Introduction
Ischemic preconditioning is an endogenous protective
mechanism whereby tissue subject to single or multiple brief
episodes of ischemia/reperfusion develops protection against
subsequent potentially lethal ischemic injury. Previous studies
have demonstrated that preconditioning is additionally triggered by
non-ischemic stress, including exposure to reactive oxygen species
(ROS) (1,2). However, the exact mechanisms
underlying preconditioning remain to be fully understood.
Ataxia-telangiectasia mutated (ATM) serine/threonine
kinase is a member of a superfamily of phosphatidylinositol (PI)
3-kinase-like kinases (3) and is
regarded as a lynchpin of cellular defenses to stress, particularly
antioxidative stress, maintaining cellular redox homeostasis
(4). Previous studies have
reported that ATM-deficient mice have increased levels of ROS,
particularly in the nervous system, leading to neuronal
degeneration (5,6). In addition, it has been reported that
activation of ATM in the cytoplasm protects neurons against
oxidative stress-induced damage (7). Patients with ataxia telangiectasia,
carrying mutations at the two ATM alleles
(ATM−/−), present with progressive cerebellar
ataxia and cerebellar degeneration (8,9).
There is accumulating evidence to suggest that ATM is a central
regulator of the response to DNA damage, including DNA repair,
telomere maintenance and regulation of the cell cycle (10–12).
Although ATM is expressed in the brain and neurons (13), its involvement in preconditioning
remains to be investigated. The present study investigated whether
H2O2 preconditioning protected against injury
induced by oxidative stress in Neuro-2a (N2a) cells, and the role
of ATM in H2O2 preconditioning.
Materials and methods
Cell culture and treatment
N2a mouse neuroblast cells (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) were cultured in high-Dulbecco's modified
Eagle's medium/OPTI-Minimal Essential Medium (1:1; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 5% (v/v)
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere of 5% CO2 at 37°C. Cells were
passaged by trypsinization and seeded at ~105 cells/ml.
When cells reached 60–80% confluence, the culture medium was
replaced with serum-free medium for 12–24 h. Cells were initially
treated with 20, 50, 100, 300, 600 and 1,000 µM
H2O2 for 12 h to assess the effect of
different doses of H2O2 on N2a cell
viability. The results of this treatment indicated that 600 uM
H2O2 was the median lethal dose. Therefore,
600 µM H2O2 was used for subsequent
experiments. Subsequently, cells were pretreated with 100 µM
H2O2 for 90 min followed by 12 h recovery and
subsequent exposure to the median lethal dose of 600 µM
H2O2 for 12 h. To evaluate the involvement of
ATM in preconditioning-induced protection, additional experiments
were performed. N2a cells were treated with 10 µM ATM-specific
inhibitor Ku55933 (Sigma-Aldrich; Merck KGaA) for 30 min or
transfected with ATM small interfering RNA (siRNA) for 36 h prior
to H2O2 preconditioning. Following
H2O2 preconditioning, these cells were
subjected to the lethal dose of 600 µM
H2O2.
Assessment of cell viability
An MTT assay was used to determine cell viability.
N2a cells were seeded at a density of 1×104 cells/well
in a 96-well culture plate. At the end of each experiment, 10 µl
MTT (0.5 mg/ml) was added to the cell medium and incubated for 4 h
at 37°C. Following incubation, MTT solutions were removed, dimethyl
sulfoxide was added, and the absorbance at 490 nm was measured
using a microplate reader. Data are expressed as a percentage of
the control, which was considered to be 100% viable.
siRNA transfection
N2a cells were transfected with 50 nM ATM siRNA or
Scramble control siRNA using Lipofectamine 2000® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The siRNA sequences utilized targeted the
following mouse ATM coding sequence: 5′-GCTTGAGGCTGATCCATATTC-3′.
To determine the effect of siRNA transfection, the N2a cells were
collected and lysed with lysis buffer [50 mM NaCl, 10 mM Tris-base,
1 mM EDTA, 2 mM sodium orthovanadate
(Na3VO4), 1 mM NaF, 1 mM
phenylmethyl-sulfonyl fluoride, 1% sodium dodecyl sulfate (SDS)] at
95°C for 10 min for western blot analysis 48 h following
transfection.
RT-qPCR
Total cellular RNA was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and cDNA was generated from 1 µg total RNA using the M-MLV
reverse transcription kit (Promega Corporation, Madison, WI, USA).
Quantification of gene copies was performed using the ABI 7300
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) with the Power SYBR® Green PCR Master Mix kit
(Promega Corporation). The primer sequences used were as follows:
Forward, 5′-GCACACGGATTGCTCAAGGA-3′ and reverse,
5′-GCCCATTCGGAATATGGATCAG-3′ for ATM (14); and forward,
5′-CAATGACCCCTTCATTGA-3′ and reverse, 5′-GACAAGCTTCCCGTTCTCAG-3′
for GAPDH (15). The following
thermocycling conditions were used: An initial predenaturation step
at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C
for 15 sec and annealing at 60°C for 60 sec. All amplification
reactions for each sample were repeated in at least triplicate, and
the relative expression values were normalized to those of GAPDH
using the 2−ΔΔCq method (16).
Western blot analysis
Cells were lysed with lysis buffer [50 mM NaCl, 10
mM Tris-base, 1 mM EDTA, 2 mM Na3VO4, 1 mM
NaF, 1 mM phenylmethyl-sulfonyl fluoride, 1% SDS], and the protein
content of the lysates was measured using the bicinchoninic acid
assay. Subsequently, 40 µg/lane protein was separated by 7.0%
SDS-polyacrylamide gel electrophoresis and electrophoretically
transferred to a nitrocellulose membrane. The membranes were
blocked with 5% bovine serum albumin (Beyotime Institute of
Biotechnology, Haimen, China) in TBS containing 1% Tween 20 at room
temperature for 1 h, and incubated with ATM (mouse monoclonal
antibody; dilution, 1:1,000; cat. no. sc-47739; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), phosphorylated (p)-ATM
antibodies (mouse monoclonal antibody; dilution, 1:500; cat. no.
sc-73615; Santa Cruz Biotechnology, Inc.) and β-actin (rabbit
polyclonal antibody; dilution, 1:1,000; cat. no. sc-130656; Santa
Cruz Biotechnology, Inc.) at 4°C overnight, followed by incubation
with a horseradish peroxidase-conjugated secondary antibody (either
goat anti-rabbit; dilution, 1:1,000; cat. no. A0208. Or goat
anti-mouse; dilution, 1:1,000; cat. no. A0216; Beyotime Institute
of Biotechnology) for 1 h at room temperature. The immunostaining
was visualized by enhanced chemiluminescence (Beyotime Institute of
Biotechnology). The blots were scanned, and the pixel count and
intensity of each band was quantified using the Scion Image
software (version 4.2.3.2; Scion Corporation, Frederick, MD, USA).
