Introduction
Cockayne syndrome (CS) is a rare autosomal recessive
multisystem disorder characterized by poor growth, neurological
abnormality and a short life span (1). It occurs at a rate of about 2.7 per
million births in western Europe (2). Typical features of CS include severe
growth failure, mental retardation, microcephaly, cutaneous
photosensitivity, dental decay and deep sunken eyes (3). A total of four subtypes have been
described according to the time of onset and rate of the
progression: Moderate type I CS, with the first symptoms appearing
from the end of the first year of life and mortality occurring
prior to the age of 20; early-onset and/or severe type II CS;
late-onset type III CS; and the most severe
cerebro-oculo-facio-skeletal (COFS) syndrome, with disease onset at
the prenatal stage (1,4). These subtypes of CS share a large
overlapping spectrum of severity.
CS is clinically diagnosed by reduced recovery of
RNA and DNA synthesis in fibroblasts following ultraviolet (UV)
irradiation (5), involving
defective nucleotide-excision repair (NER) as the molecular
pathogenesis (6). The
transcription-coupled NER (TCR) system allows RNA polymerase
II-blocking lesions to be rapidly removed from the transcribed
strand of active genes. Mutations in the excision repair
cross-complementation group 6 (ERCC6) and ERCC8 genes are primarily
responsible for CS, of which a mutation in ERCC6, the CS type B
(CSB) gene, accounts for two thirds of all cases (3). Although clinical symptoms are
indistinguishable between CSA and CSB patients, CSA mutations have
not been previously reported in severe CS subgroups.
Chinese patients with CS patients have rarely been
reported, rendering it difficult for clinicians to make a clear
diagnosis based on the clinical symptoms of CS. Based on whole
exome sequencing (WES), the present study identified two novel
mutations causing compound heterozygous ERCC6 in a Chinese family
with two brothers suffering from premature aging. CSB was diagnosed
according to clinical characteristics and genetic analysis.
Depending on the diagnosis, this study successfully performed a
molecular prenatal diagnosis of CSB on amniotic fluid sampling
obtained from the family.
Materials and methods
Ethical approval
The present study was conducted in accordance with
the guiding principles of the Declaration of Helsinki. Informed
written consent for the collection of samples, images, tests and
inclusion of data were obtained from the parents and grandparents
of the patients. Written consents on behalf of the patients were
obtained from their parents as the patients did not understand
consent and lacked the ability to write. Use of all human materials
used in this study were approved by the Institutional Review Ethics
Board of Xijing Hospital, Fourth Military Medical University
(Xi'an, China).
Genomic DNA isolation and WES
Genomic DNA was isolated from peripheral blood
samples using standard methods. WES of genomic DNA from the two
patients was subsequently performed to identify potential
disease-causing gene mutations shared by the two patients.
Sequencing was performed using a Genome Analyzer HiSeq2000 system
(Illumina, Inc., San Diego, CA, USA) following enrichment of exonic
and adjacent intronic sequences using a NimbleGen 44M capture
platform (SeqCap EZ Human Exome Library v2.0; NimbleGen; Roche
Sequencing, Madison, WI, USA).
ERCC6 sequencing
The mutation in ERCC6 identified by WES was verified
by Sanger sequencing in two orientations of the polymerase chain
reaction (PCR) product. The primers used are as follows: Forward,
5′-CAGCGTTTACTACTTGCCAG-3′ and reverse, 5′-CCACTTGGAAATCTCCCTT-3′
for c.1834C>T; forward, 5′-CTGGGAGTGACAGGTAGTGA-3′ and reverse,
5′-CAACTCACAGTAAGACATCTAAGC-3′ for c.2923C>T. Primers were
designed to amplify other protein-coding exons of the ERCC6 gene
and their flanking intronic sequences as previously reported
(7). Direct sequencing of the PCR
products was performed using an ABI PRISM® 310 Genetic
Analyzer system (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The base-pair number of the mutation site
was determined according to the GenBank mRNA reference sequence
NM_000124.
Prenatal diagnosis
Fetal DNA was obtained from amniocytes by
amniocentesis at 6 months of gestation and further used for genetic
testing of the ERCC6 mutations by Sanger sequencing.
