Introduction
Astragalus, known as Huang Qi in China,
comprises the dry roots of Astragalus membranaceus (Hsiao)
Bge or Astragalus membranaceus (Fisch) Bge. It has been used
as an important component of herbal prescriptions to reduce
swelling, drain pus and eradicate toxins for thousands of years
(1). Several biological active
ingredients have been obtained from Astragalus membranaceus,
including polysaccharides, saponins, flavonoids, amino acids and
trace elements. However, modern pharmacological investigations have
demonstrated that the primary pharmacological ingredients of
Astragalus membranaceus in wound healing are polysaccharides
and saponins, which have effects on the improvement of immune
function and the stimulation of cell physiology metabolism
(2). Yang et al (3) reported that Astragalus
polysaccharide (APS) has an effect on diabetic skin wounds.
Therefore, APS is considered to be important in promoting wound
healing.
To improve current understanding of APS on wound
healing properties and the possible mechanisms, the present study
purified a fraction, APS2-1, from a type of commercial
Astragali. Following purification, the effects of APS2-1 on
the proliferation, migration and cell cycle progression of human
fibroblasts were examined. In addition, the mRNA and protein
expression levels of cyclin D1 and IκBα in the presence of APS2-1
were evaluated. The secretion of transforming growth factor
(TGF)-β1, basic fibroblast factor (bFGF), and epidermal growth
factor (EGF) were also measured. Ultimately, the wound healing
properties of APS2-1 were evaluated in a scalded mouse model. The
aims of the present study were to provide valuable data for
evaluating the wound healing effect of APS2-1, in order to
effectively and appropriately exploit APS2-1 for treating wounds in
the future.
Materials and methods
Materials and chemicals
The Astragalus membranaceus roots were
purchased from Guangyi Chinese Herbal Cultivation Co., Ltd
(Fengzhen, China). DEAE-cellulose 52 and Sephadex G-100 were
purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany)
and Pharmacia; GE Healthcare Life Sciences (Uppsala, Sweden),
respectively. 3-(4,5)-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) and dimethylsulfoxide (DMSO) were purchased from
Sigma-Aldrich; Merck Millipore. DMEM, streptomycin and penicillin
were obtained from Hyclone; GE Healthcare Life Sciences (Logan, UT,
USA). ELISA kits for TGF-β1, bFGF, EGF and cyclin D1 were purchased
from Shanghai Honsun Biological Co., Ltd. (Shanghai China). TRIzol
reagent and SYBR Green I detection reagents were purchased from
Bio-Rad Laboratories, Inc., (Hercules, CA, USA). Bovine serum
albumin (BSA) was purchased from Sangon Biotech Co., Ltd.
(Shanghai, China). All primary antibodies were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The secondary
antibody was purchased from Wuhan Boster Biological Technology,
Ltd. (Wuhan, China). All other reagents were of analytical grade
and purchased from local chemical suppliers in China.
Extraction and purification of
APS
The Astragalus membranaceus roots were dried
at 50°C, broken and passed through a 100-eye mesh, followed by
defatting with 95% ethanol at room temperature for 48 h to remove
the majority of the polyphenols, pigments and monosaccharides
(performed three times) (4). The
defatted powder (100 g) was then extracted with distilled water
(m:v=1:5) at 80°C for 2 h, and centrifuged at 13,400 × g for 10 min
at room temperature. Following centrifugation, the supernatant was
concentrated in a rotary evaporator to 100 ml and then precipitated
by adding four times the volume of anhydrous ethanol for 12 h at
4°C. The precipitate was dissolved in 200 ml distilled water and
deproteinized using the Sevag method (5), producing the crude APS solution. The
crude APS solution was dialyzed against double-distilled water for
3 days and lyophilized to crude APS.
The crude APS (50 mg) was dissolved in 1 ml
distilled water, and fractionated on a DEAE-cellulose
anion-exchange column (1.6×25.0 cm) with double distilled water and
0.1 M NaCl at a flow rate of 2 ml/min, producing the two fractions
of APS-1 and APS-2, respectively. APS-2 was further purified on a
Sephadex G-100 column (1.6×50.0 cm) with double distilled water at
a flow rate of 1.00 ml/min, producing a homogeneous fraction,
APS2-1.
Infared (IR) and ultraviolet (UV)
spectroscopy
APS2-1 (3.0 mg) was ground with KBr and pressed into
a 1 mm pellet. The IR spectrum between 4,000 and 400
cm−1 was recorded on a Perkin-Elmer spectrometer. For
the UV spectrum, the aqueous solution of APS2-1 at 1.0 mg/ml was
scanned with wavelengths between 190 and 400 nm on a UV-vis
spectrophotometer.
In vitro wound healing assay
Analysis of cell viability
Human skin fibroblast cells (CCC-HSF-1; HSF) were
seeded at a density of 3,000 cells per well in a 96-well plate
containing DMEM with 10% (v/v) FBS. The cells were then incubated
in 1, 5 and 25 mg/l concentrations of APS2-1 for 48 h at 37°C. An
additional culture of cells in DMEM in the absence of APS2-1 was
used as control.
At the end of culture, 30 µl MTT-PBS solution (5
mg/ml) was added directly into each well. The plates were further
incubated for 4 h at 37°C, followed by the addition of 100 µl DMSO
to each well. The absorbance was recorded at 540 nm and measured
using a spectrophotometer (6).