The results were normalized to β-actin expression.
Flow cytometric analysis of
apoptosis
Flow cytometry was performed as described in a
previous study by Tang et al (1). Briefly, treated N2a cells
(2×106) were collected and centrifuged at 5,000 ×
g at 4°C for 10 min. The cell pellet was resuspended in cold
PBS and fixed using 70% ethanol at 4°C for 1 h. The cells were then
centrifuged at 5,000 × g for 10 min, and resuspended in PBS.
DNase-free RNaseA (100 µl, 200 µg/ml) was added to the cells and
incubated at 37°C for 10 min. Cells were subsequently incubated
with propidium iodide (PI) at a final concentration of 100 mg/l,
filtered and incubated in the dark at room temperature for 10 min
prior to flow cytometric analysis. The PI fluorescence of
individual nuclei was measured using a flow cytometer
(Beckman-Coulter, Inc., Brea, CA, USA). DNA labeling data were
analyzed using CellQuest v.3.0 sampling software (BD Biosciences,
Franklin, NJ, USA) for flow cytometry.
Caspase-3 activity assay
Caspase-3 activity was measured using a colorimetric
CaspACE kit (Promega Corporation) according to the manufacturer's
protocol. Cells were lysed using the kit lysis buffer (Promega
Corporation) and centrifuged for 5 min at 5,000 × g and 4°C.
The supernatant was used for the measurement of caspase-3
activity.
Determination of
8-hydroxy-2′-deoxyguanosine (8-OHdG) in DNA
DNA was extracted from N2a cells with the DNA
Extractor kit (Wako Pure Chemical Industries, Ltd., Osaka, Japan)
according to the manufacturer's protocol. The extracted DNA was
digested with 8 units nuclease P1 (Cell Biolabs, Inc., San Diego,
CA, USA) for 2 h at 37°C in a final concentration of 20 mM sodium
acetate (pH 5.2), followed by treatment of 6 units alkaline
phosphatase for 1 h at 37°C in a final concentration of 100 mM Tris
(pH 7.5). The reaction mixture was centrifuged for 5 min at 6,000 ×
g and 4°C, and the supernatant was used for the 8-OHdG