Results
Case presentation
The two patients were brothers born at full-term by
spontaneous vaginal delivery to non-consanguineous healthy Chinese
parents. The birth weight and height were not available; however,
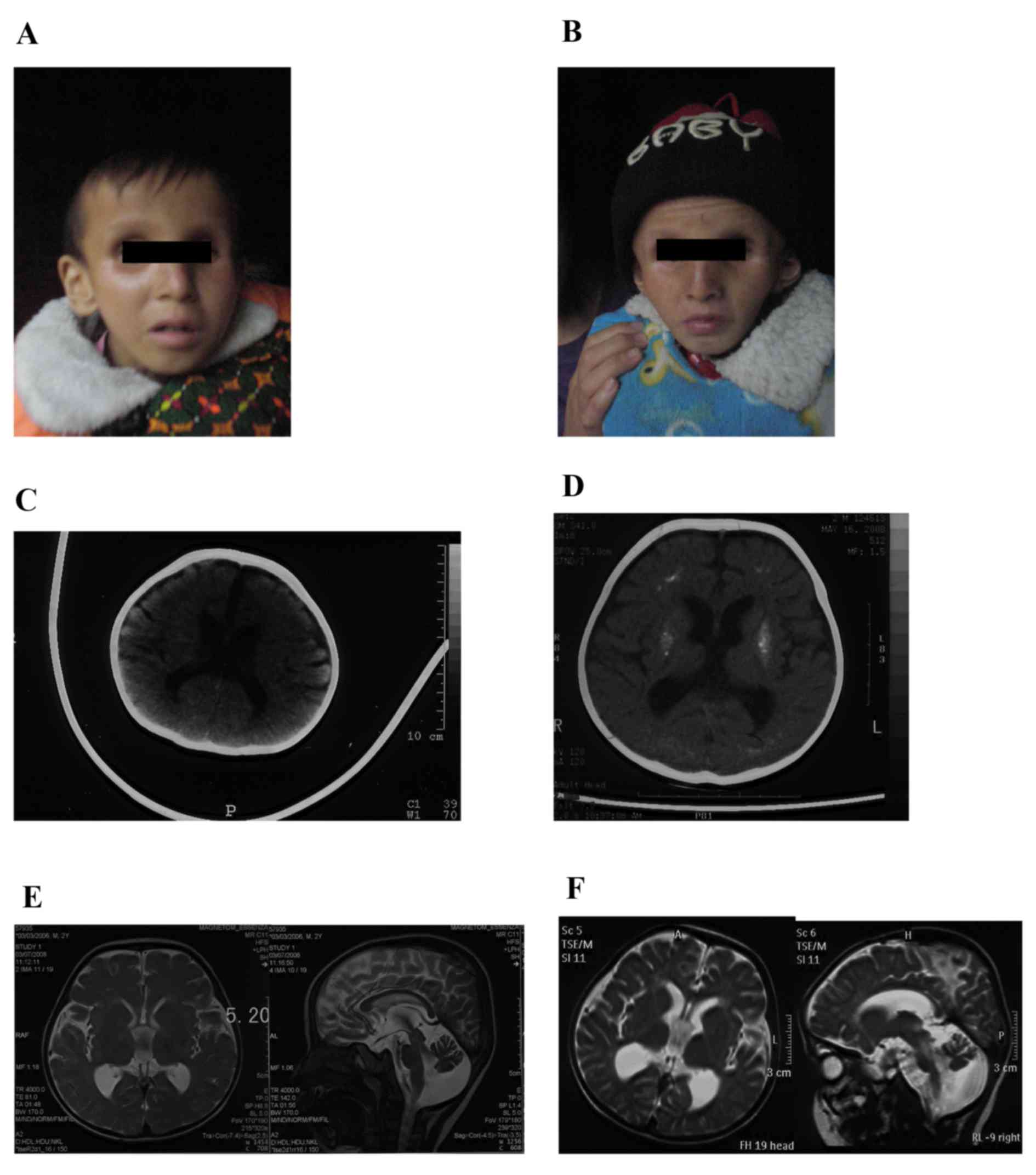
they were typical according to the mother. The proband (III-2;
Fig. 1A) was healthy during the
neonatal period. Development was delayed and regression was
observed. He was able to hold his head steady at 9 months, and roll
over and sit erect without support when over 1 year old. He was
able to speak certain simple words at ~18 months. However, at 2
years old, he lost the ability to speak and climb, and to sit or
stand without support. He was able to walk landing on the toes,
with support. Clinic features at 3 years old included cutaneous
photosensitivity that was observed at ~6 months old and
subsequently disappeared until 4 years old, bird-like features with
deep-set eyes, a beaked nose, large and protruding ears, a small
lower jaw, dedentition, low muscle tone of lower limbs, a head
circumference of 43.5 cm, dysphagia, joint contracture and a
pear-shaped chest. The proband additionally exhibited thinning
hair, healthy hearing ability and cataracts. The proband died at
the age of 9 years and 4 months.
