was
performed using the nuclear and cytoplasmic protein extraction kit
according to the manufacturer's protocol. Cells were detached with
cold PBS and centrifuged at 1,000 × g for 5 min at 4°C, and the
pellet was dissolved with cytoplasmic protein extraction agent A
supplemented with PMSF. Subsequent to mixing by vortex for 5 sec,
the tubes were incubated for 10–15 min on ice to promote lysis.
Cytoplasmic protein extraction agent B was added and mixing by
vortex was performed for 5 sec, followed by incubation on ice for
60 sec. The samples were centrifuged for 5 min at 14,000 × g at 4°C
and the supernatant, consisting of the cytosolic fraction, was
immediately frozen for further analysis. The pellet was
re-suspended in nuclear protein extraction agent supplemented with
PMSF. Subsequently, the tubes were mixed by vortex 15–20 times over
the course of 30 min and centrifuged for 10 min at 14,000 × g at
4°C to obtain supernatants containing the nuclear extracts. Protein
concentration was measured using the BCA protein assay kit. Levels
of NF-κB p65 in the nuclear protein extract were determined by
western blot analysis as described above.
Statistical analysis
All data represent the mean of samples from three
separately-performed experiments. Results are expressed as mean ±
standard deviation. One-way analysis of variance and
Student-Newman-Keuls tests were used for statistical evaluation
with SPSS 14.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
GW attenuates LPS-induced
cytotoxicity
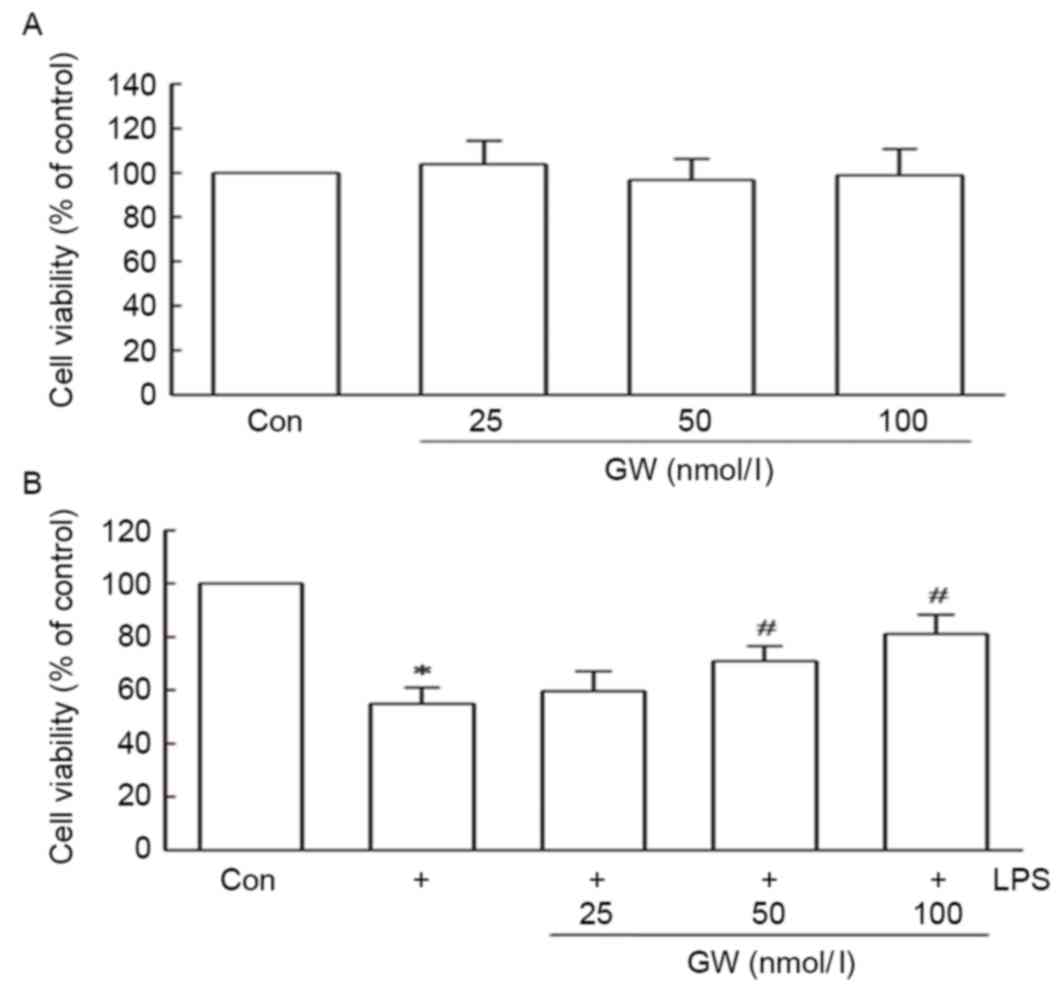
Using the MTT assay, a possible cytoprotective
effect of GW against LPS-induced cell death was detected. To
evaluate whether GW was cytotoxic to H9c2 cells, the cells were
first treated with concentrations of GW ranging between 25 and 100
nmol/l for 24 h. A reduction in cell viability was not observed
(Fig. 1A). Subsequent analyses
tested whether GW was able to protect against LPS-induced
cytotoxicity. When cells were pretreated for 24 h with growing
concentrations of GW (25–100 nmol/l), prior to treatment with 1
µg/ml LPS for 24 h, significant increases in cell viability were
observed compared with treatment with LPS alone. As exhibited in
Fig. 1B, pretreatment with 50 and
100 nmol/l GW significantly increased cell viability to 70.89±5.61
and 81.16±7.12 % of the control, respectively, compared with
treatment with LPS alone. These results indicate that GW may have a
protective role against LPS-induced cell cytotoxicity.