Quantitation ELISA assay (catalog no. STA-320; Cell Biolabs, Inc.),
according to the manufacturer's protocol.
Statistical analysis
SPSS statistical software was used for statistical
analysis (version 18.0; SPSS, Inc., Chicago, IL, USA). The data are
expressed as the mean ± standard error of at least 3 replicate
experiments. Comparisons among multiple groups were performed using
one-way analysis of variance followed by the Student-Newman-Keuls
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of H2O2
preconditioning on cell viability following oxidative stress
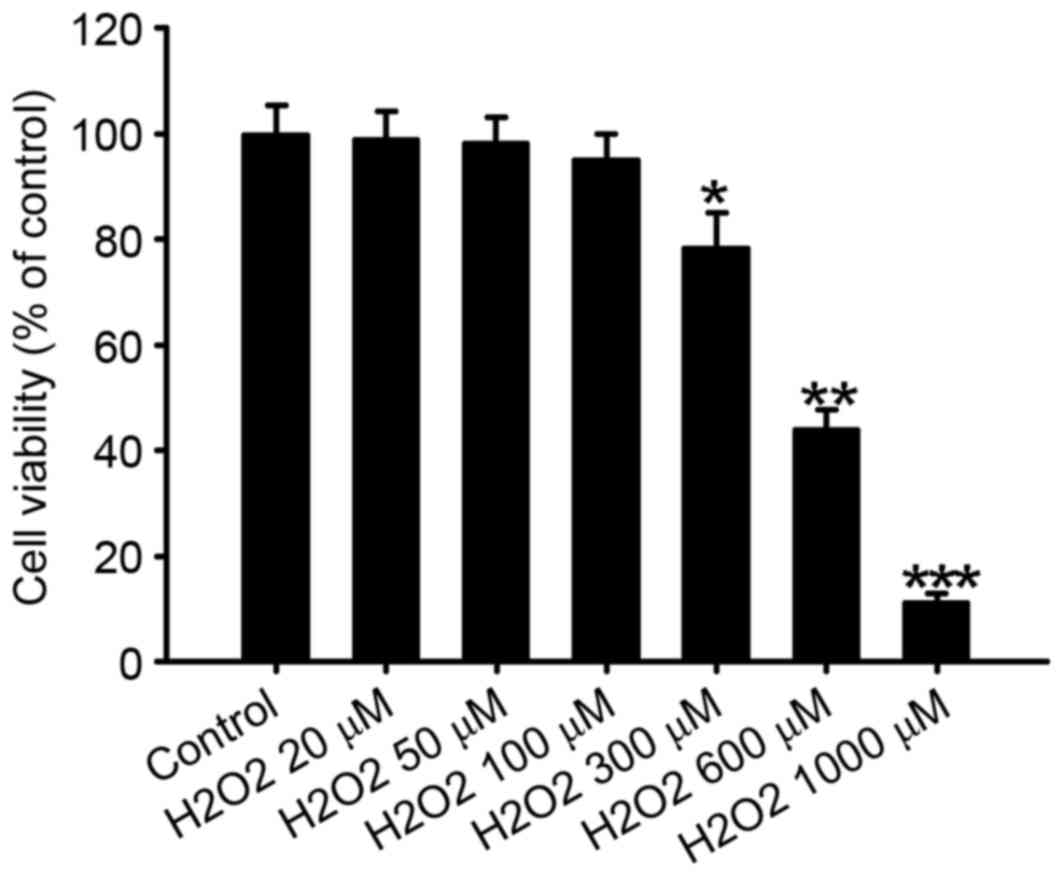
The effect of different concentrations of
H2O2 on N2a cell viability was evaluated by
MTT assay (Fig. 1). Treatment with
20–100 µM H2O2 did not significantly affect
N2a cell viability; however, concentrations of 300, 600 and 1,000
µM significantly decreased N2a cell viability compared with the
control, to 78.5±6.5 (P<0.05), 44.2±3.5 (P<0.01) and
11.4±1.4% (P<0.001), respectively. In addition, an MTT assay
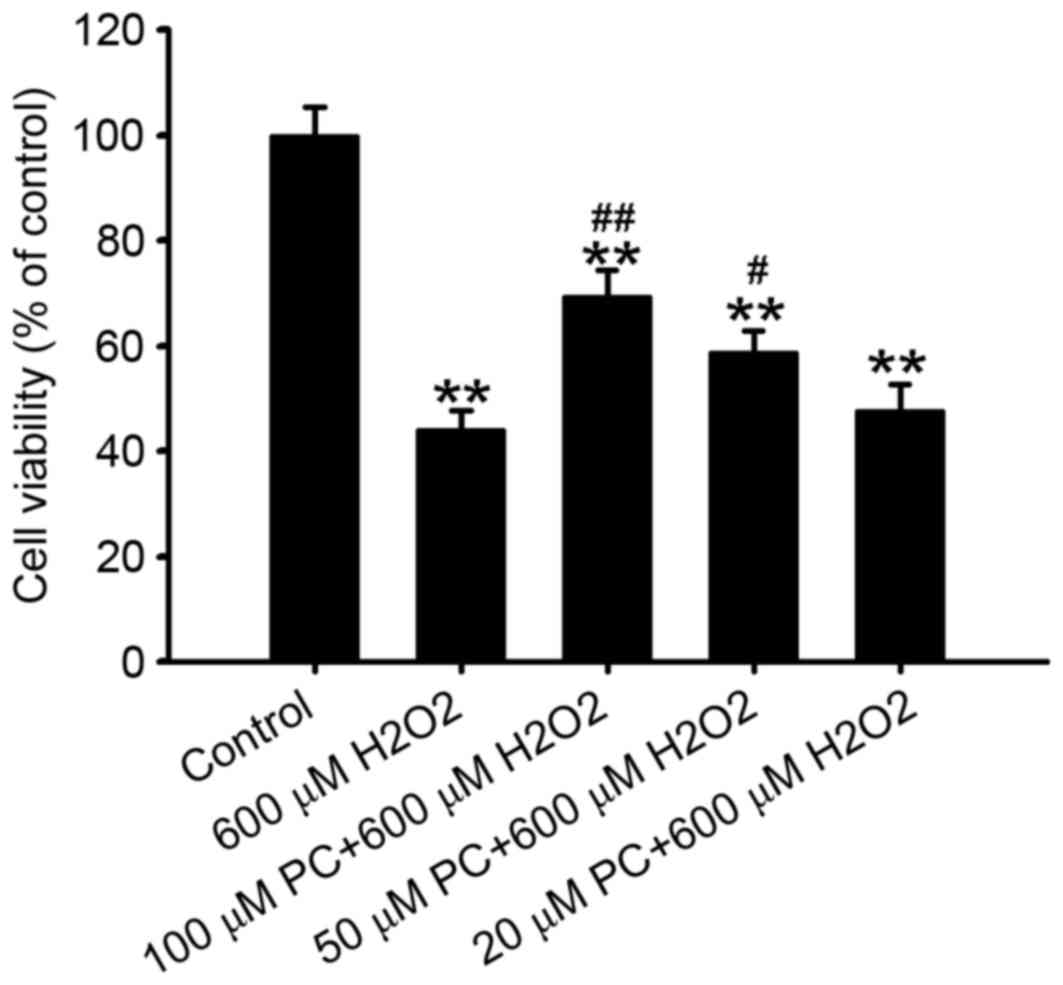
demonstrated that preconditioning cells with 50 or 100 µM
H2O2 attenuated the reduction of N2a cell
viability induced by 600 µM H2O2, compared
with the non-preconditioned group (P<0.05 and P<0.01,
respectively; Fig. 2), with 100 µM
H2O2 most effective. Preconditioning with 20
µM H2O2 failed to significantly attenuate the
reduction in cell viability induced by treatment with 600 µM
H2O2 (P>0.05; Fig. 2). Therefore, 100 µM
H2O2 was selected for preconditioning in
subsequent experiments.
H2O2
preconditioning decreases neuronal apoptosis, caspase-3 activity
and 8-OHdG content
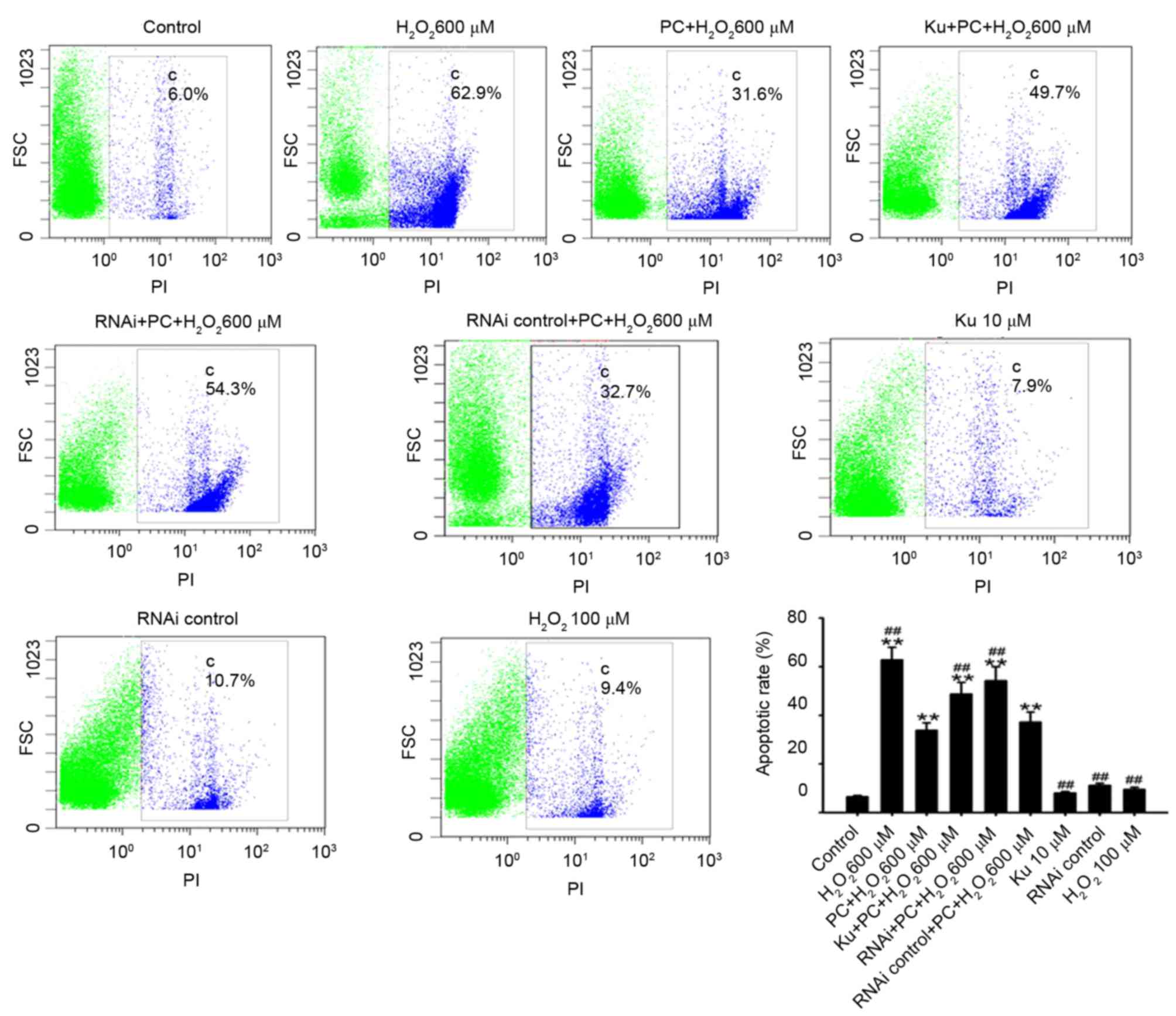
Following exposure of N2a cells to 600 µM
H2O2 for 12 h, the percentage of apoptotic
N2a cells increased significantly compared with the control
(62.8±5.2 vs. 6.5±0.5%; P<0.01; Fig. 3). Preconditioning with 100 µM
H2O2 for 90 min did not significantly alter
the apoptotic rate compared with the control (9.5±0.89 vs.