Analysis of CCC-HSF-1 migration and cell
cycle
The effect of APS2-1 on CCC-HSF-1 migration was
measured using a Transwell migration assay (8.0 µm pore size;
Corning Incorporated, Corning, NY, USA) according to the
manufacturer's protocol. In brief, 1×104 CCC-HSF-1 cells
were seeded in the upper chamber of 24-well plates and cultured in
serum-free DMEM supplemented with 0.5% of BSA. APS2-1 was added
directly to the cell suspension at a final concentration of 25 mg/l
for 24, 48 and 72 h. Cells cultured in the same medium without
APS2-1 were used as the normal control., As a chemoattractant, 10%
FBS was added into the lower well of the culture plate. The cell
suspension was left in the inside of each insert to migrate at
37°C. After 24 h, the migrated cells on the bottom side of the
insert were fixed with paraformaldehyde and stained with
hematoxylin, following which images were captured. The cells were
counted from eight randomly-selected regions per well using a Nikon
E200® photomicroscope (Nikon Corporation, Tokyo, Japan),
equipped with the Moticam 2300® image capture system
(version 3.2; Motic Deutschland GmbH, Wetzlar, Germany). The data
were from three independent experiments performed in duplicate
(7).
In order to investigate the effect of APS2-1 on HSF
cell proliferation, the effect of APS2-1 on progression of the cell
cycle was evaluated. The HSF cells, which were in the exponential
phase of growth, were treated with APS2-1 (final concentration, 25
mg/l) for 24 h at 3×104 cells/well. Cells cultured in
DMEM in the absence of APS 2-1 were used as the normal control.
Subsequently, the cell supernatants were collected and the cells
were stained with propidium iodide. Following FACSCalibur flow
cytometry (BD Biosciences Franklin Lakes, NJ, USA), Mod-Fit LT™
version 3.2 analytic software (Verity Software House, Topsham, ME,
USA) was used to determine the DNA content of cells (8).
Reverse transcription-quantitative polymerase
chain reaction (RT-qPCR) analysis and western blot analysis
Following incubation of the HSF cells
(1×106 cells/well in a 96-well plate) with APS2-1 for 48
h at 37°C in a humidified incubator with 5% CO2, the
cells were collected to prepare total RNA and total protein. Total
RNA was extracted from the HSF cells, which had been cultured with
APS2-1 for 48 h, using TRIzol reagent (Bio-Rad Laboratories, Inc.).
The concentration of extracted RNA was determined using a UV
spectrophotometer. cDNA was synthesized from 500 ng total RNA using
PrimeScript™ RT reagent (Takara Bio, Inc., Otsu, Japan). The
primers used for qPCR were as follows: Cyclin D1, forward
5′-GAGACCATCCCCCTGACGGC-3′ and reverse 3′-TCTTCCTCCTCCTCGGCGGC-5′;
β-actin, forward 5′-CTGTCCCTGTATGCCTCTG-3′ and reverse
5′-ATGTCACGCACGATTTCC-3′. The reactions (containing 10 µl SYBR
Green Master Mix, 1 µl primers, 1 µl cDNA sample liquid, and 8 µl
diethyl pyrocarbonate) were performed using a SYBR PrimeScript
RT-PCR kit (Takara Bio, Inc.) with an ABI 7500 sequence detection
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
amplifying conditions were as follows: 12 min predenaturation at
95°C, and then 30 cycles of 94°C for 20 sec, 60°C for 30 sec, 72°C
for 30 sec. Each experiment was performed with three biological
replicates. As an internal control, levels of β-actin were
quantified in parallel with the target genes. Normalization and
fold changes were calculated using the 2−ΔΔCq method
(9).
The proteins in the nucleus and cytoplasm were
extracted using Sangon nuclear and cytoplasmic extraction reagents,
respectively, according to the manufacturer's protocol (Sangon
Biotech Co., Ltd.), and then quantified using a bicinchoninic acid
Protein Assay kit (Pierce; Thermo Fisher Scientific, Inc.). The
nuclear and cytoplasmic extracts (40 µg) were separated on a 10%
polyacrylamide gel and transferred onto a polyvinylidene difluoride
membrane. The blot was subsequently blocked with 5% non-fat milk in
TBST for 2 h at room temperature, and then incubated with the
following primary antibodies for 12 h at 4°C: Anti-nuclear factor
(NF)-κB p65 (#8242; 1:500), anti-inhibitor of NF-κBα (IκBα) (#4812;
1:1000), anti-cyclin D1 (#2978; 1:1,000), anti-β-actin (#4970;
1:1,000) and anti-histone H3 (#5748; 1:500), all purchased from
Cell Signaling Technology, Inc. This was followed by incubation
with goat anti-rabbit IgG-horseradish peroxidase (#BA1054; 1:200,
Wuhan Boster Biological Technology, Ltd.) for 2 h at room
temperature. The immunoreactive bands were visualized using ECL
western blot detection reagents (Thermo Fisher Scientific, Inc.).
The optical densities of bands were measured and quantified using
an image analysis system (ImageQuant LAS4000mini; GE Healthcare
Life Sciences).
In vivo wound healing assay
Animal experiments
Specific pathogen-free male C57BL/6 mice (age, 10–11
weeks; weight, 20±2 g) used in the wound healing assessment were
provided by Shanghai SLRC Laboratory Animal Co., Ltd.
(SCXK2003-0003; Shanghai, China). All mice were acclimatized under
conditions of 22–25°C and a 12-h light-dark cycle, with free access
to common rod-like diets and water. Following acclimation, the mice
were randomized into three groups (13 mice per group). Following
hair removal from the dorsal surface, the mice were anesthetized
with sodium pentobarbital (0.5 mg/g), and a 10-mm full-thickness
excisional skin wound was made on the back of each mouse. Each
wound was treated with 0.5 g ointment (APS2-1:
Vaseline/SDS/H2O=2:60:2:65) as the experimental group,
PBS: Vaseline/SDS/H2O=2:60:2:65 as the control group and
Jingwanhong ointment (Tianjin Darentang Jingwanhong Pharmaceutical
Co., Ltd, Tianjin, China) as the positive control for 3 weeks and
dressed with fresh gauze. All mice were housed individually.