GW inhibits LPS-induced cell
apoptosis
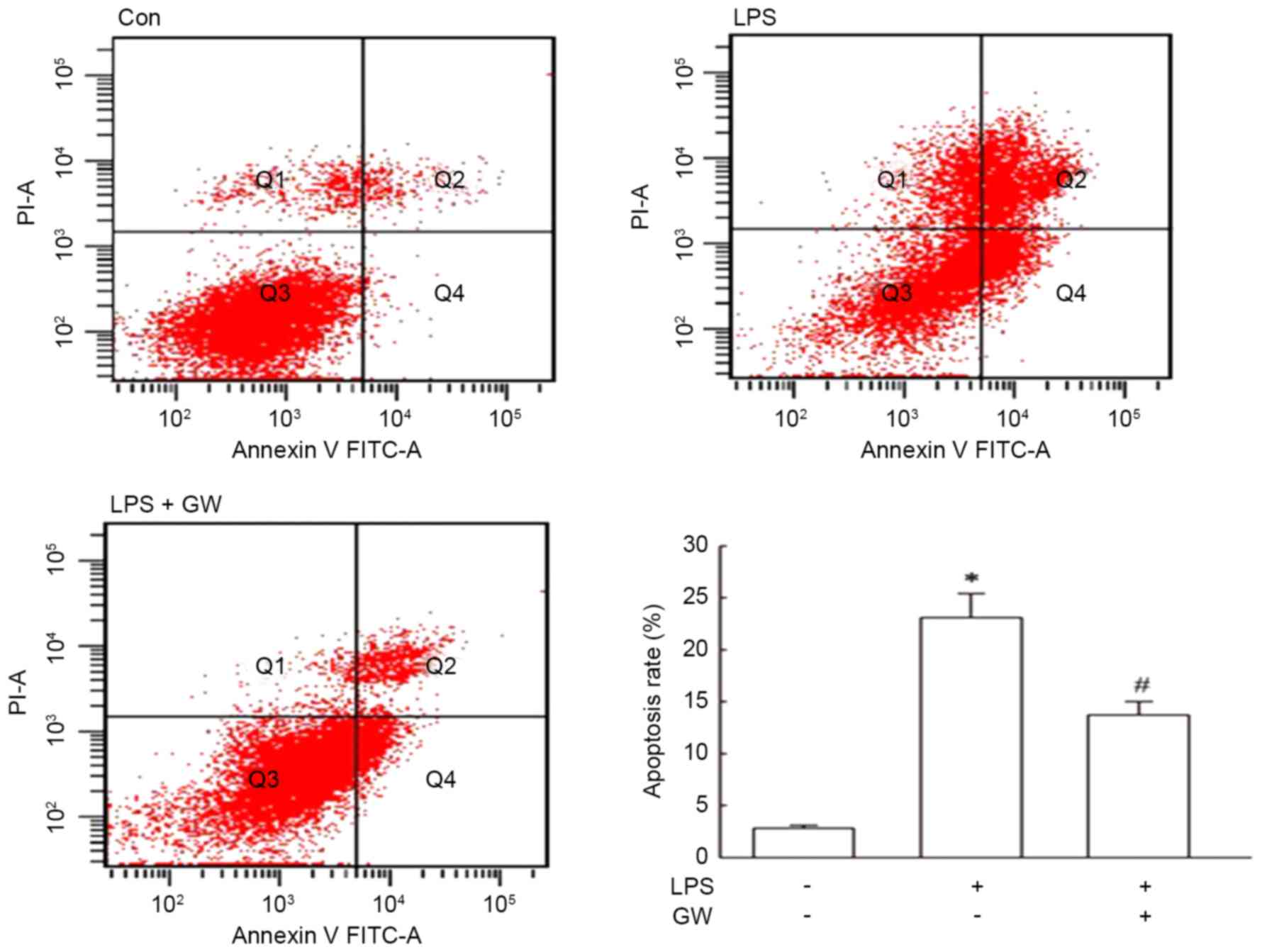
Apoptosis was quantitated by flow cytometry using
Annexin V-FITC/PI staining. As presented in Fig. 2, LPS led to a significant increase
in the apoptotic rate of H9c2 cells compared with the control
group, and the apoptotic rate was decreased markedly by
pretreatment with GW compared with the LPS alone group. Subsequent
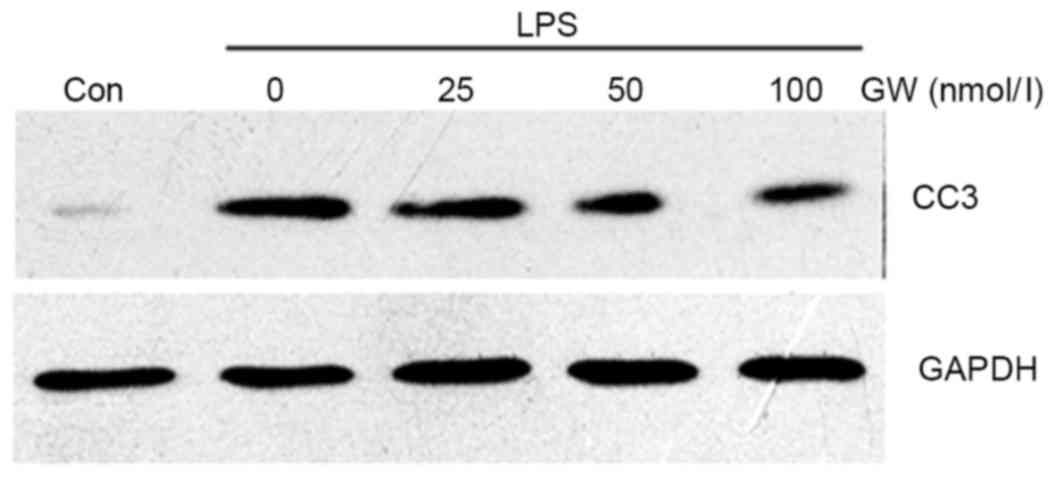