6.5±0.5%; n=5; P>0.05; Fig. 3);
however, subsequent 600 µM H2O2-induced
apoptosis was significantly inhibited following preconditioning
compared with the non-preconditioned group (33.8±3.1 vs. 62.8±5.2%,
respectively; n=5; P<0.01; Fig.
3).
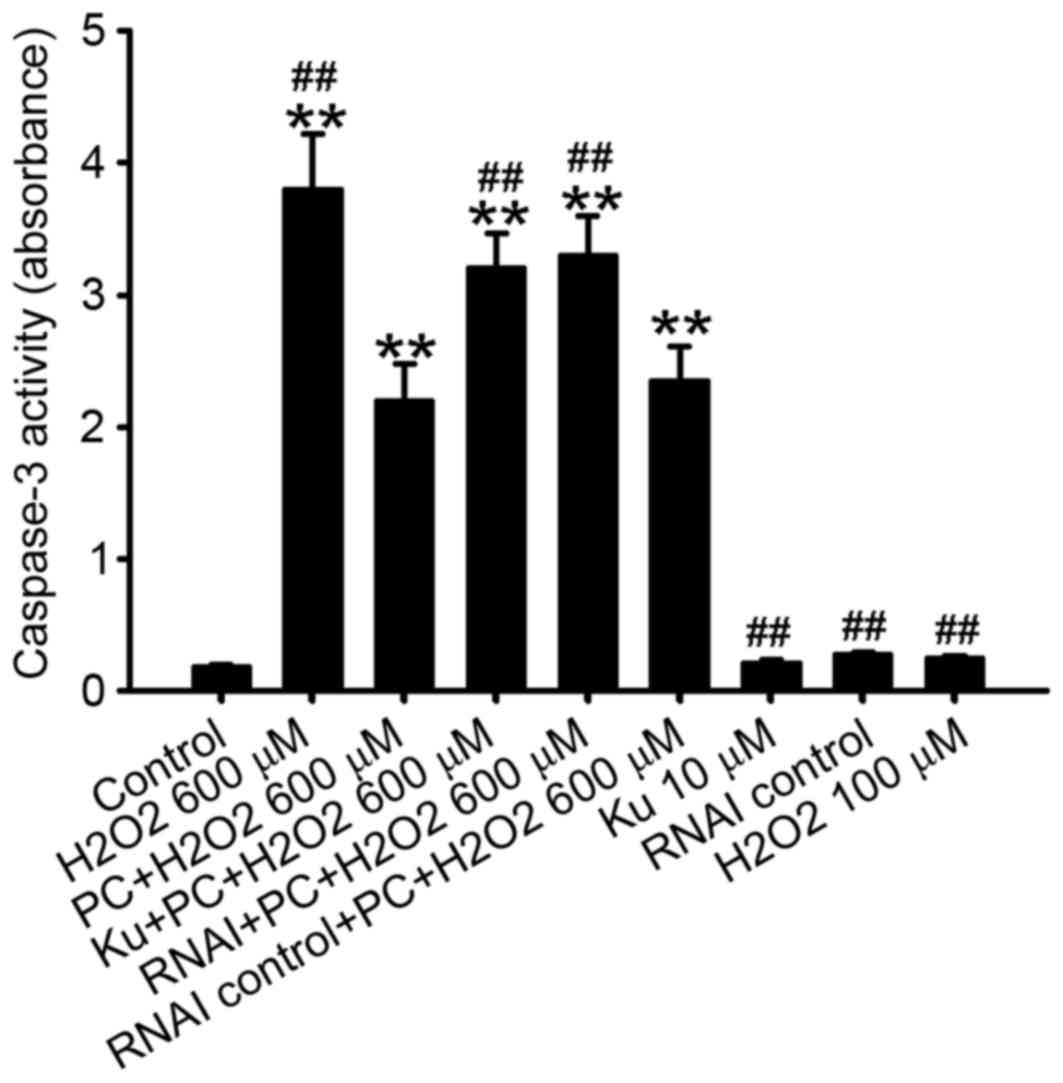
The caspase 3 protein is a member of the
cysteine-aspartic acid protease (caspase) family (17). Sequential activation of caspases
serves a central role in the execution-phase of cell apoptosis,
thus caspase-3 activity is a marker of cell apoptosis (18). Consistent with the results of flow
cytometric analysis, caspase-3 activity was significantly decreased
in N2a cells preconditioned with 100 µM H2O2
and exposed to 600 µM H2O2 compared with the
non-preconditioned group (P<0.01; Fig. 4).