The elder brother (Fig.
1B) had very similar symptoms as the proband; however, without
cutaneous photosensitivity. He had earlier disease onset at 3
months old, and died at the age of 8 years and 5 months.
A computed tomography (CT) scan of the proband brain
obtained at 6 months old (Fig. 1C)
revealed a mildly enlarged ventricular system and sulci. Cerebral
white matter and gray matter were blurred, and a mottled
high-density signal was visible in the bilateral basal ganglia
region due to mild calcification. No other abnormalities were
indicated. A CT scan of the brain at 3 years old (Fig. 1D) revealed progressive
deterioration of the disease, and diffuse and symmetric
calcifications of the bilateral basal ganglia region, centrum
semiovale, frontal and parietal lobes, and generally enlarged
ventricular system and sulci. Magnetic resonance imaging (MRI) of
the brain (Fig. 1E) demonstrated
broadened cisterna magna, thinning corpus callosum, and signal
contraction of white and gray matter behind the eyes. A follow-up
MRI scan of the brain at four years old (Fig. 1F) revealed marked sulci and gyri
enlargement, ventricular system expansion, cerebral and cerebellar
atrophy, and severe calcifications of bilateral basal ganglia
region.
WES analysis
A recessive file including 790 genes with 1081
homozygous or compound heterozygous variants shared by the two
subjects was generated. Excluding the variants with a minor allele
frequency >0.005 in dbSNP (ncbi.nlm.nih.gov/projects/SNP/) 126, 129 and 131 or
1,000 Genomes Project.15, and synonymous mutations, 58 candidate
genes were selected. Of the candidate genes, 16 genes were recorded
in the Human Ageing Genomic Resources (HAGR) database
(genomics.senescence.info/), concerning function on gene
transcription, signal pathways, cell cycles, development, or as
nuclear receptor (Tables I and
II). According to the description
in HAGR, ERCC6 and transcription factor 3 (TCF3) were selected as
the most likely candidates.
 | Table I.Categorization of variants from whole
exome sequencing. |
Table I.
Categorization of variants from whole
exome sequencing.
| Category | Number |
|---|
| Detected
variants | 3,619 |
| Detected genes | 2,564 |
| Shared variants | 1,081 |
| Shared genes | 790 |
| Candidate
variants | 173 |
| Candidate genes |
58 |
| Table II.Age-associated candidate genes. |
Table II.
Age-associated candidate genes.