Histopathological examination
Images of the wounds were captured with a
10-megapixel digital camera (Sony Corporation, Tokyo, Japan) at 7,
14 and 21 days, and an image analyzer (Image-Pro Plus 6.0; Media
Cybernetics, Inc., Rockville, MD, USA) was used to obtain
wound-size measurements. At the end of 7, 14 and 21 days, wound
healing was quantified by calculating the remaining wound area for
each group. The percentage of wound closure was calculated as
follows: wound closure (%)=(area of original wound-area of actual
wound)/area of original wound ×100. To assess the wound
histologically, mice at day 14 were sacrificed by cervical
dislocation for histological assessment. The harvested wound areas,
including a border of normal tissue, were immediately fixed in 10%
neutral-buffered formalin. The specimens were embedded in paraffin,
sectioned into 5 µm slices, and stained with hematoxylin and eosin
to determine the quality of wound healing using a Nikon
E200® photomicroscope equipped with the Moticam
2300® image capture system (10). The scalded tissue samples at 7 days
were collected and maintained at −80°C for further evaluation.
Biochemical analysis
The scalded tissue samples at 7 days were prepared
through homogenization in PBS, and the supernatants were collected
by centrifugation of the tissue homogenate at 9,300 × g under 4°C
for 10 min. The levels of TGF-β1, bFGF, EGF and cyclin D1 in the
scalded tissue were determined using commercial kits according to
the manufacturer protocols.
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analyzed using one-way analysis of variance
followed by a LSD-T range test using Graphpad Prism 5 Software
(GraphPad Software, Inc., San Diego, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Purification and spectra analysis of
APS2-1
The crude polysaccharide isolated from the root of
Astragalus by a series of experimental procedures, including
ethanol infusion, water extraction, deproteination, dialysis,
ethanol precipitation, and lyophilization, was termed APS.
Following purification via DEAE-cellulose 52 ion-exchange
charomatography, the polysaccharide aqueous solution was separated
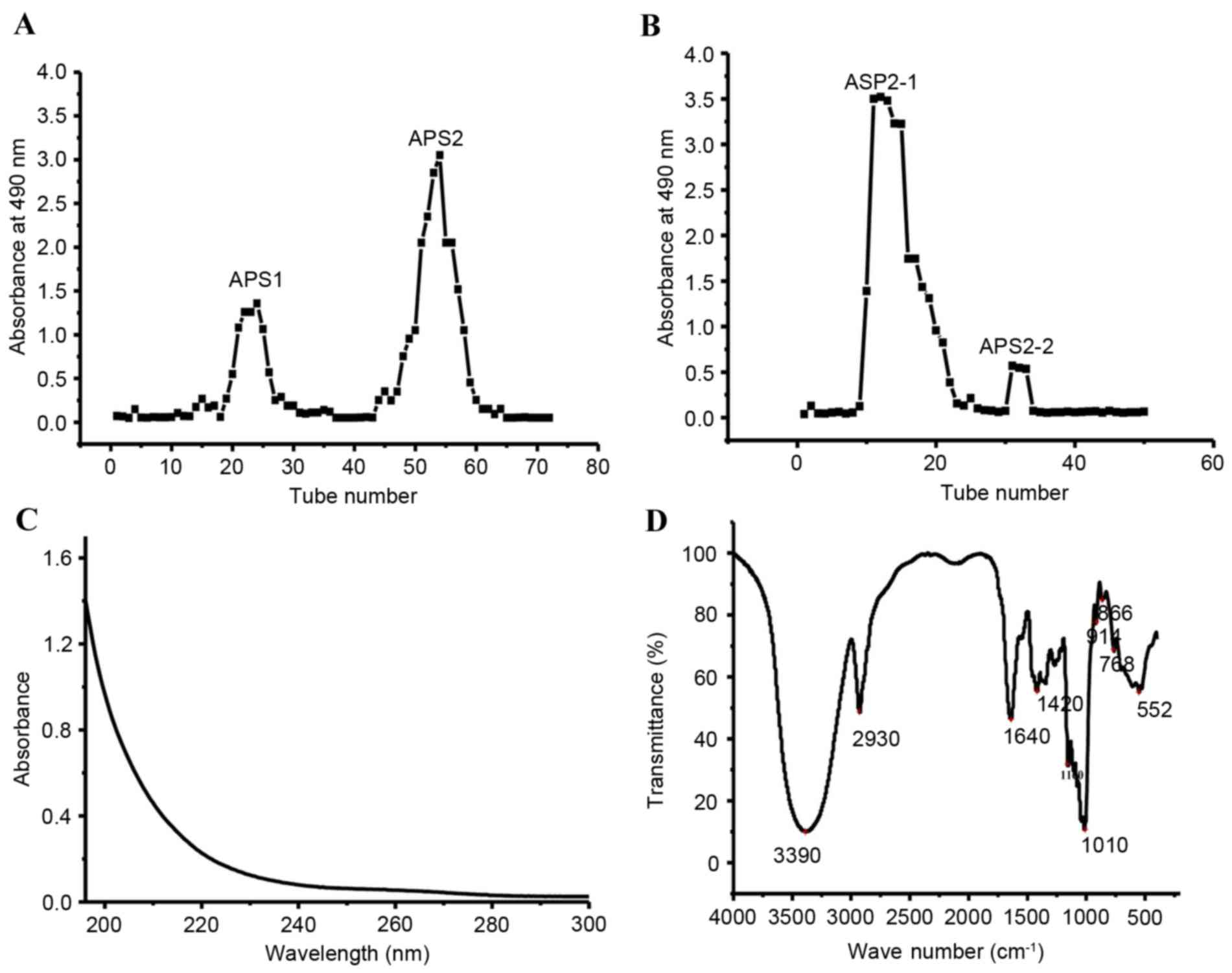
into two fractions, designated as APS1 and APS2 (Fig. 1A). The primary fraction, APS2, was
then collected and purified with Sephadex G-100 charomatography. As
a result, two purified fractions were generated, termed APS2-1 and
APS2-2 (Fig. 1B). Considering the
ease of obtaining APS2-1, the subsequent experiments focused on the
APS2-1 fraction.