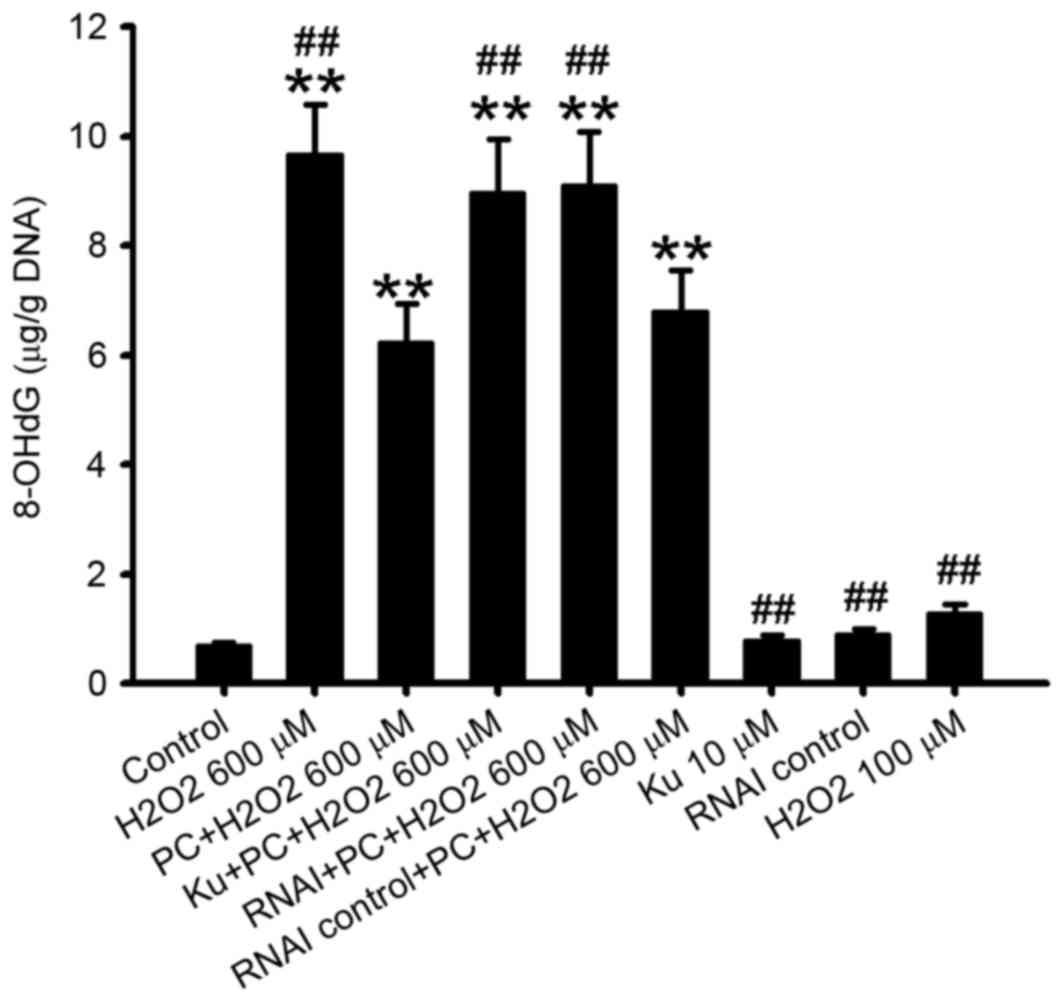
8-OHdG is a marker of oxidative stress (19). The present study observed that
8-OHdG content was additionally significantly decreased in N2a
cells preconditioned with 100 µM H2O2 and
exposed to 600 µMH2O2 compared with the
non-preconditioned group (P<0.01; Fig. 5).
Effect of H2O2
preconditioning on ATM expression
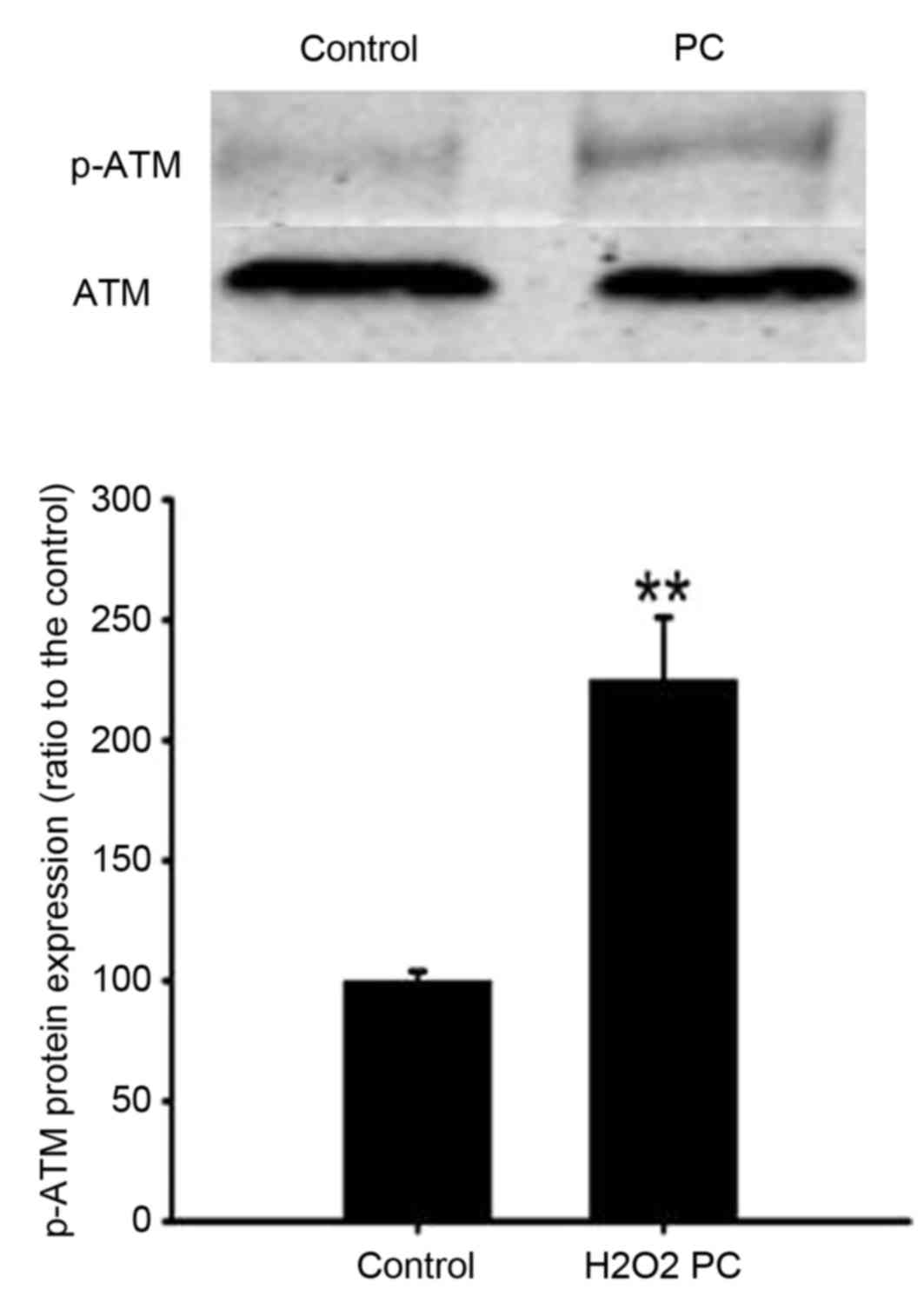
The effect of 100 µM H2O2
preconditioning on ATM mRNA and protein expression levels was
determined. Preconditioning of N2a cells with 100 µM
H2O2 for 90 min significantly increased p-ATM
protein expression levels compared with the control (P<0.01;
Fig. 6). Preconditioning with 100
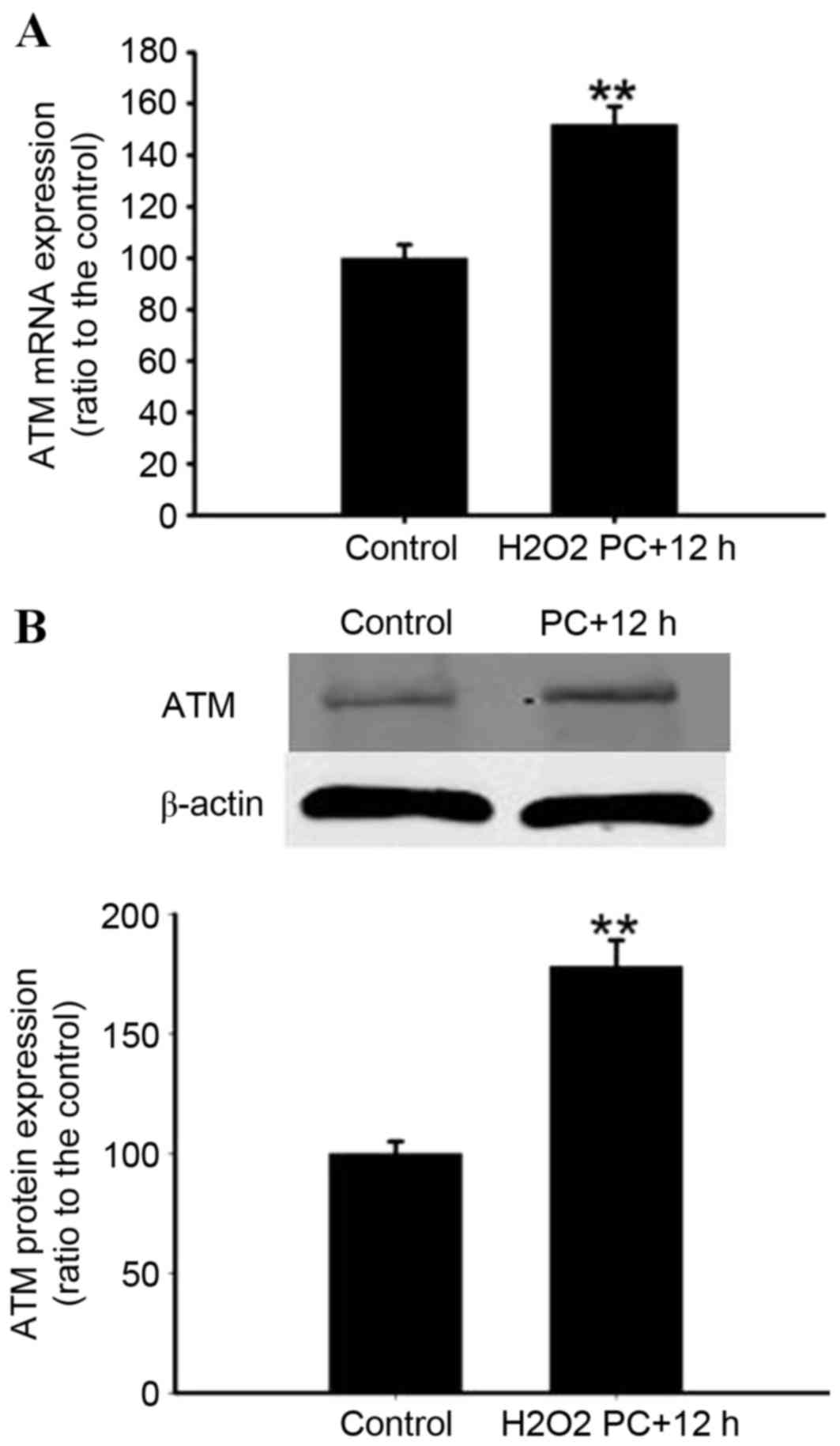
µM H2O2 for 90 min, then 12 h later,
increased the expression of ATM mRNA and protein when compared with
the control (P<0.01; Fig. 7A and
B).
ATM inhibition or knockdown attenuates
the protective effect of H2O2
preconditioning
To determine the involvement of ATM in
H2O2 preconditioning, RNA interference (RNAi)
with siRNA, and treatment with an ATM inhibitor, was performed.
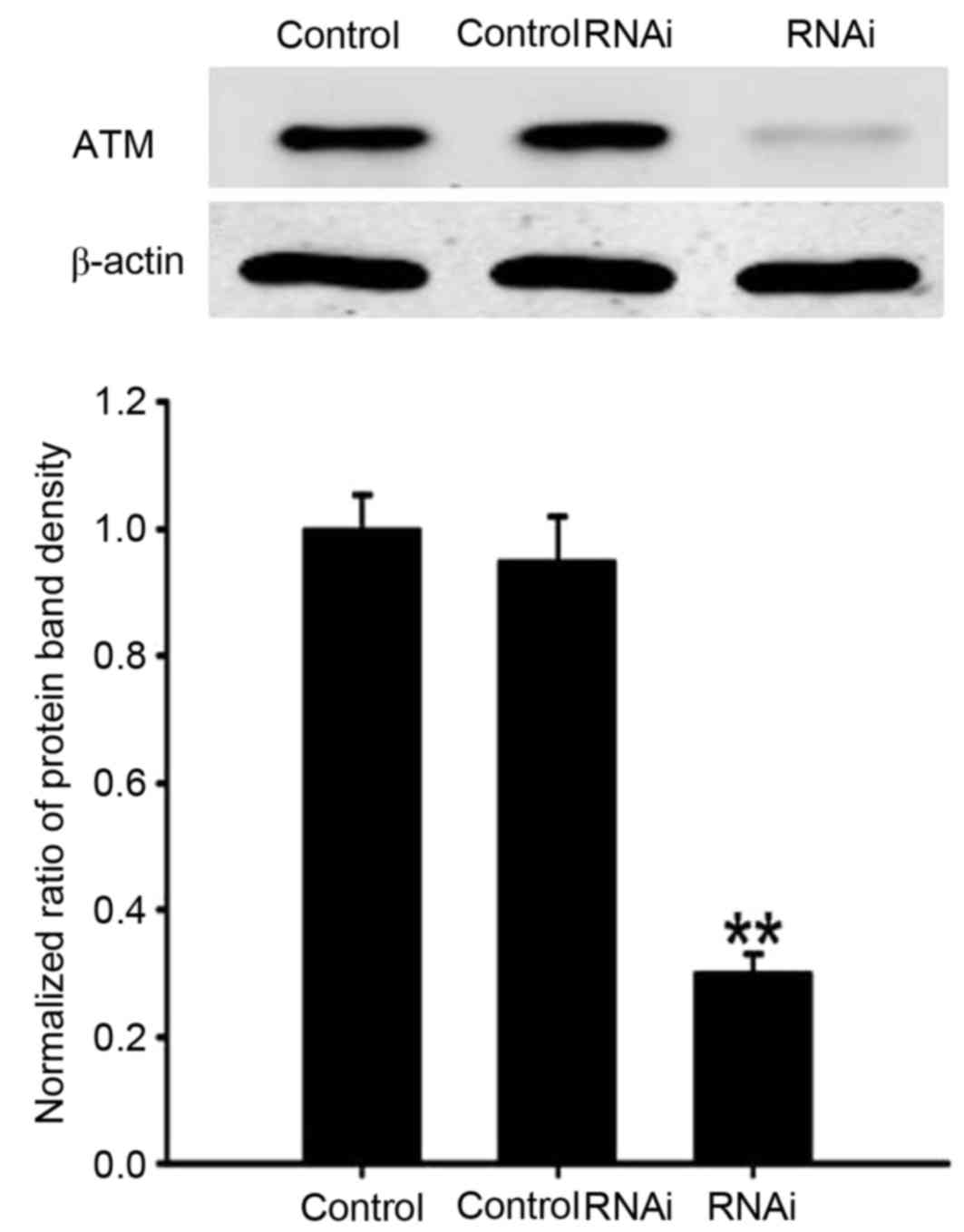
siRNA-mediated knockdown of ATM resulted in reduction of ATM
protein expression compared with the untransfected control and
scramble control groups (P<0.01; Fig. 8). When N2a cells were incubated
with 10 µmol/l Ku55933 for 36 h or transfected with 50 nM control
siRNA, the percentage of apoptotic cells was 8.0±0.68 and
11.1±0.96%, respectively, and were not significantly different
compared with the control group (6.5±0.5%; P>0.05; Fig. 3). However, the anti-apoptotic
effect of preconditioning with 100 µM H2O2
was decreased by pretreatment with the ATM inhibitor Ku55933 or
silencing of ATM with RNAi compared with the preconditioned group
(P<0.01 and P<0.01, respectively; Fig. 3). In addition, the decreased
caspase-3 activity observed following preconditioning with 100 µM
H2O2 was inhibited by the pretreatment of
cells with the ATM inhibitor Ku55933 or silencing of ATM with RNAi
compared with the preconditioned group (P<0.01 and P<0.01,
respectively; Fig. 4) and the
decrease in 8-OHdG content observed following preconditioning with
100 µM H2O2 was also inhibited by
pretreatment with the ATM inhibitor Ku55933 or silencing of ATM
with RNAi compared with the preconditioned group (P<0.01 and
P<0.01, respectively; Fig.
5).
Discussion
The results of the present study revealed that