| Function | Genes |
|---|
|
Transcription-associated (including
nucleotide-excision repair proteins) | POU1F1, GTF2H2, SP1,
STAT5A, ERCC6, TCF3, POLA1 |
| Signaling
pathways | DLL3, STAT5A,
PTK2 |
| Cell cycle | BUB3, CHEK2,
POU1F1 |
| Nuclear receptor | NCOR1, NCOR2 |
| Development | VEGFA |
| Others | HSPD1 |
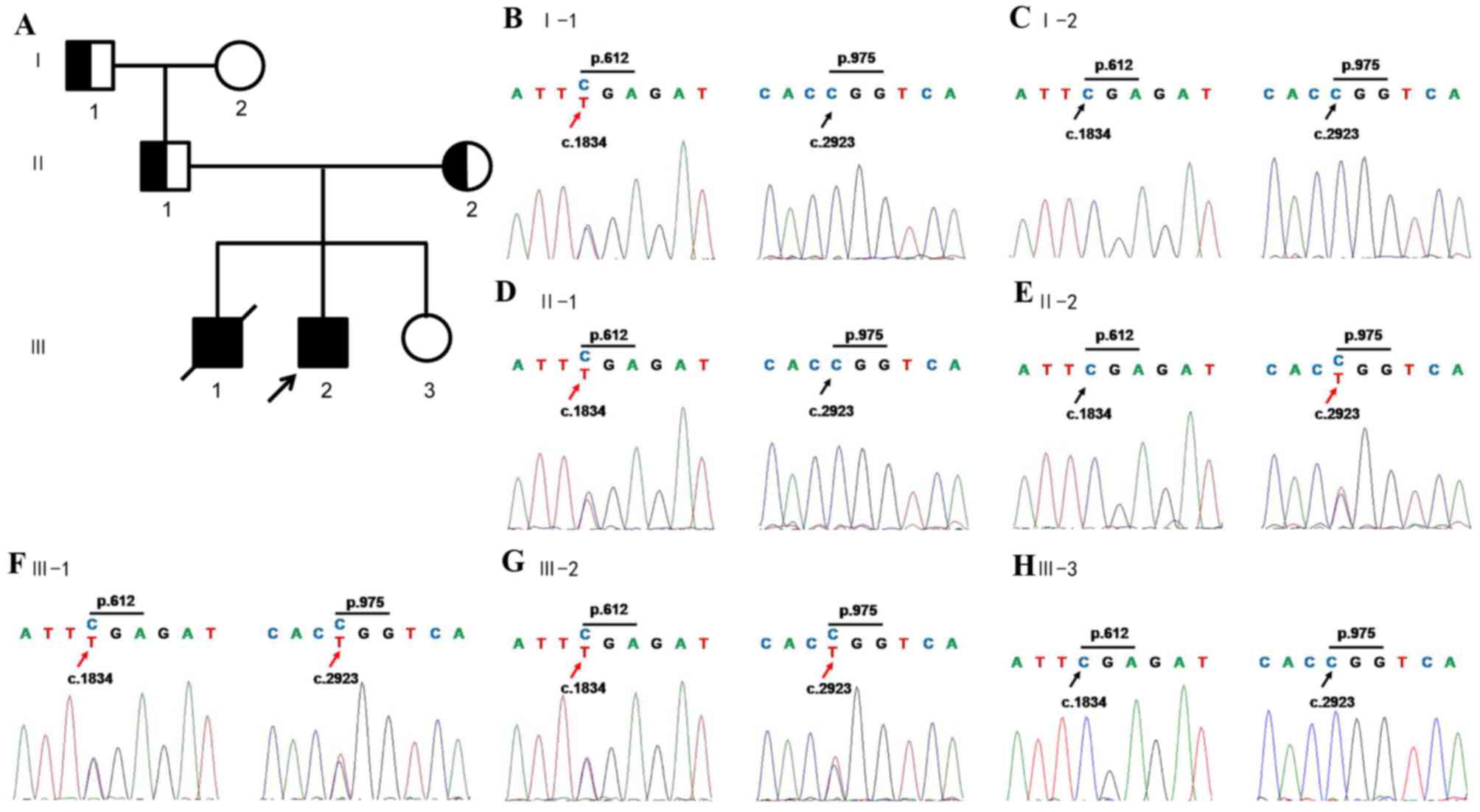
ERCC6 mutation inheritance in the
family
The variations of TCF3 were proved to be a false
positive of WES by Sanger sequencing. ERCC6 contained compound
heterozygous mutations according to WES results; one was a nonsense
mutation in exon9 c.1834C>T, p.Arg612X, leading to a truncated
protein, and the other was a missense mutation in exon16,
c.2923C>T, p.Arg975Trp, causing amino acid transition of
arginine to tryptophan. The compound heterozygous mutations were
confirmed by Sanger sequencing of amplicons of patients. By the
following co-segregation of the two ERCC6 alleles in the pedigree
the present study demonstrated that the c.1834C>T allele was
paternal, and the c.2923C>T allele was maternal (Fig. 2). Sequencing of other exons was
additionally performed and no other variations were detected (data
not shown).
