Introduction
Temporomandibular disorder (TMD) comprises a series
of conditions, which cause pain and dysfunction in the muscles and
joints between the lower jaw and the base of the skull. TMD is a
complex and multifactorial disease, with physical, social and
psychological factors usually associated with its etiology
(1,2). Although progress has been made, the
precise pathogenesis of TMD remains to be fully elucidated, which
leads to difficulty in diagnosis and therapy. Usually, the
temporomandibular cartilage withstands pressures of frequent jaw
movement and biting hard food (3).
Abnormal elevated intra-articular pressure and local inflammatory
response are important in TMD. The differential expression of
inflammatory cytokines has been found in patients with TMD,
including monocyte chemotactic protein 1, interleukin (IL)-1 and
IL-8 (4). Inflammation can also
stimulate the production of matrix metalloproteinases (MMPs),
downregulate the expression of tissue inhibitor of
metalloproteinases (TIMPs) and accelerate the degradation of
cartilage matrix (5). An imbalance
between MMPs and TIMPs has been reported to be important in the
progression of TMD (6).
IL-1 is a potent pro-inflammatory cytokine, which
functions as the gatekeeper for inflammation (7). It is produced by a variety of cells
and acts on almost every organ system of the body. IL-1 consists of
two distinct proteins (IL-1α and IL-1β), which bind to the type 1
IL-1 receptor to activate a downstream pro-inflammatory pathway
(8). IL-1β, a cytokine released by
synovial cells and macrophages, causes inflammation of the
articular cartilage (9).
The Notch signaling pathway consists of a family of
four type I transmembrane receptors (Notch1-4), which undergo
proteolytic processing by a furin-like convertase during transit to
the cell surface (10). It is
important in the growth, differentiation and survival of various
cell types in diverse tissues (11). In the canonical signaling pathway,
binding of a ligand triggers sequential receptor cleavage, and
results in the release and translocation of the Notch1
intracellular domain (NICD) to the nucleus, where it functions as a
transcriptional activator (12,13).
Several lines of evidence have demonstrated that Notch is important
in regulating the responsiveness of immune cells to stimulation and
infection (14,15). Furthermore, inflammatory mediators
can increase the expression of Notch (16), and the upregulation of Notch
signaling elevates responses to interferon-γ, leading to increased
expression of inflammatory cytokines in macrophages (17,18).
However, the role of Notch1 signaling in IL-1β-stimulated
temporomandibular chondrocytes remains to be elucidated.
The present study investigated whether the
inhibition of Notch1 is able to inhibit inflammatory responses and
cartilage destruction in TMD by establishing an in vitro
model using temporomandibular chondrocytes.
Materials and methods
Isolation and culture of
chondrocytes
The temporomandibular chondrocytes were isolated
from the articular cartilage of 15 male Sprague-Dawley rats
(6-week-old; 140–160 g). Animal care and all procedures were
performed according to institutional guidelines and were approved
by the Ethics Committee of Shandong University (Shangdong, China).
All animals were housed in the same room on a 12 h light:dark cycle
at 22°C and had full access to standard chow and water. All
surgical procedures were performed under sodium pentobarbital
anesthesia and all efforts were made to minimize suffering.
Briefly, the harvested cartilage was minced and incubated in a
trypsin-containing solution for 2 h at 37°C. The minced cartilage
was then washed with phosphate-buffered saline (PBS; Sigma-Aldrich;
KGaA Millipore, Darmstadt, Germany) and incubated with 0.2%
collagenase (Sigma-Aldrich; KGaA Millipore) at 37°C overnight.
Following digestion, the chondrocytes were collected via
centrifugation at 800 × g for 15 min at 37°C and cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal calf serum
(Gibco; Thermo Fisher Scientific, Inc.), 100 IU/ml penicillin and
100 µg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.). The
cells were serum-starved overnight prior to treatment. Cells in
passages 1–3 were used in all experiments.