western blot analysis demonstrated that CC3 protein expression was
markedly increased in LPS-stimulated cells compared with the
control group, while GW pretreatment decreased CC3 protein
expression in a dose-dependent manner compared with the LPS group
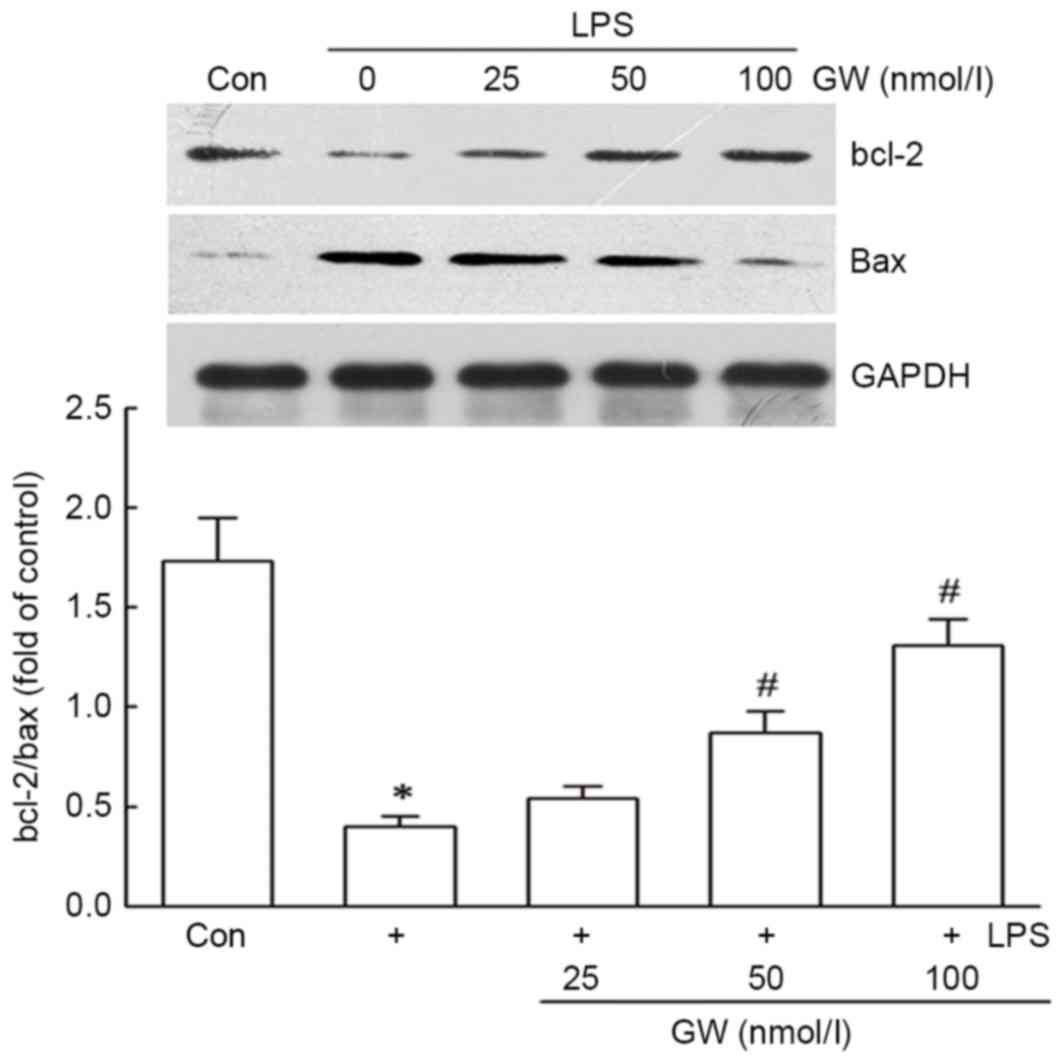
(Fig. 3). Western blot analysis
also indicated that GW notably inhibited the decrease of the
bcl-2/bax ratio induced by LPS in a dose-dependent manner (Fig. 4).