H2O2 preconditioning protects N2a cells
against oxidative stress-induced injury, H2O2
preconditioning upregulates ATM mRNA and protein expression levels,
and pretreatment with an ATM inhibitor or knockdown of ATM
abrogates the protective effects of H2O2
preconditioning against lethal H2O2-induced
cell injury. This demonstrated that ATM may mediate the protective
effects of H2O2 preconditioning.
Oxidative stress induced by ROS is a primary cause
of ischemia/reperfusion injury; however, previous studies have
reported that ROS generated from brief ischemia/reperfusion events
triggers preconditioning-like protection. Brief exposure to
exogenous oxygen species protected PC12 cells and neurons against
subsequent serious oxidative stress injury via opening of surface
KATP channels (20),
increasing expression of Bcl-2 (1)
and hypoxia-inducible factor-1α protein (21), or enhancing the expression and
functional activities of volume-activated chloride channels
(22). The present study observed
that H2O2 preconditioning protected against
oxidative stress-induced injury in N2a cells, as assessed by MTT
assays, flow cytometry, and analysis of capase-3 activity and
8-OHdG content.
Although numerous previous studies have been
performed, the cellular and molecular mechanisms underlying
preconditioning remain to be fully clarified. A previous study
reported that activation of ATM regulates cell redox homeostasis in
various w ays, including the enhancement of glucose-6-phosphate
dehydrogenase activity, thereby increasing the intracellular
nicotinamide adenine dinucleotide phosphate and glutathione content
significantly (10). ATM-deficient
lymphoid stem cells exhibit mitochondrial dysfunction and a
significant increase in ROS; exogenous ATM restores mitochondrial
function and reduces the generation of ROS (23). ATM is additionally present in the
peroxisomes, regulating catalase activity (24). Previous studies have reported that
when PC12 cells or neurons were subjected to metabolic stress
including serum starvation, ATM regulated the insulin-associated
signaling pathway and inhibited neuronal apoptosis (25,26).
In addition, a previous study indicated that histone
acetyltransferase 4 accumulates more readily in the nuclei of
ATM-deficient neurons, and inhibits myocyte enhancer factor
2A/cyclic adenosine monophosphate response element
binding-dependent transcription to promote neurodegeneration
(27). Based on these findings,