Cell treatment and gene
inhibition
The primary temporomandibular chondrocytes were
cultured at a density of 5,000 cells/well in DMEM at 37°C in an
atmosphere of 5% CO2 and treated with 10 ng/ml IL-1β
(Sigma-Aldrich; KGaA Millipore). The cells were harvested following
incubation for 24 h. To inhibit the expression of Notch1, the cells
were transfected with Notch1 small interfering (s)iRNA (GenePharma,
Shanghai, China) or negative control siRNA using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Experiments were
performed 24 h following transfection. The cells in the Notch1
group were stimulated with IL-1β following inhibition of Notch1 by
siRNA.
Nuclear protein extraction
The nuclear proteins of the chondrocytes were
extracted using an NE-PER Nuclear and Cytoplasmic Extraction
Reagent kit (Pierce; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. The protein was quantified using the
bicinchoninic acid (BCA) method (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). All extracts were stored at −80°C until
use.
Enzyme-linked immunosorbent assay
(ELISA)
The inflammatory cytokines in the cell culture
supernatant were measured using ELISA kits for tumor necrosis
factor (TNF)-α and IL-6 (R&D Systems, Inc., Minneapolis, MN,
USA), as described previously (19). All spectrophotometric readings were
performed using an absorption spectrometer (Thermo Fisher
Scientific, Inc.).
Measurement of mRNA expression using
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analysis
Total RNA was extracted from the cultured cells
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) and
reverse transcribed to cDNA using SYBR-Green Supermix (Bio-Rad
Laboratories, Hercules, CA, USA). The 1 µg mRNA levels were
measured using qPCR analysis. The thermocycling conditions for PCR
amplification were as follows: 95°C for 5 min, 36 cycles at 95°C
for 10 sec, annealing at 56°C for 30 sec and elongation at 72°C for
30 sec. The primer sequences were as follows: Notch1, forward
5′-CGGGTCCACCAGTTTGAATG-3′ and reverse 5′-GTTGTATTGGTTCGGCACCAT-3′;
intercellular adhesion molecule 1 (ICAM-1), forward
5′-TTGGAAGCC-TCATCCG-3′ and reverse 5′-CAATGTTGCGAGACCC-3′;
inducible nitric oxide synthase (iNOS), forward
5′-GTTCTCAGCCCAACAATACAAGA-3′ and reverse
5′-GTGGACGGGTCGATGTCAC-3′; GAP DH, forward
5′-GGATGACCTTGCCCACAGCCT-3′ and reverse
5′-ATCTCTGC-CCCCTCTGCTGA-3′. qPCR was performed on the iCycler
real-time PCR system (Bio-Rad Laboratories, Inc.) and relative
quantification was performed using the comparative quantification
(Cq) method (20).
Western blot analysis
The concentrations of protein extracted from
chondrocytes were quantified using the BCA method with a protein
assay kit (Beyotime Institute of Biotechnology, Nantong, China).
The samples (50 µg) were separated on 10% SDS-PAGE and
electrophoretically transferred onto a polyvinylidene difluoride
membrane (GE Healthcare Life Sciences, Piscataway, NJ, USA). The
blots were blocked for 2 h with 5% non-fat dry milk in
Tris-buffered saline at room temperature and then incubated
overnight with anti-Notch1 (catalog no. 36085; 1:1,000), anti-NICD
(catalog no. 4147; 1:1,000), anti-ICAM (catalog no. 4915; 1:1,000),
anti-iNOS (catalog no. 13120; 1:1,000) obtained from Cell Signaling
Technology, Inc., Danvers, MA, USA. Additional antibodies used were
anti-MMP-1 (catalog no. ab137332; 1:500, Abcam, Cambridge, MA,
USA), anti-MMP-9 (catalog no. 2270; 1:2,000), anti-TIMP-1 (catalog
no. 8946; 1:1,000), anti-nuclear factor (NF)-κB p65 (catalog no.
8242; 1:1,000), anti-phosphorylated (p)-NF-κB p65 (catalog no.