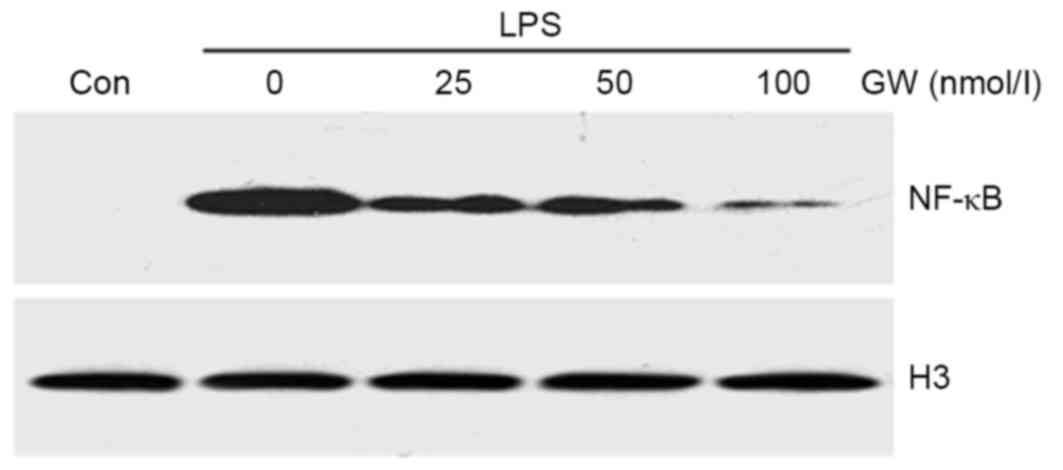
GW inhibits LPS-induced NF-κB nuclear
translocation
To investigate the molecular mechanisms by which GW
protects H9c2 cells from LPS-induced apoptosis, the nuclear NF-κB
protein expression level was examined to determine whether the
regulation of NF-κB is responsible for the anti-apoptotic effect of
GW. As displayed in Fig. 5, when
cells were pretreated for 24 h with increasing concentrations of GW
prior to incubation with LPS, NF-κB protein expression was
decreased in a dose-dependent manner compared with the LPS group.
These results suggest that GW prevents LPS-induced apoptosis,
possibly through inhibition of NF-κB activation.
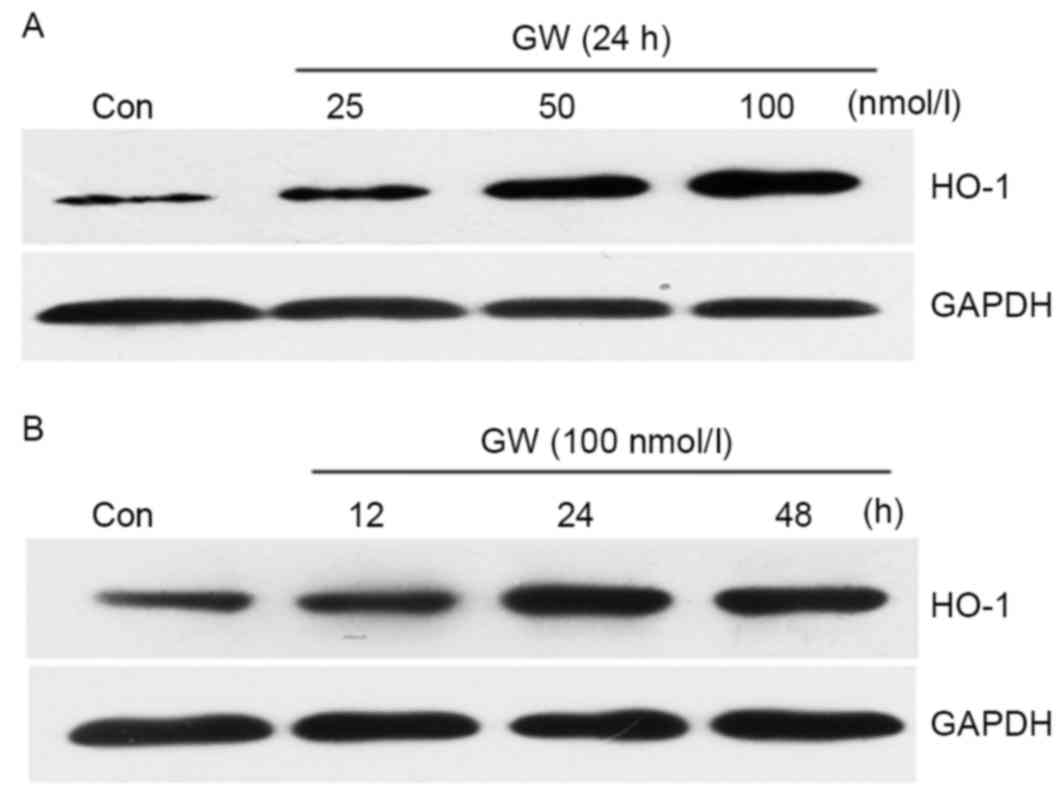
GW induces HO-1 protein
expression
The cells were treated with increasing
concentrations of GW (25–100 nmol/l). Treatment with GW for 24 h
increased HO-1 expression in a dose-dependent manner (Fig. 6A). HO-1 expression was subsequently
determined by treating the cells with 100 nmol/l GW for 12, 24 and
48 h. As presented in Fig. 6B, a
maximum increase of HO-1 protein expression was observed following
treatment for 24 h.
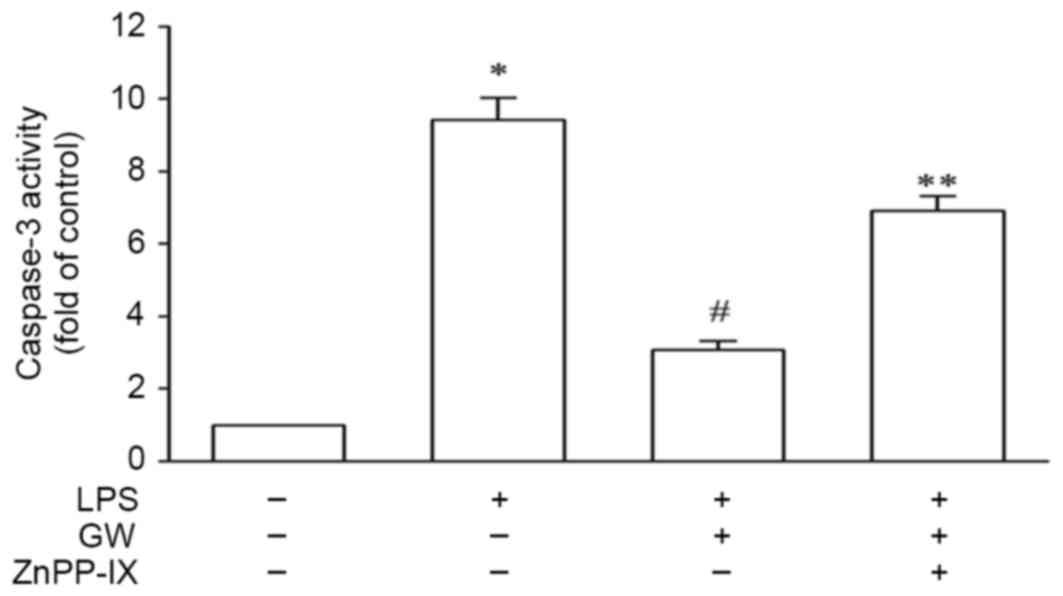
HO-1 mediates the protective effect of