The UV spectrum of the APS2-1 is shown in Fig. 1C. No peak was observed at 280 nm,
suggesting that APS2-1 carried no protein or polypeptide. The FT-IR
spectrum of APS2-1 showed the characteristic polysaccharide OH
(3,390 cm−1) and CO stretch (1,010 cm−1), as
shown in Fig. 1D (11). The band at 2,930 cm−1
was attributed to asymmetrical stretching vibration of the
CH2-group (12).
Absorption at 1,640 cm−1 was due to being associated
with water. The band at 1,420 cm−1 was assigned to the
C-OH deformation vibration with the contribution of C-O-C symmetric
stretching vibration of the carboxylate group. The band at ~1,160
cm−1 was associated with C-O-C asymmetric stretching
vibration in the glycosidic groups of polysaccharides (13). The absorption peak at 914
cm−1 was caused by D-glucopyranose, and the presence of
a β-glycosidic bond was indicated by the band at ~760
cm−1 (14).
Effect of APS2-1 on HSF cell
proliferation and migration
Fibroblast proliferation is an important step in
wound healing for granulation formation. The proliferation rate
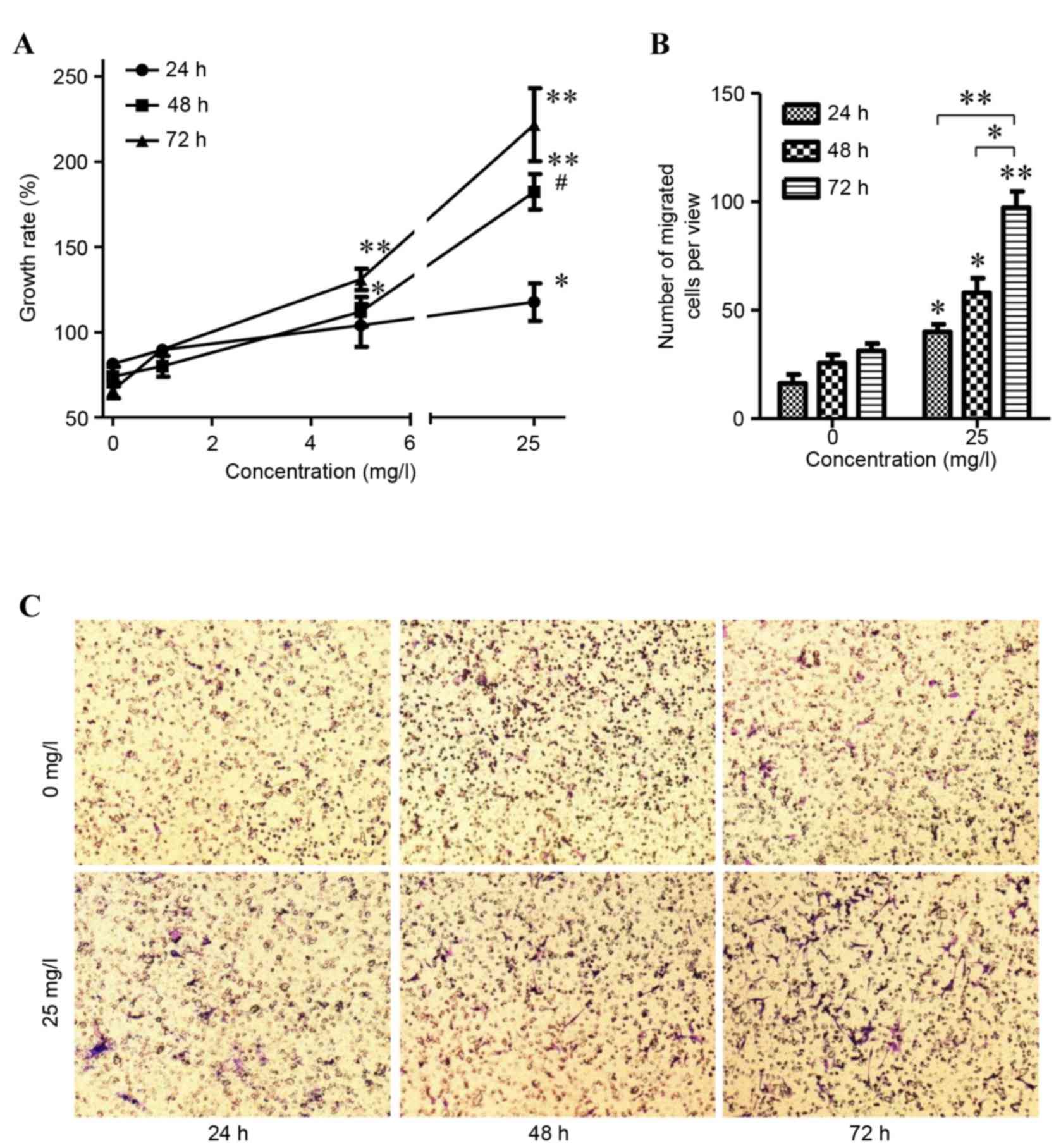
affects the wound healing rate. As shown in Fig. 2, APS2-1 concentrations of 1, 5 and
25 mg/l stimulated the proliferation of the HSF cells in a
dose-dependent manner, compared with 0 mg/l. As the concentration
of APS2-1 increased between 5 and 25 mg/l, it significantly
stimulated the proliferation of the HSF cells (P<0.01; Fig. 2A). The difference in proliferation
rates between HSF cells treated for 48 and 72 h with APS2-1 was not
significant at a concentration of 25 mg/ml (P>0.05; Fig. 2B). Therefore, in the following
experiments, the HSF cells treated with 25 mg/l APS2-1 for 48 h
were used in the cell migration assay.
Effect of 25 mg/l APS2-1 on HSF cell
migration
The effect of ASP2-1 on HSF cell migration was
determined, the results of which are shown in Fig. 2B and C. The numbers of migrated
cells following treatment with 25 mg/l APS2-1 for 24, 48 and 72 h
were higher, compared with those in the control. As the culture
duration increased, the number of migrated cells also increased,
and there were significant differences between treatment durations
(P<0.05 and P<0.01; Fig.