4806; 1:1,000), anti-histone (catalog no. 7631; 1:1,000) and
anti-β-actin antibodies (catalog no. 3700; 1:1,000) all obtained
from Cell Signaling Technology, Inc., at 4°C. Following washing
with 0.1% TBS-Tween 20, the membranes were incubated with secondary
horseradish peroxidase-conjugated IgG (catalog nos. ZB-2301 and
ZB-2305; 1:1,000; ZSGB-BIO, Beijing, China) at room temperature for
2 h. The blots were then visualized using enhanced
chemiluminescence with ECL reagents (Pierce; Thermo Fisher
Scientific, Inc.).
Statistical analysis
Data are presented as the mean ± standard deviation
from three independent experiments. SPSS software, version 13.0
(SPSS, Inc., Chicago, IL, USA) was used for statistical analysis.
Intergroup comparisons were performed using Student's t-test
(two-tailed) or one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
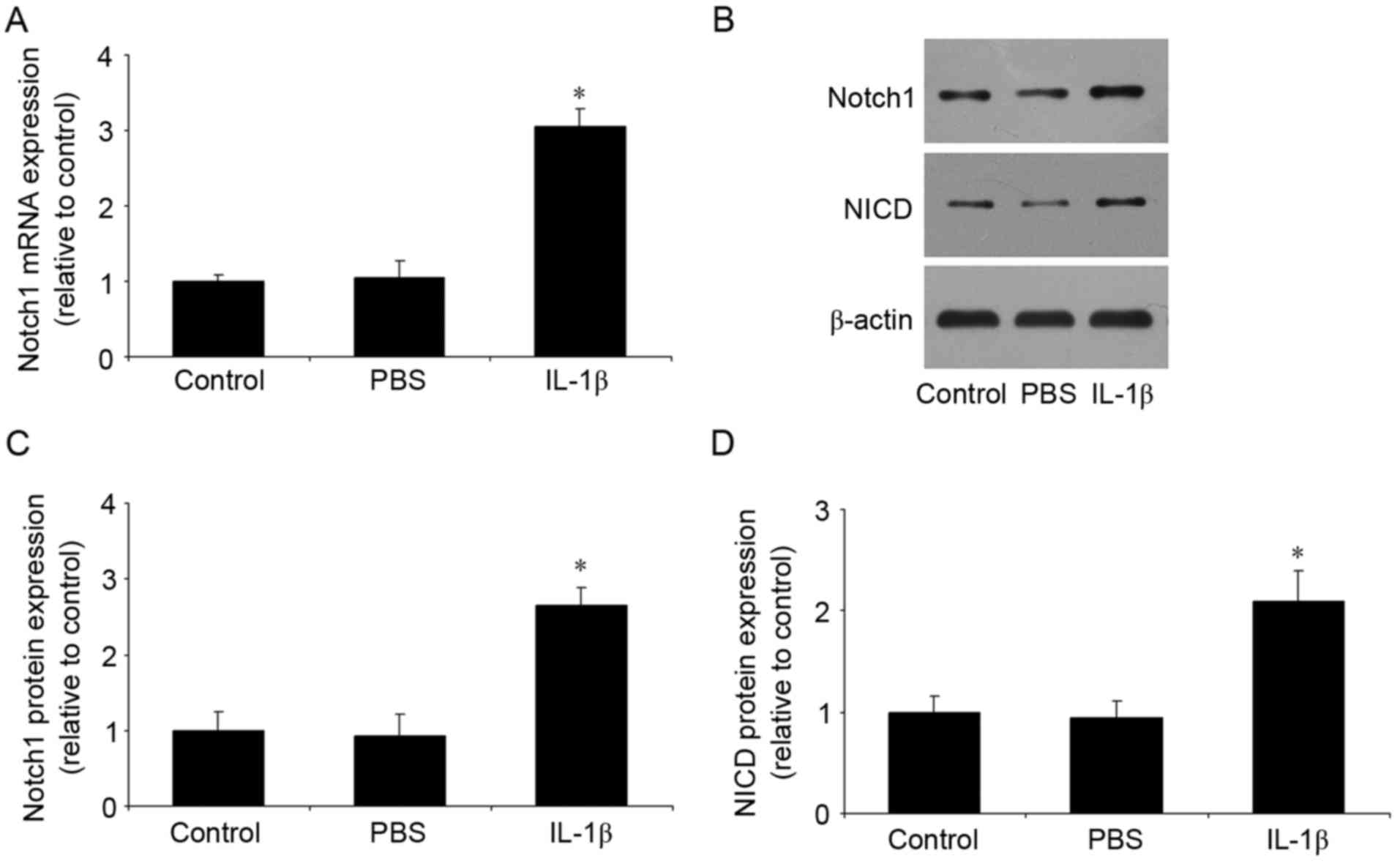
IL-1β increases the expression of
Notch1 and NICD
Following stimulation of the temporomandibular
chondrocytes with IL-1β, the expression levels of Notch1 and NICD
were measured using RT-qPCR and western blot analyses. As shown in
Fig. 1A-D, no differences were
found between the control group and the PBS group. However,
compared with the control group, IL-1β significantly increased the
mRNA and protein expression levels of Notch1, and the protein
expression of NICD.