GW against LPS-induced cell apoptosis
In order to validate the protective role of HO-1,
H9c2 cells exposed to LPS were pretreated with 100 nmol/l GW for 24
h in the absence or the presence of 10 µM ZnPP-IX. As exhibited in
Fig. 7, the caspase-3 activity was
increased by 9.42±0.61-fold that of the control in the LPS group,
whereas it was reduced to 3.07±0.24-fold that of the control
following pretreatment with GW. Co-incubation with ZnPP-IX, which
partly negated the effect of GW, increased the caspase-3 activity
to 6.91±0.40-fold that of the control. These results suggest that
the protective effect of GW on LPS-induced cell apoptosis is
HO-1-dependent.
HO-1 mediates the inhibitory effect of
GW on NF-κB activation
To determine whether the upregulation of HO-1 by GW
is important to the survival of H9c2 cells, due to the modulation
of NF-κB, the effects of ZnPP-IX on the expression of NF-κB were
tested. H9c2 cells exposed to LPS were pretreated with 100 nmol/l
GW for 24 h in the absence or the presence of 10 µM ZnPP-IX. As
presented in Fig. 8, ZnPP-IX
co-incubation reversed the inhibitory effect of GW on LPS-induced
NF-κB activation, suggesting that HO-1 may mediate the inhibitory
effect of GW on NF-κB activation in LPS-stimulated H9c2 cells.