2B). These results showed that APS2-1 may be a stimulus for HSF
cell propagation and migration.
Effect of APS2-1 on cell cycle
progression of HSF cells
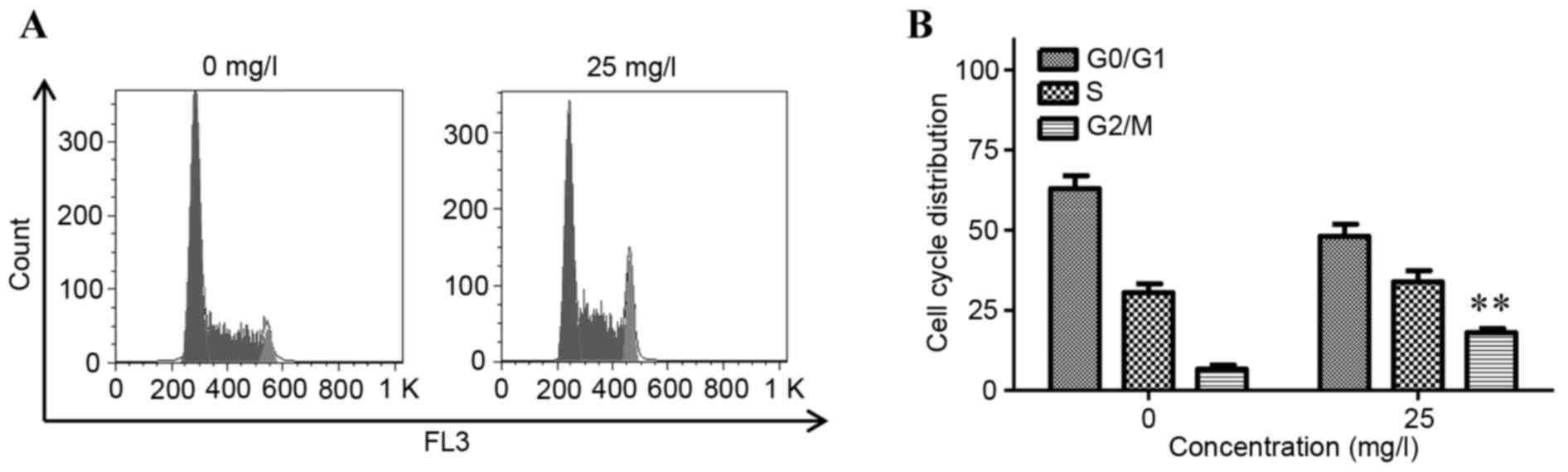
As shown in Fig. 3A and
B, a statistically significant increase was demonstrated in the
proportion of cells in the G2/M phase from 5.23±1.36 to 16.07±1.02%
in the 25 mg/l group (P<0.01). These results suggested that
APS2-1 promoted cell cycle progression by increasing the percentage
of cells in the S and G2/M phases, and decreasing the percentage of
cells in the G0/G1 phase, leading to promotion of cell division
cycle at the concentration of 25 mg/l.
Effect of APS2-1 on the expression of
IκBα and cyclin D1
Cyclin D1, an important cell cycle regulatory factor
at the G1 phase, can regulate E2F target genes, which assist cells
through the G1 phase and in entering the S phase. Downregulated
cyclin D1 promotes the transition of G1/S phase in cells, which
shortens the G1/S stage and accelerates cell proliferation
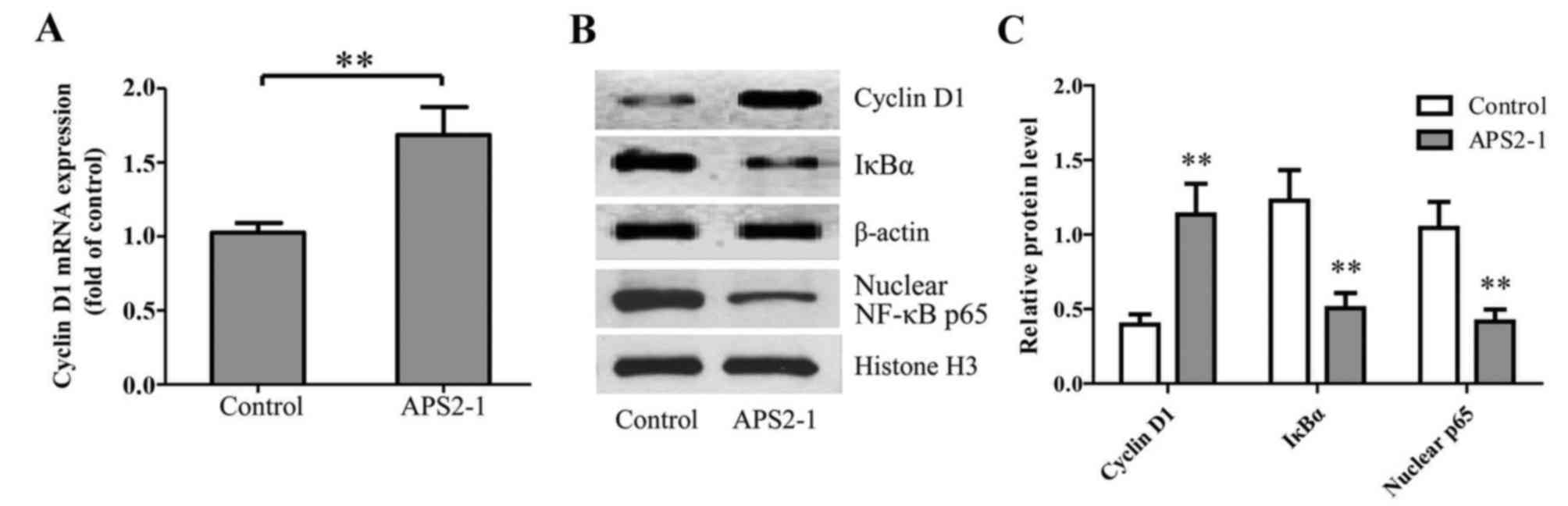
(15). As shown in Fig. 4A, 25 mg/l APS2-1 significantly
increased the mRNA expression of cyclin D1 in the HSF cells,
leading to accelerated cell cycle progression and promoting cell
proliferation.