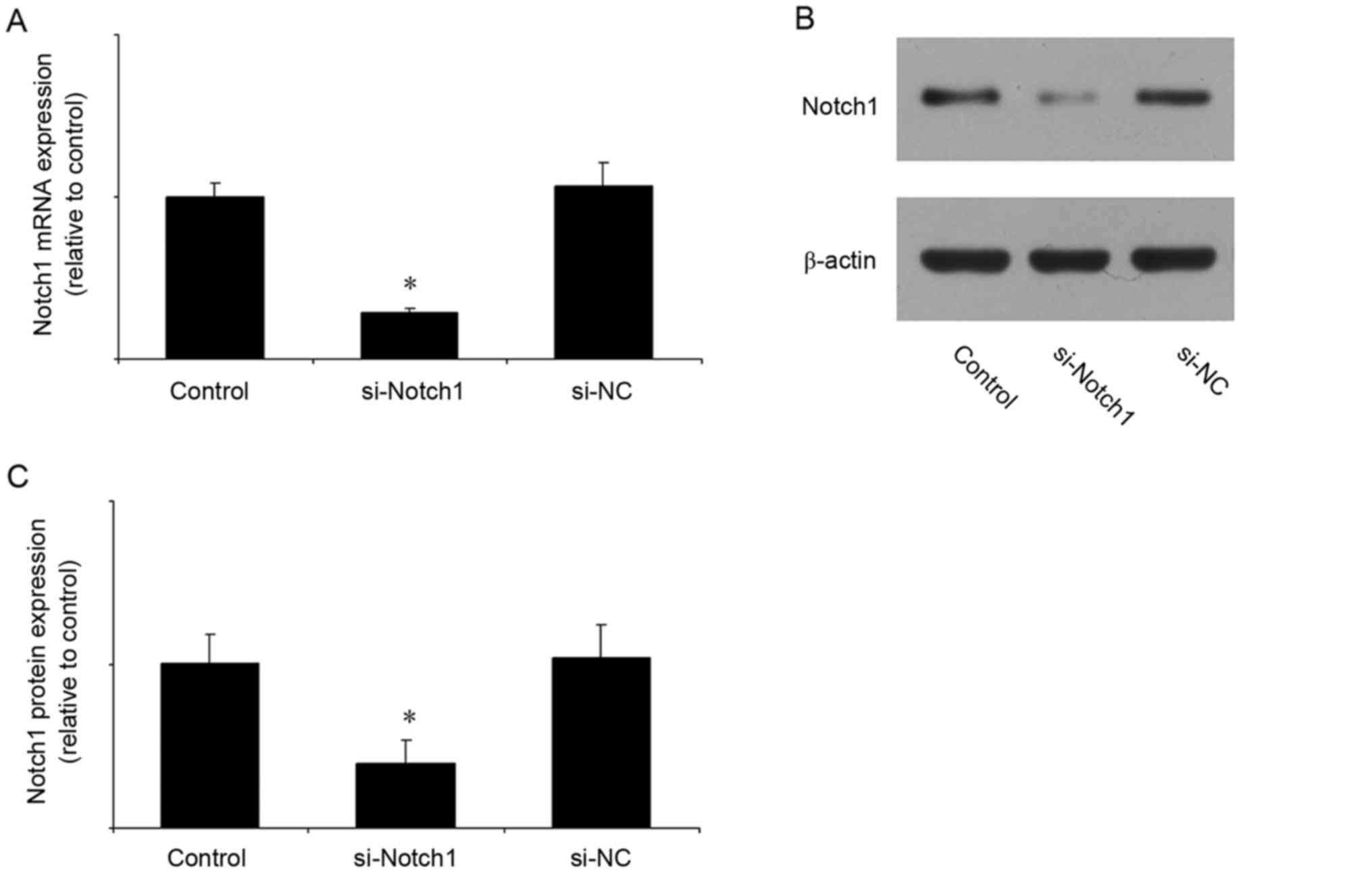
Notch1 siRNA reduces the mRNA and
protein expression of Notch1
In the subsequent experiments, the expression of
Notch1 was inhibited by siRNA. The efficiency of Notch1 siRNA was
first verified. Compared with the control group, Notch1 siRNA
significantly reduced the mRNA (Fig.