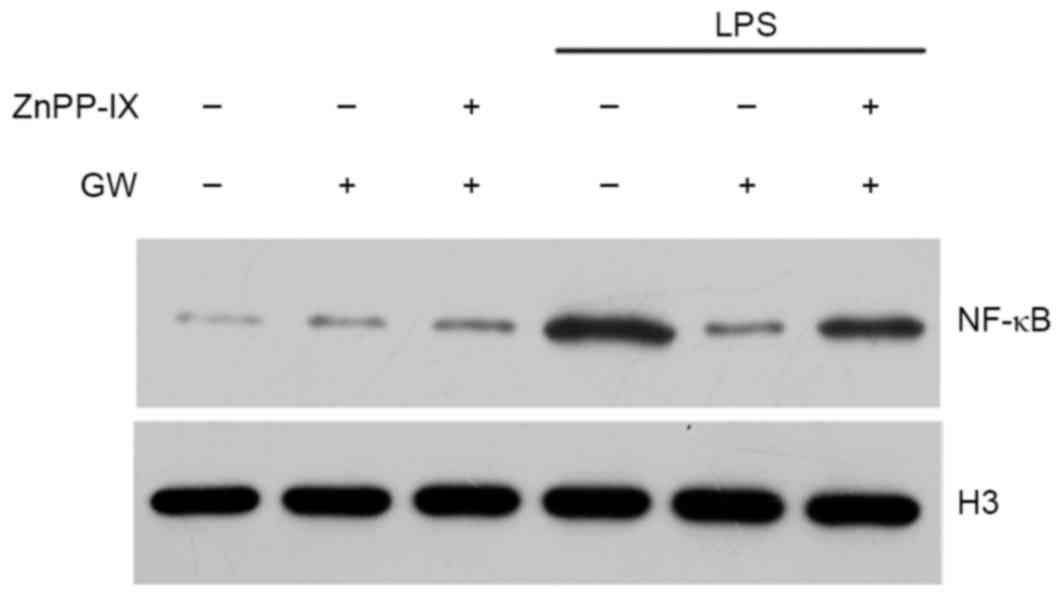 | Figure 8.HO-1 mediates the inhibitory effect of
GW on NF-κB activation in LPS-stimulated cells. Cells were
pretreated with GW, ZnPP-IX, or LPS as indicated and NF-κB was
determined by western blot analysis. Cells were incubated for 24 h
in the absence or presence of 10 µmol/l of ZnPP-IX, which was added
1 h before GW. Following pretreatment, cells were incubated for
another 24 h with or without LPS. GW, GW501516; HO-1, heme
oxygenase-1; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB;
ZnPP-IX, zinc protoporphyrin-IX; H3, histone 3. |
Discussion
The present study investigated the anti-apoptotic
effect elicited by the selective PPARδ agonist GW in LPS-stimulated
H9c2 cardiomyoblasts. The possible mechanism underlying this
anti-apoptotic effect was also investigated. It was demonstrated
that the activation of NF-κB, induced by LPS, was inhibited
significantly by pretreatment with GW. GW also induced HO-1 protein
expression in a dose-dependent manner. In addition, it was revealed
that HO-1 mediated the inhibitory effect of GW on NF-κB activation
in LPS-stimulated H9c2 cells.
The protective effect of GW against LPS-induced cell
toxicity was investigated by MTT assay. The results demonstrated
that GW protected H9c2 cells from LPS-induced cytotoxicity in a
dose-dependent manner. It is known that LPS is a major feature of
Gram-negative septic shock, and it has been tested as a stress
model for inducing apoptosis in vitro (15). Previously, the activation of PPARδ
was demonstrated to be implicated in the suppression of apoptosis
in human umbilical vein endothelial cells (16). Therefore, the present study
examined whether GW has a protective effect against LPS-induced
apoptosis in H9c2 cells.
Apoptosis is typically associated with the
regulation of caspase-3 (17) and
the family of bcl-2 proteins which include pro-apoptotic protein
bax and anti-apoptotic protein bcl-2 (18). The present study provided evidence
that pretreatment with GW significantly inhibited the LPS-induced
increase in the rate of apoptosis as measured by Annexin V-FITC/PI
staining. It was also observed that GW may significantly decrease
the expression of CC3, and caspase-3 activity, which are
accompanied by a marked increase in the bcl-2/bax ratio. These
results demonstrated that the activation of PPARδ may protect H9c2
cells from LPS-induced apoptosis.
LPS is a potent pro-inflammatory cytokine stimulator
in cardiomyocytes. A large body of evidence suggests that NF-κB
activation, mediated by pro-inflammatory cytokines such as TNF-α or
IL-1, serves an important role in the LPS-induced apoptosis of
cardiomyocytes (19). A previous
report indicated that PPARδ regulated inflammation via NF-κB
signaling in sepsis (20).