Wound formation is coupled with the overproduction
of proinflammatory signaling cytokines, including, NF-κB and IκBα,
which have important pathological roles in the progression of wound
healing (16). NF-κB remains in an
inactive form when combined with inhibitory proteins (IκBs). When
cells are stimulated, IκBs are phosphorylated by IκB kinase,
leading to IκB degradation. As shown in Fig. 4B and C, APS2-1 significantly
(P<0.01) decreased the level of phosphorylation of IκBα, and
inhibited the translocation of the NF-κB p65 subunit from the
cytoplasm to the nucleus.
Wound healing effect of APS2-1 on the
C57BL/6 mice wound model
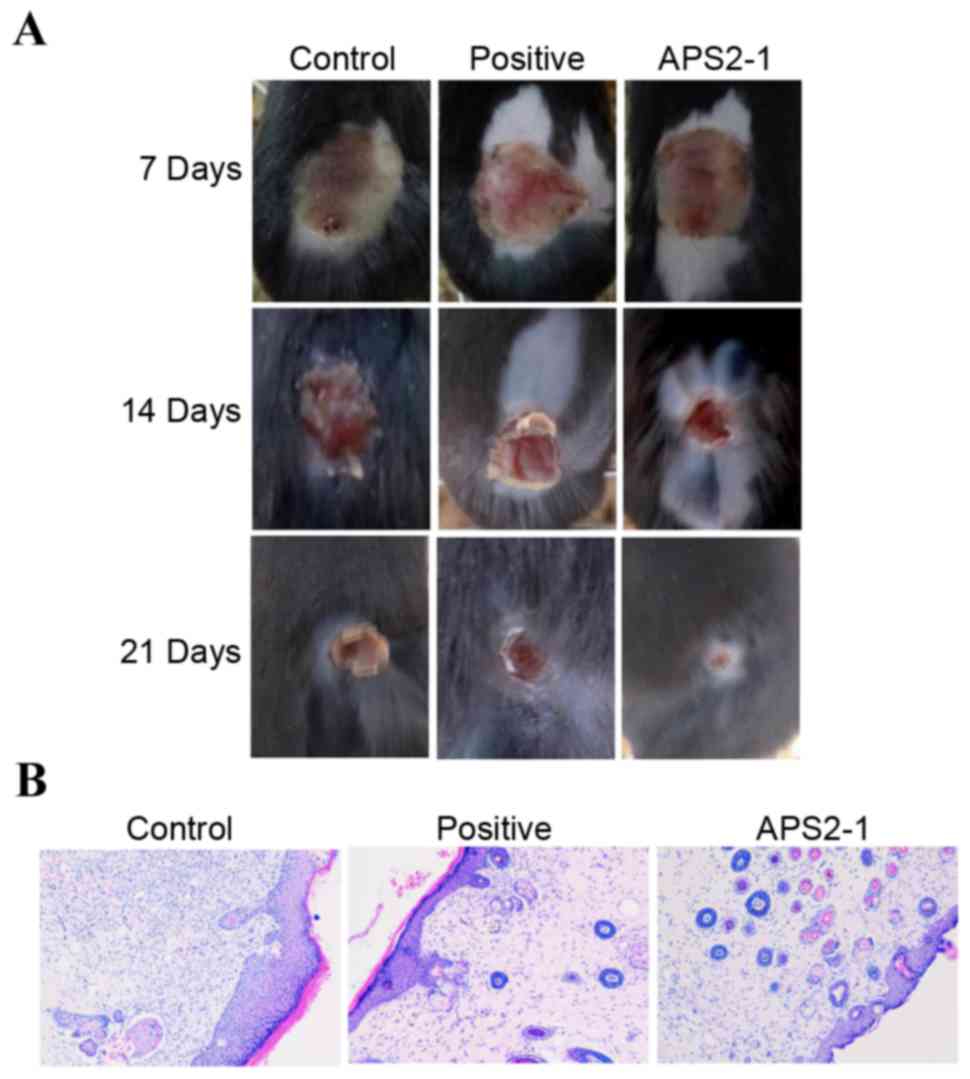
As shown in Fig.
5A, the APS2-1-treated wound groups exhibited accelerated wound
closure, compared with the control group and showed no significant
difference, compared with the positive group. This enhancement
appearance was observed at 7 days post-surgery, and became more
evident on day 14. At 21 days, complete wound closure was observed
in the APS2-1 treated mice, whereas no completion occurred in the
control groups (Fig. 5A). In order
to further examine the difference between the three groups,
histopathological examination was performed to determine the wound
tissue at 14 days. As shown in Fig.
5B, mass inflammatory cell infiltration was observed in the
control group. In the APS2-1-treated mice, the infiltration was
reduced and fibroblasts appeared (Fig.
5B). In addition, new blood vessels and skin appeared in the
APS2-1 and positive groups, suggesting that APS2-1 was capable of
healing wounds by promoting re-epithelialization and
revascularization.