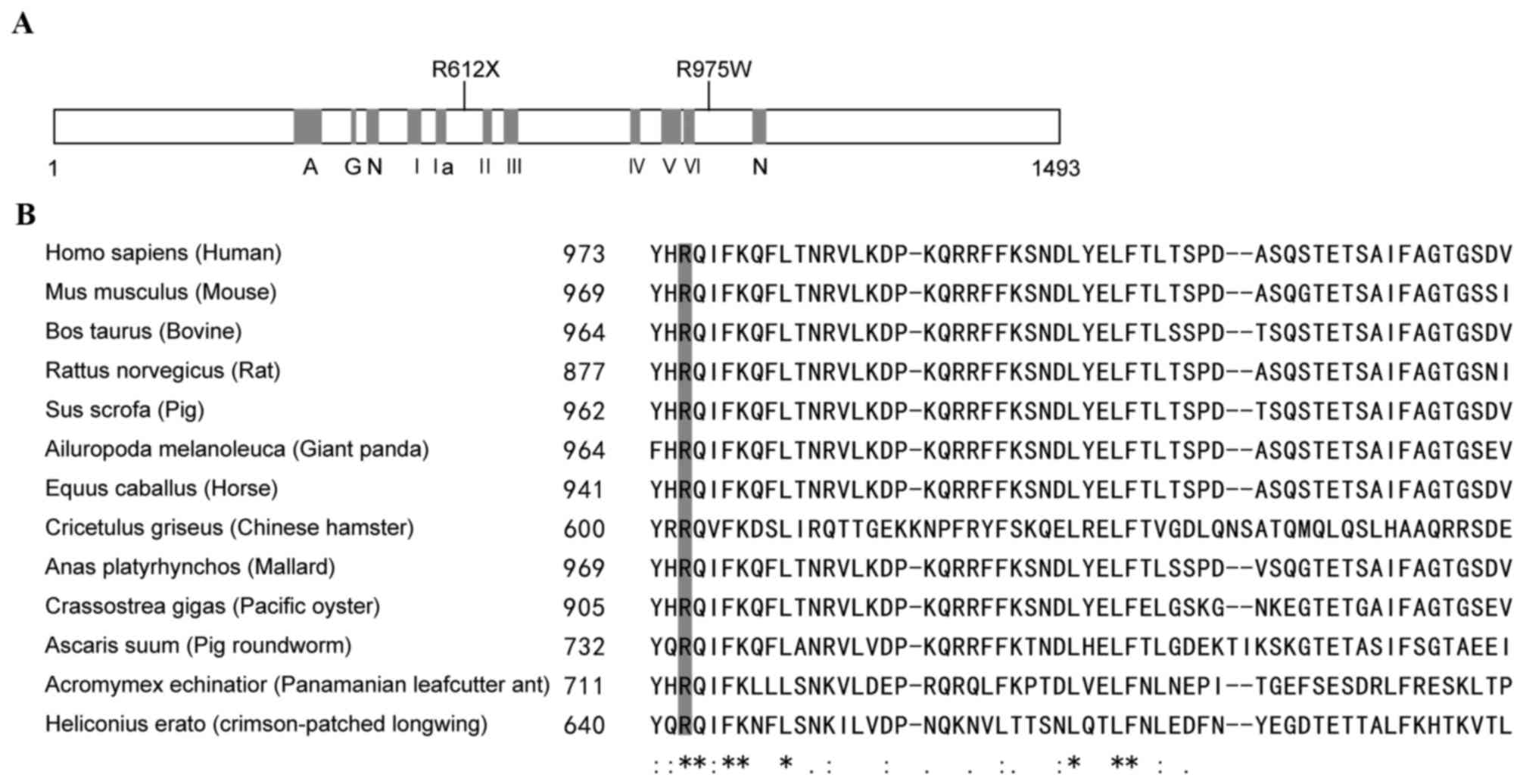
Structure-function correlations of the
ERCC6 mutation
ERCC6 encodes a 1493-amino acid protein which
contains seven helicase-like motifs that are divided into a
helicase ATP-binding domain and a helicase C-terminal domain. The
nonsense mutation, p.R612X, caused protein truncation at motif II
(Fig. 3A). The other mutation, an
amino acid substitution of Arg with Trp at codon 975, which was
highly conserved in mammals (Fig. 3A
and B), was characterized as ‘damaging’ using SIFT (sift.jcvi.org/) and Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) online
prediction tools.