ATM is regarded as an essential endogenous protective protein
against stress (4).
As the preconditioning process induces endogenous
protective mechanisms, it was hypothesized that ATM may be involved
in H2O2 preconditioning. Therefore, the
effect of H2O2 preconditioning on the
expression levels of ATM was measured. Notably,
H2O2 preconditioning was observed to increase
the protein expression levels of p-ATM, which indicated that
H2O2 preconditioning activated ATM. Following
H2O2 preconditioning for 12 h, ATM mRNA and
protein expression levels increased, which supported this
hypothesis. Additionally, the ATM inhibitor Ku55933, or knockdown
of ATM using RNAi, attenuated the protective effect of
H2O2 preconditioning against oxidative
stress-induced injury. These data suggested that ATM is involved in
H2O2 preconditioning.
In conclusion, the results of the present study
demonstrated, to the best of our knowledge for the first time, that
H2O2 preconditioning activates ATM and
upregulates ATM mRNA and protein expression levels in N2a cells.
Treatment with the ATM inhibitor, Ku55933, or silencing of ATM with
RNAi attenuated the protective effect of H2O2
preconditioning in N2a cells. These results provide insight into
the mechanisms underlying the involvement of ATM in
H2O2 preconditioning. In addition, the
present study highlights the potential of the ATM protein as a key
therapeutic target for the prevention and treatment of ischemic
brain damage.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (grant no. 81301057 to J.H.W.
and grant no. 30971428 to D.F.W), the Foundation of Health and
Family Planning Commission of Hubei province (grant no. WJ2015MB029
to D.F.W) and the Chenguang Plan of Wuhan Municipal Science and
Technology Bureau (grant no. 2014070404010226 to J.L.).
References
|
1
|
Tang XQ, Feng JQ, Chen J, Chen PX, Zhi JL,
Cui Y, Guo RX and Yu HM: Protection of oxidative preconditioning
against apoptosis induced by H2O2 in PC12 cells: Mechanisms via
MMP, ROS, and Bcl-2. Brain Res. 1057:57–64. 2005. View Article : Google Scholar
|
|
2
|
León OS, Menéndez S, Merino N, Castillo R,
Sam S, Pérez L, Cruz E and Bocci V: Ozone oxidative
preconditioning: A protection against cellular damage by free
radicals. Mediators Inflamm. 7:289–294. 1998. View Article : Google Scholar :
|
|
3
|
Shiloh Y: ATM and related protein kinases:
Safeguarding genome integrity. Nat Rev Cancer. 3:155–168. 2003.