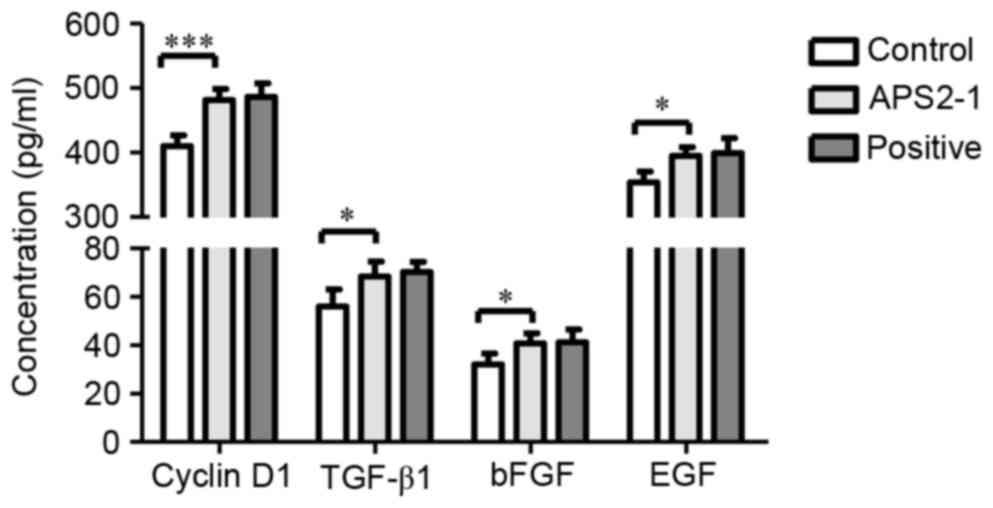
Effect of APS2-1 on levels of cyclin
D1, TGF-β1, bFGF and EGF
Fibroblasts, an important type of stromal cell,
appear in the process of wound healing and secrete numerous
cytokines, including TGF-β1, bFGF, EGF and insulin-like growth
factor 1. TGF-β1 is also important in wound repair, which can
promote fibroblast proliferation and the secretion of extracellular
matrix, and inhibit its degradation (17). As shown in Fig. 6, the levels of cyclin D1 and TGF-β1
in the APS2-1-treated mice were significantly higher (P<0.001
for cyclin D1, and P<0.05 for TGF-β1), compared with those in
the control mice, and no significant difference (P>0.05) was
found between the APS2-1 and positive groups.
EGF and bFGF are important stimulators in the
formation of re-epithelialization and keratinocyte migration in
wound healing. During the first 7 days post-wounding, the wounds
treated with APS2-1 had significantly increased EGF and bFGF,
compared with those in the control group (P<0.05; Fig. 6).
TGF-β1 is a member of the TGF-β family, which is
closely associated with anti-inflammatory effects, the regulation
of differentiation, chemostasis to macrophages and the control of
matrix synthesis in wound repair (18). As shown in Fig. 6, TGF-β1 was significantly increased
in the APS2-1-treated mice, compared with the mice in the control
group.
Discussion
Wound healing is a complex and ordered sequence of
events, which involves cell migration, proliferation, inflammation,
synthesis of extracellular matrix and wound remodeling (19). Generally, wound healing rate, wound
healing duration and pathological analysis of skin wounds are
direct and effective evaluation indices of wound healing effects
(20). The ordered sequence of
wound healing involves cell proliferation and migration, synthesis
of extracellular matrix, angiogenesis and remodeling (7).
When a wound occurs, there are several types of
cells recruited for involvement in the healing process (21). For example, neutrophils, monocytes
and mast cells infiltrate the site of injury and produce cytokines
(22). Fibroblasts, endothelial
cells and keratinocytes then proliferate and migrate due to the
released cytokines stimulated. Fibroblasts, one of the most common
connective tissue cells, are critical in wound healing. During
wound healing, fibroblasts interact with surrounding cells,
including fat cells, mast cells and keratinocytes, but also produce
extracellular matrix, glycoprotein, adhesive molecules and various
cytokines. Through cell-to-cell direct contact, and
cell-to-cytokine indirect contact, fibroblasts contribute to repair
wounds.
In addition, fibroblasts in wound edges begin to
grow and migrate into the provisional matrix of the wound to form
granulation tissue (7,21,23).
The roles of the exogenous growth factors associated with wound
repair have been demonstrated to vary, and include bFGF, EGF, TGF-α
and TGF-β (21,24,25).
Zhang et al (7) reported
that a type of traditional Chinese medicine exertes wound-healing
effects on the Hs27 human skin fibroblast cell line via activation
of the TGF-β1 pathway. In addition, Telgenhoff and Shroot (26) reported that chronic wound fluid
rapidly degrades exogenous growth factors and decreases the
production of cyclin D1. Thus, the evaluation of the expression of
cyclin D1 is also required for further investigation of the wound
healing properties of active components.
In the present study, the APS2-1 fraction was
purified from a type of commercial Astragali. It was then
demonstrated that APS2-1 inhibited the expression of IκBα and
cyclin D1 of HSF cells, and promoted the proliferation, migration
and cycle progression of the HSF cells. On further investigation,
in vitro experiments revealed the mRNA and protein
expression levels of IκBα and cyclin D1 also decreased in the
presence of APS2-1. APS2-1 was capable of promoting the
re-epithelialization, revascularization and cytokine secretion of
TGF-β1, bFGF and EGF. Ultimately, the mechanism underlying the
wound healing properties of APS2-1 may be associated with reducing
the inflammatory response, promoting cell cycle progression and the
secretion of cytokines.
References
|
1
|
Yao X, Wu G and Yu WM: The effect of
huangqi injection on immune function in rats with scald. Chin
Tradit Pat Med. 7:494–496. 2000.
|
|
2
|
Yi Z, Zhang M, Shi S and Wang S: Effects
of astragalus extract combined with rhEGF on scalded wound healing
and vascularization in rats. China Pharmacist. 1:9–12. 2012.(In
Chinese).
|
|
3
|
Yang Y, Wang F, Yin D, Fang Z and Huang L:
Astragulus polysaccharide-loaded fibrous mats promote the
restoration of microcirculation in/around skin wounds to accelerate
wound healing in a diabetic rat model. Colloids and surfaces B
Biointerfaces. 136:111–118. 2015. View Article : Google Scholar
|
|
4
|
Cui J, Gu X, Wang F, Ouyang J and Wang J:
Purification and structural characterization of an α-glucosidase
inhibitory polysaccharide from apricot (Armeniaca sibirica L. Lam.)
pulp. Carbohyd Polym. 121:309–314. 2015. View Article : Google Scholar
|
|
5
|
Staub A: Removal of protein-Sevag method.