ERCC6 prenatal diagnosis
Based on the molecular characterization of the
probands, a prenatal diagnosis on a 6-month old fetus of the family
was performed. Analysis of ERCC6 exons 9 and 16 revealed that the
fetus did not inherit any of the mutations present in the proband
(Fig. 2). A follow-up study of the
baby 2 years after birth did not reveal any CS symptoms.
Discussion
CS may be diagnosed by clinical characterization and
in vitro assays on fibroblasts by experienced clinicians.
Chinese patients with CS have rarely been reported before;
therefore, the final clinical diagnosis for our cases by clinicians
was progeroid syndrome. Thus, WES or targeted approaches by
next-generation sequencing for progeroid syndrome may be effective
to make a clear genetic diagnosis. The present study selected WES
in consideration of time and cost. The results demonstrated that
WES was an effective method of genetic testing case, and may be
applied to other undiagnosed inherited diseases.
The criteria for CS diagnosis was initially defined
by Nance and Berry in 1992 (1).
Developmental delay, growth failure and microcephaly were the
mandatory criteria, with at least three out of the five minor
criteria: Cutaneous photosensitivity, pigmentary retinopathy and/or
cataracts, sensorineural hearing loss, dental caries and cachectic
dwarfism (1). Recently, certain
modifications were proposed to improve the specificity of criteria:
Progressive courses of growth failure, microcephaly and
sensorineural hearing loss were specifically defined, enamel
hypoplasia was proposed to replace dental caries, and cachectic
dwarfism was replaced by enophthalmia (distinctive facial features)
(3). Brain imaging allows for a
more reliable diagnosis of CS: White matter hypomyelination,
atrophy of the cortex and cerebellum, and bilateral calcifications
of the putamen. Patients in the present study fulfilled these
clinical diagnostic criteria. In addition, the patients exhibited
other CS characteristics; cutaneous photosensitivity was initially
observed at disease onset and lasted ~4 years, and language ability
was lost during disease progression. However, hypodontia, a type of
hearing loss, which is considered a constant feature in patients
with CS, was not observed at 3 years old. Brain imaging by CT and
MRI scanning of the proband revealed the abnormalities mentioned
above. These were detectable at the disease onset, and
progressively worsened over 2 years. Based on these phenotypes,
type II CS was suggested as the diagnosis for these patients.
Previously, related or non-related patients with the same mutations
have been reported. These patients often exhibit characteristic
heterogeneities, including cataracts and deafness (4,8,9). The
siblings in the present report additionally had varying degrees of
cutaneous photosensitivity, suggesting that certain reported
characteristics are not inevitable in CS. Further investigation of
the patients carrying the same pathogenic mutations will facilitate
improvement of the criteria for CS diagnosis.
ERCC6, or CSB, is a component of the NER pathway and
primarily causes type II CS. There are numerous other CS subtypes
and NER dysfunction diseases involving ERCC6: COFS1 (MIM#
214150) (10), UV-sensitive
syndrome (UVSS) 1 (MIM# 600630) (11) and De Sanctis-Cacchione syndrome
(MIM# 609413) (12).
However, no clear phenotype-genotype correlations have been
reported. There have been >80 different mutations reported in
the CSB gene, with almost all types of mutations distributed along
the whole gene sequence, including short insertions and deletions,
nonsense, missense, splice and promoter mutations, and chromosomal
microdeletions including CSB (4,13–16).
Of these mutations, nonsense and splice mutations causing truncated
proteins account for a large proportion, and 12 missense mutations
have been reported (4,16). Previous studies have investigated
CSB-piggyBac3 (PGBD3) fusion protein interactions with functional
CSB on TCR, to elucidate genotype-phenotype correlations (4,17–19).
It has been proposed that CSB-PGBD3, consisting of the first five
exons of CSB and the PGBD3 transposon, may serve a deleterious role
in the absence of functional CSB and trigger the classical CS
phenotype. As a result, mutations downstream of intron 5 may cause
CS whereas mutations upstream of intron 5 may cause milder forms of
CS or UVSS (4,17). This was consistent with the
biallelic mutations [NM_000124.2: Exon9 c.1834C>T (p.Arg 612X),
and exon16 c.2923C>T (p. Arg975Tyr)] of the siblings in the
present study. Previous reports of CS patients additionally support
this hypothesis (13–16,20,21).