View Article : Google Scholar
|
|
4
|
Bhatti S, Kozlov S, Farooqi AA, Naqi A,
Lavin M and Khanna KK: ATM protein kinase: The linchpin of cellular
defenses to stress. Cell Mol Life Sci. 68:2977–3006. 2011.
View Article : Google Scholar
|
|
5
|
Kuang X, Yan M, Ajmo JM, Scofield VL,
Stoica G and Wong PK: Activation of AMP-activated protein kinase in
cerebella of Atm-/-mice is attributable to accumulation of reactive
oxygen species. Biochem Biophys Res Commun. 418:267–272. 2012.
View Article : Google Scholar :
|
|
6
|
Kamsler A, Daily D, Hochman A, Stern N,
Shiloh Y, Rotman G and Barzilai A: Increased oxidative stress in
ataxia telangiectasia evidenced by alterations in redox state of
brains from Atm-deficient mice. Cancer Res. 61:1849–1854. 2001.
|
|
7
|
Kim TS, Kawaguchi M, Suzuki M, Jung CG,
Asai K, Shibamoto Y, Lavin MF, Khanna KK and Miura Y: The ZFHX3
(ATBF1) transcription factor induces PDGFRB, which activates ATM in
the cytoplasm to protect cerebellar neurons from oxidative stress.
Dis Model Mech. 3:752–762. 2010. View Article : Google Scholar
|
|
8
|
Frappart PO and McKinnon PJ:
Ataxia-telangiectasia and related diseases. Neuromolecular Med.
8:495–511. 2006. View Article : Google Scholar
|
|
9
|
Hoche F, Seidel K, Theis M, Vlaho S,
Schubert R, Zielen S and Kieslich M: Neurodegeneration in ataxia
telangiectasia: What is new? What is evident? Neuropediatrics.
43:119–129. 2012.
|
|
10
|
Cosentino C, Grieco D and Costanzo V: ATM
activates the pentose phosphate pathway promoting anti-oxidant
defence and DNA repair. EMBO J. 30:546–555. 2011. View Article : Google Scholar
|
|
11
|
Dobbin MM, Madabhushi R, Pan L, Chen Y,
Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y and Tsai LH: SIRT1
collaborates with ATM and HDAC1 to maintain genomic stability in
neurons. Nat Neurosci. 16:1008–1015. 2013. View Article : Google Scholar :
|
|
12
|
Khanna KK, Lavin MF, Jackson SP and
Mulhern TD: ATM, a central controller of cellular responses to DNA
damage. Cell Death Differ. 8:1052–1065. 2001. View Article : Google Scholar
|
|
13
|
Ditch S and Paull TT: The ATM protein
kinase and cellular redox signaling: Beyond the DNA damage
response. Trends Biochem Sci. 37:15–22. 2012. View Article : Google Scholar
|
|
14
|
Li MJ, Wang WW, Chen SW, Shen Q and Min R:
Radiation dose effect of DNA repair-related gene expression in
mouse white blood cells. Med Sci Monit. 17:BR290–BR297. 2011.
View Article : Google Scholar :
|
|
15
|
Li JZ, Wu JH, Yu SY, Shao QR and Dong XM:
Inhibitory effects of paeoniflorin on
lysophosphatidylcholine-induced inflammatory factor production in
human umbilical vein endothelial cells. Int J Mol Med. 31:493–497.
2013.
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Alnemri ES, Livingston DJ, Nicholson DW,
Salvesen G, Thornberry NA, Wong WW and Yuan J: Human ICE/CED-3
protease nomenclature. Cell. 87:1711996. View Article : Google Scholar
|
|
18
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ and
Los M: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar
|
|
19
|
Zhang N, Komine-Kobayashi M, Tanaka R, Liu
M, Mizuno Y and Urabe T: Edaravone reduces early accumulation of
oxidative products and sequential inflammatory responses after
transient focal ischemia in mice brain. Stroke. 36:2220–2225. 2005.
View Article : Google Scholar
|
|
20
|
Tang XQ, Chen J, Tang EH, Feng JQ and Chen
PX: Hydrogen peroxide preconditioning protects PC12 cells against
apoptosis induced by oxidative stress. Sheng Li Xue Bao.
57:211–216. 2005.
|
|
21
|
Chang S, Jiang X, Zhao C, Lee C and
Ferriero DM: Exogenous low dose hydrogen peroxide increases
hypoxia-inducible factor-1alpha protein expression and induces
preconditioning protection against ischemia in primary cortical
neurons. Neurosci Lett. 441:134–138. 2008. View Article : Google Scholar :
|
|
22
|
Zhu L, Zuo W, Yang H, Zhang H, Luo H, Ye
D, Lin X, Mao J, Feng J, Chen L and Wang L: Involvement of
volume-activated chloride channels in H2O2 preconditioning against
oxidant-induced injury through modulating cell volume regulation
mechanisms and membrane permeability in PC12 cells. Mol Neurobiol.
48:205–216. 2013. View Article : Google Scholar
|
|
23
|
Ambrose M, Goldstine JV and Gatti RA:
Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid
cells. Hum Mol Genet. 16:2154–2164. 2007. View Article : Google Scholar
|
|
24
|
Watters D, Kedar P, Spring K, Bjorkman J,
Chen P, Gatei M, Birrell G, Garrone B, Srinivasa P, Crane DI and
Lavin MF: Localization of a portion of extranuclear ATM to
peroxisomes. J Biol Chem. 274:34277–34282. 1999. View Article : Google Scholar
|
|
25
|
Li Y, Xiong H and Yang DQ: Functional
switching of ATM: Sensor of DNA damage in proliferating cells and
mediator of Akt survival signal in post-mitotic human neuron-like
cells. Chin J Cancer. 31:364–372. 2012. View Article : Google Scholar :
|
|
26
|
Yang DQ, Halaby MJ, Li Y, Hibma JC and
Burn P: Cytoplasmic ATM protein kinase: An emerging therapeutic
target for diabetes, cancer and neuronal degeneration. Drug Discov
Today. 16:332–338. 2011. View Article : Google Scholar
|
|
27
|
Li J, Chen J, Ricupero CL, Hart RP,
Schwartz MS, Kusnecov A and Herrup K: Nuclear accumulation of HDAC4
in ATM deficiency promotes neurodegeneration in ataxia
telangiectasia. Nat Med. 18:783–790. 2012. View Article : Google Scholar :
|