Meth Carbohyd Chem. 5:5–6. 1965.
|
|
6
|
Lau KM, Lai KK, Liu CL, Tam JC, To MH,
Kwok HF, Lau CP, Ko CH, Leung PC, Fung KP, et al: Synergistic
interaction between Astragali Radix and Rehmanniae Radix in a
Chinese herbal formula to promote diabetic wound healing. J
Ethnopharmacol. 141:250–256. 2012. View Article : Google Scholar
|
|
7
|
Zhang Q, Fong CC, Yu WK, Chen Y, Wei F,
Koon CM, Lau KM, Leung PC, Lau CB, Fung KP and Yang M: Herbal
formula Astragali Radix and Rehmanniae Radix exerted wound healing
effect on human skin fibroblast cell line Hs27 via the activation
of transformation growth factor (TGF-beta) pathway and promoting
extracellular matrix (ECM) deposition. Phytomedicine. 20:9–16.
2012. View Article : Google Scholar
|
|
8
|
Gu M, Xu J, Han C, Kang Y, Liu T, He Y,
Huang Y and Liu C: Effects of berberine on cell cycle, DNA,
reactive oxygen species and apoptosis in L929 murine fibroblast
cells. Evid Based Complement Alternat Med. 2015:7963062015.
View Article : Google Scholar :
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
10
|
Liu X, Wang Z, Wang R, Zhao F, Shi P,
Jiang Y and Pang X: Direct comparison of the potency of human
mesenchymal stem cells derived from amnion tissue, bone marrow and
adipose tissue at inducing dermal fibroblast responses to cutaneous
wounds. Int J Mol Med. 31:407–415. 2013.
|
|
11
|
O'Connor NA, Abugharbieh A, Yasmeen F,
Buabeng E, Mathew S, Samaroo D and Cheng HP: The crosslinking of
polysaccharides with polyamines and dextran-polyallylamine
antibacterial hydrogels. Int J Biol Macromol. 72:88–93. 2015.
View Article : Google Scholar
|
|
12
|
Zhang S, He B, Ge J, Li H, Luo X, Zhang H,
Li Y, Zhai C, Liu P, Liu X and Fei X: Extraction, chemical analysis
of Angelica sinensis polysaccharides and antioxidant activity of
the polysaccharides in ischemia-reperfusion rats. Int J Biol
Macromol. 47:546–550. 2010. View Article : Google Scholar
|
|
13
|
Hosseini M Sadat, Hemmati K and Ghaemy M:
Synthesis of nanohydrogels based on tragacanth gum biopolymer and
investigation of swelling and drug delivery. Int J Biol Macromol.
82:806–815. 2016. View Article : Google Scholar
|
|
14
|
Xu JK, Li MF and Sun RC: Identifying the
impact of ultrasound-assisted extraction on polysaccharides and
natural antioxidants from Eucommia ulmoides Oliver. Process
Biochem. 50:473–481. 2015. View Article : Google Scholar
|
|
15
|
Coller HA: What's taking so long? S-phase
entry from quiescence versus proliferation. Nat Rev Mol Cell Biol.
8:667–670. 2007. View
Article : Google Scholar
|
|
16
|
Thomasova D, Mulay SR, Bruns H and Anders
HJ: p53-independent roles of MDM2 in NF-κB signaling: Implications
for cancer therapy, wound healing, and autoimmune diseases.
Neoplasia. 14:1097–1101. 2012. View Article : Google Scholar :
|
|
17
|
Thorey IS, Hinz B, Hoeflich A, Kaesler S,
Bugnon P, Elmlinger M, Wanke R, Wolf E and Werner S: Transgenic
mice reveal novel activities of growth hormone in wound repair,
angiogenesis, and myofibroblast differentiation. J Biol Chem.
279:26674–26684. 2004. View Article : Google Scholar
|
|
18
|
Barrientos S, Stojadinovic O, Golinko MS,
Brem H and Tomic-Canic M: Growth factors and cytokines in wound
healing. Wound Repair Regen. 16:585–601. 2008. View Article : Google Scholar
|
|
19
|
Peppa M, Stavroulakis P and Raptis SA:
Advanced glycoxidation products and impaired diabetic wound
healing. Wound Repair Regen. 17:461–472. 2009. View Article : Google Scholar
|
|
20
|
Ali A, Herndon DN, Mamachen A, Hasan S,
Andersen CR, Grogans RJ, Brewer JL, Lee JO, Heffernan J, Suman OE
and Finnerty CC: Propranolol attenuates hemorrhage and accelerates
wound healing in severely burned adults. Crit Care. 19:2172015.
View Article : Google Scholar :
|
|
21
|
Werner S and Grose R: Regulation of wound
healing by growth factors and cytokines. Physiol Rev. 83:835–870.
2003.
|
|
22
|
Trautmann A, Toksoy A, Engelhardt E,
Brocker EB and Gillitzer R: Mast cell involvement in normal human
skin wound healing: Expression of monocyte chemoattractant
protein-1 is correlated with recruitment of mast cells which
synthesize interleukin-4 in vivo. J Pathol. 190:100–106. 2000.
View Article : Google Scholar
|
|
23
|
Farmer DR and Nelson DM: A fibrin matrix
modulates the proliferation, hormone secretion and morphologic
differentiation of cultured human placental trophoblast. Placenta.
13:163–177. 1992. View Article : Google Scholar
|
|
24
|
Guo S and Dipietro LA: Factors affecting
wound healing. J Dent Res. 89:219–229. 2010. View Article : Google Scholar :
|
|
25
|
Schlingemann RO: Role of growth factors
and the wound healing response in age-related macular degeneration.
Graefes Arch Clin Exp Ophthalmol. 242:91–101. 2004. View Article : Google Scholar
|
|
26
|
Telgenhoff D and Shroot B: Cellular
senescence mechanisms in chronic wound healing. Cell Death Differ.
12:695–698. 2005. View Article : Google Scholar
|