However, certain patients do not follow this paradigm (4), suggesting that the mutation spectrum
is further expanded; thus, additional evidence on correlations of
protein expression levels and disease severity is required.
Furthermore, structure-function correlations require investigation,
as biallelic missense mutations cause severe CS (4,16).
Prenatal diagnosis for CS has been performed using
various methods (9). In the
present study, sequencing of the ERCC6 amplicons for the causative
mutations from DNA of amniocytes was performed; the fetus did not
inherit pathogenic alleles from the parents. Follow-up studies for
the baby over 2 years additionally confirmed the prenatal
diagnosis. Although tetramer assays of cultured amniocytes are the
primary method for CS clinical confirmation, in a family with known
mutations, direct testing for the causative mutations will be
efficient and reliable.
In conclusion, CS is a rare multisystem disorder
that encompasses a wide spectrum of clinical phenotypes, from the
most severe prenatal subtype to the mildest late-onset subtype, or
UVSS. To date, ~130 CS patients have been genetically confirmed and
reported in the literature (4,13–16).
The present study reported two novel causative mutations on ERCC6
loci, and the clinical characteristics are described. These results
add to clinical and molecular data for elucidating
genotype-phenotype correlations in CS; however, further
investigations are required.
Acknowledgements
The authors would like to thank Professor J. Fan
(Fourth Military Medical University, Xi'an, China) for editing this
manuscript. The present study was supported by the China
Postdoctoral Science Foundation (grant no. 2014M562545) and the Key
Innovation Project of Shaanxi Province (grant no. 2013FWPT-06).
Glossary
Abbreviations
Abbreviations:
|
CS
|
cockayne syndrome
|
|
ERCC
|
excision repair cross-complementation
group
|
|
COFS
|
cerebro-oculo-facio-skeletal
|
|
NER
|
nucleotide-excision repair
|
|
WES
|
whole exome sequencing
|
|
TCR
|
transcription-coupled
nucleotide-excision repair
|
|
PCR
|
polymerase chain reaction
|
References
|
1
|
Nance MA and Berry SA: Cockayne syndrome:
Review of 140 cases. Am J Med Genet. 42:68–84. 1992. View Article : Google Scholar
|
|
2
|
Kleijer WJ, Laugel V, Berneburg M, Nardo
T, Fawcett H, Gratchev A, Jaspers NG, Sarasin A, Stefanini M and
Lehmann AR: Incidence of DNA repair deficiency disorders in western
Europe: Xeroderma pigmentosum, Cockayne syndrome and
trichothiodystrophy. DNA Repair (Amst). 7:744–750. 2008. View Article : Google Scholar
|
|
3
|
Laugel V: Cockayne syndrome: The expanding
clinical and mutational spectrum. Mech Ageing Dev. 134:161–170.
2013. View Article : Google Scholar
|
|
4
|
Laugel V, Dalloz C, Durand M, Sauvanaud F,
Kristensen U, Vincent MC, Pasquier L, Odent S, Cormier-Daire V,
Gener B, et al: Mutation update for the CSB/ERCC6 and CSA/ERCC8
genes involved in Cockayne syndrome. Hum Mutat. 31:113–126. 2010.
View Article : Google Scholar
|
|
5
|
Rapin I, Lindenbaum Y, Dickson DW, Kraemer
KH and Robbins JH: Cockayne syndrome and xeroderma pigmentosum.
Neurology. 55:1442–1449. 2000. View Article : Google Scholar :
|
|
6
|
Cleaver JE, Lam ET and Revet I: Disorders
of nucleotide excision repair: The genetic and molecular basis of
heterogeneity. Nat Rev Genet. 10:756–768. 2009. View Article : Google Scholar
|
|
7
|
Laugel V, Dalloz C, Stary A, Cormier-Daire
V, Desguerre I, Renouil M, Fourmaintraux A, Velez-Cruz R, Egly JM,
Sarasin A and Dollfus H: Deletion of 5′ sequences of the CSB gene
provides insight into the pathophysiology of Cockayne syndrome. Eur
J Hum Genet. 16:320–327. 2008. View Article : Google Scholar
|
|
8
|
Colella S, Nardo T, Mallery D, Borrone C,
Ricci R, Ruffa G, Lehmann AR and Stefanini M: Alterations in the
CSB gene in three Italian patients with the severe form of Cockayne
syndrome (CS) but without clinical photosensitivity. Hum Mol Genet.