However, whether activation of PPARδ may inhibit apoptosis in
LPS-stimulated H9c2 cells via NF-κB signaling is still unknown.
NF-κB is a nuclear transcription factor that
regulates expression of a number of genes that are critical for the
regulation of tumorigenesis, inflammation and apoptosis. NF-κB is
activated by a variety of stimuli that include ischemia, cytokines,
hypoxia, free radicals and LPS. In the inactive state, NF-κB is
retained in the cytosol through complexation with inhibitor of
NF-κB (IκB) proteins. Upon LPS-induced phosphorylation, IκBα is
degraded, and NF-κB is released from the cytosolic complex with IκB
proteins and translocates to the nucleus, where it promotes the
transcription of both pro- and anti-apoptotic proteins (21). The association between cardiac
NF-κB activity and apoptosis remains unclear; in certain cellular
systems, it has been proven to be detrimental, and in others,
protective (22). It has been
suggested that the inhibition of NF-κB activation in H9c2 cells may
be useful in the prevention of apoptosis induced by LPS (15). In the present study, it was
demonstrated that pretreatment with GW inhibited the activation of
NF-κB induced by LPS in a dose-dependent manner. These results
suggest that GW prevents LPS-induced apoptosis through inhibition
of NF-κB activation.
Pharmacological and genetic induction of HO-1 has
been demonstrated to exert an anti-apoptotic effect in various
cardiovascular diseases (23,24).
A previous study demonstrated that HO-1 was upregulated in
endothelial cells and vascular smooth muscle cells by GW in
vitro (25), and to the best
of our knowledge the present study demonstrates for the first time
that GW induces HO-1 protein expression in a dose-dependent manner
in H9c2 cells. In addition, pretreatment with GW significantly
inhibited the LPS-induced activation of caspase-3. This effect was
demonstrated to be partly attributed to the induction of HO-1, as
the inhibitor of HO-1 (ZnPPIX) markedly reversed the protective
effect afforded by GW. These results suggest that the induction of
HO-1 may serve a marked role in mediating the anti-apoptotic effect
of GW in LPS-stimulated H9c2 cells. Additionally, it was identified
in this study that ZnPP-IX co-incubation reversed the inhibitory
effect of GW on LPS-induced NF-κB nuclear translocation, which
suggests that HO-1 may mediate the inhibitory effect of GW on NF-κB
activation in LPS-stimulated H9c2 cells. Therefore, it was
concluded that PPARδ activation protects H9c2 cells from
LPS-induced apoptosis probably through the HO-1 mediated
suppression of NF-κB activation. These results are in agreement
with previous reports demonstrating that HO-1 activation attenuates
the apoptosis of cardiomyocytes by inhibiting NF-κB activity
(24).
A previous study demonstrated that CO, a product of
heme metabolism by HO-1, exhibited a potent anti-apoptotic effect
in LPS-stimulated endothelial cells (26); however, whether CO is involved in
the cytoprotection afforded by GW remains unknown. Therefore,
experiments aimed at broadening understanding of the more detailed
mechanisms will be the subject of future studies.
In conclusion, the present study demonstrates that
GW protects H9c2 cardiomyoblasts against LPS-induced apoptosis. The
protective effect of GW was demonstrated to be associated with the
HO-1 mediated suppression of NF-κB activation. The results suggest
that PPARδ is a promising therapeutic target for the treatment of
sepsis-associated cardiac dysfunction.
References
|
1
|
Czaja AS, Zimmerman JJ and Nathens AB:
Readmission and late mortality after pediatric severe sepsis.
Pediatrics. 123:849–857. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Watson RS, Carcillo JA, Linde-Zwirble WT,
Clermont G, Lidicker J and Angus DC: The epidemiology of severe
sepsis in children in the United States. Am J Respir Crit Care Med.
167:695–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maeder M, Fehr T, Rickli H and Ammann P:
Sepsis-associated myocardial dysfunction: Diagnostic and prognostic
impact of cardiac troponins and natriuretic peptides. Chest.
129:1349–1366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ayala A, Perl M, Venet F, Lomas-Neira J,
Swan R and Chung CS: Apoptosis in sepsis: Mechanisms, clinical
impact and potential therapeutic targets. Curr Pharm Des.
14:1853–1859. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ehrenborg E and Krook A: Regulation of
skeletal muscle physiology and metabolism by peroxisome
proliferator-activated receptor delta. Pharmacol Rev. 61:373–393.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barish GD, Narkar VA and Evans RM: PPAR
delta: A dagger in the heart of the metabolic syndrome. J Clin
Invest. 116:590–597. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li AC, Binder CJ, Gutierrez A, Brown KK,
Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum
JL, et al: Differential inhibition of macrophage foam-cell
formation and atherosclerosis in mice by PPARalpha, beta/delta, and
gamma. J Clin Invest. 114:1564–1576. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liou JY, Lee S, Ghelani D,
Matijevic-Aleksic N and Wu KK: Protection of endothelial survival
by peroxisome proliferator-activated receptor-delta mediated 14-3-3
upregulation. Arterioscler Thromb Vasc Biol. 26:1481–1487. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piqueras L, Reynolds AR, Hodivala-Dilke
KM, Alfranca A, Redondo JM, Hatae T, Tanabe T, Warner TD and
Bishop-Bailey D: Activation of PPARbeta/delta induces endothelial
cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol.