8:935–941. 1999. View Article : Google Scholar
|
|
9
|
Falik-Zaccai TC, Laskar M, Kfir N, Nasser
W, Slor H and Khayat M: Cockayne syndrome type II in a Druze
isolate in Northern Israel in association with an insertion
mutation inERCC6. Am J Med Genet A. 146A:1423–1429. 2008.
View Article : Google Scholar
|
|
10
|
Jaakkola E, Mustonen A, Olsen P, Miettinen
S, Savuoja T, Raams A, Jaspers NG, Shao H, Wu BL and Ignatius J:
ERCC6 founder mutation identified in Finnish patients with COFS
syndrome. Clin Genet. 78:541–547. 2010. View Article : Google Scholar
|
|
11
|
Horibata K, Iwamoto Y, Kuraoka I, Jaspers
NG, Kurimasa A, Oshimura M, Ichihashi M and Tanaka K: Complete
absence of Cockayne syndrome group B gene product gives rise to
UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci
USA. 101:pp. 15410–15415. 2004; View Article : Google Scholar :
|
|
12
|
Colella S, Nardo T, Botta E, Lehmann AR
and Stefanini M: Identical mutations in the CSB gene associated
with either Cockayne syndrome or the DeSanctis-cacchione variant of
xeroderma pigmentosum. Hum Mol Genet. 9:1171–1175. 2000. View Article : Google Scholar
|
|
13
|
Ghai SJ, Shago M, Shroff M and Yoon G:
Cockayne syndrome caused by paternally inherited 5Mb deletion of
10q11.2 and a frameshift mutation of ERCC6. Eur J Med Genet.
54:272–276. 2011. View Article : Google Scholar
|
|
14
|
Zhang H, Gao J, Ye J, Gong Z and Gu X:
Maternal origin of a de novo microdeletion spanning the ERCC6 gene
in a classic form of the Cockayne syndrome. Eur J Med Genet.
54:e389–e393. 2011. View Article : Google Scholar
|
|
15
|
Xin B and Wang H: Identification of two
novel ERCC6 mutations in old order amish with cockayne syndrome.
Mol Syndromol. 3:288–290. 2013.
|
|
16
|
Yu S, Chen L, Ye L, Fei L, Tang W, Tian Y,
Geng Q, Yi X and Xie J: Identification of two missense mutations of
ERCC6 in three Chinese sisters with Cockayne syndrome by whole
exome sequencing. PLoS One. 9:e1139142014. View Article : Google Scholar :
|
|
17
|
Newman JC, Bailey AD, Fan HY, Pavelitz T
and Weiner AM: An abundant evolutionarily conserved CSB-PiggyBac
fusion protein expressed in Cockayne syndrome. PLoS Genet.
4:e10000312008. View Article : Google Scholar :
|
|
18
|
Bailey AD, Gray LT, Pavelitz T, Newman JC,
Horibata K, Tanaka K and Weiner AM: The conserved Cockayne syndrome
B-piggyBac fusion protein (CSB-PGBD3) affects DNA repair and
induces both interferon-like and innate antiviral responses in
CSB-null cells. DNA Repair (Amst). 11:488–501. 2012. View Article : Google Scholar :
|
|
19
|
Weiner AM and Gray LT: What role (if any)
does the highly conserved CSB-PGBD3 fusion protein play in Cockayne
syndrome? Mech Ageing Dev. 134:225–233. 2013. View Article : Google Scholar :
|
|
20
|
Xin B and Wang H: Identification of two
novel ERCC6 mutations in old order amish with cockayne syndrome.
Mol Syndromol. 3:288–290. 2013.
|
|
21
|
Shehata L, Simeonov DR, Raams A, Wolfe L,
Vanderver A, Li X, Huang Y, Garner S, Boerkoel CF, Thurm A, et al:
ERCC6 dysfunction presenting as progressive neurological decline
with brain hypomyelination. Am J Med Genet A. 164:2892–2900. 2014.
View Article : Google Scholar :
|