27:63–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pesant M, Sueur S, Dutartre P, Tallandier
M, Grimaldi PA, Rochette L and Connat JL: Peroxisome
proliferator-activated receptor delta (PPARdelta) activation
protects H9c2 cardiomyoblasts from oxidative stress-induced
apoptosis. Cardiovasc Res. 69:440–449. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carlson D, Maass DL, White DJ, Tan J and
Horton JW: Antioxidant vitamin therapy alters sepsis-related
apoptotic myocardial activity and inflammatory responses. Am J
Physiol Heart Circ Physiol. 291:H2779–H2789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Otterbein LE and Choi AM: Heme oxygenase:
Colors of defense against cellular stress. Am J Physiol Lung Cell
Mol Physiol. 279:L1029–L1037. 2000.PubMed/NCBI
|
|
13
|
Ali F, Ali NS, Bauer A, Boyle JJ, Hamdulay
SS, Haskard DO, Randi AM and Mason JC: PPARdelta and PGC1alpha act
cooperatively to induce haem oxygenase-1 and enhance vascular
endothelial cell resistance to stress. Cardiovasc Res. 85:701–710.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zabalgoitia M, Colston JT, Reddy SV, Holt
JW, Regan RF, Stec DE, Rimoldi JM, Valente AJ and Chandrasekar B:
Carbon monoxide donors or heme oxygenase-1 (HO-1) overexpression
blocks interleukin-18-mediated NF-kappaB-PTEN-dependent human
cardiac endothelial cell death. Free Radic Biol Med. 44:284–298.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tien YC, Lin JY, Lai CH, Kuo CH, Lin WY,
Tsai CH, Tsai FJ, Cheng YC, Peng WH and Huang CY: Carthamus
tinctorius L. Prevents LPS-induced TNFalpha signaling activation
and cell apoptosis through JNK1/2-NFkappaB pathway inhibition in
H9c2 cardiomyoblast cells. J Ethnopharmacol. 130:505–513. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang B, Liang P, Zhang B, Song J, Huang X
and Xiao X: Role of PPAR-beta in hydrogen peroxide-induced
apoptosis in human umbilical vein endothelial cells.
Atherosclerosis. 204:353–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cryns V and Yuan J: Proteases to die for.
Genes Dev. 12:1551–1570. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Zingarelli B, O'Connor M, Zhang P,
Adeyemo A, Kranias EG, Wang Y and Fan GC: Overexpression of Hsp20
prevents endotoxin-induced myocardial dysfunction and apoptosis via
inhibition of NF-kappaB activation. J Mol Cell Cardiol. 47:382–390.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zingarelli B, Piraino G, Hake PW, O'Connor
M, Denenberg A, Fan H and Cook JA: Peroxisome
proliferator-activated receptor {delta} regulates inflammation via
NF-{kappa}B signaling in polymicrobial sepsis. Am J Pathol.
177:1834–1847. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abdulkhalek S, Guo M, Amith SR, Jayanth P
and Szewczuk MR: G-protein coupled receptor agonists mediate Neu1
sialidase and matrix metalloproteinase-9 cross-talk to induce
transactivation of TOLL-like receptors and cellular signaling. Cell
Signal. 24:2035–2042. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Delhalle S, Blasius R, Dicato M and
Diederich M: A beginner's guide to NF-kappaB signaling pathways.
Ann N Y Acad Sci. 1030:1–13. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang G, Hamid T, Keith RJ, Zhou G,
Partridge CR, Xiang X, Kingery JR, Lewis RK, Li Q, Rokosh DG, et
al: Cardioprotective and antiapoptotic effects of heme oxygenase-1
in the failing heart. Circulation. 121:1912–1925. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yeh CH, Chen TP, Wang YC, Lin YM and Lin
PJ: HO-1 activation can attenuate cardiomyocytic apoptosis via
inhibition of NF-kappaB and AP-1 translocation following cardiac
global ischemia and reperfusion. J Surg Res. 155:147–156. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim HJ, Ham SA, Paek KS, Hwang JS, Jung
SY, Kim MY, Jin H, Kang ES, Woo IS, Kim HJ, et al: Transcriptional
up-regulation of antioxidant genes by PPARδ inhibits angiotensin
II-induced premature senescence in vascular smooth muscle cells.
Biochem Biophys Res Commun. 406:564–569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bernardini C, Zannoni A, Bacci ML and
Forni M: Protective effect of carbon monoxide pre-conditioning on
LPS-induced endothelial cell stress. Cell Stress Chaperones.
15:219–224. 2010. View Article : Google Scholar : PubMed/NCBI
|