2A) and protein (Fig. 2B and
C) expression levels of Notch1, whereas the negative control
siRNA had no effect.
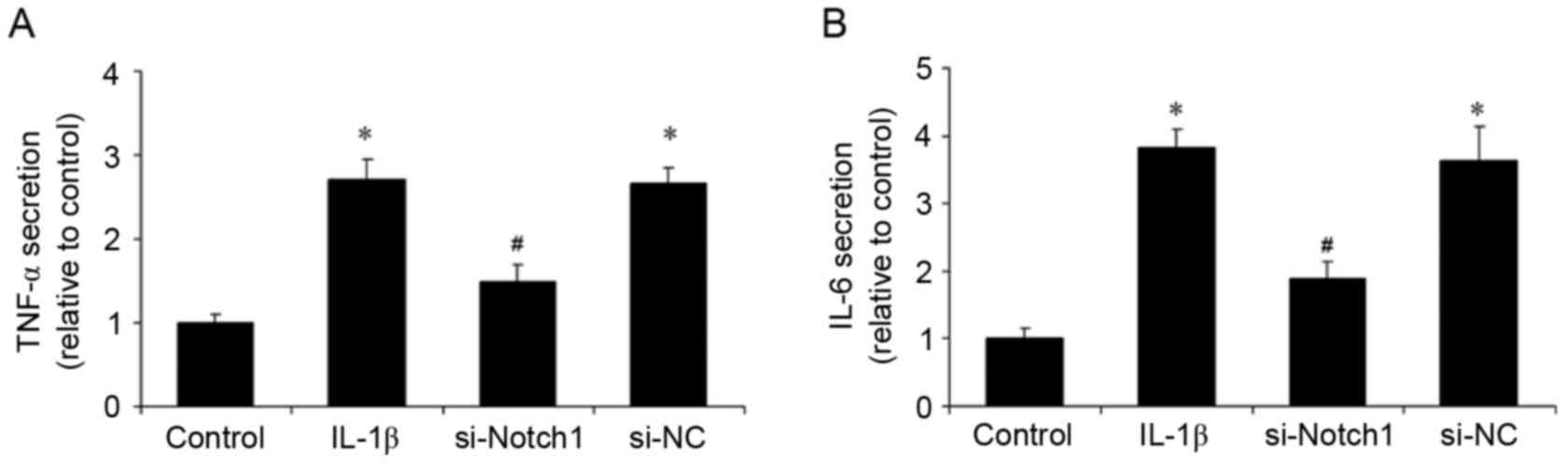
Notch1 inhibition reduces the
IL-1β-induced inflammatory response
Inflammation is important in the pathogenesis of
TMD. In the present study, following stimulation of the
chondrocytes with IL-1β, the secretion of TNF-α and IL-6 were
examined, as was the mRNA and protein expression levels of ICAM-1
and iNOS. Stimulation with IL-1β increased the levels of TNF-α and
IL-6 in the supernatants, whereas the inhibition of Notch1 by siRNA
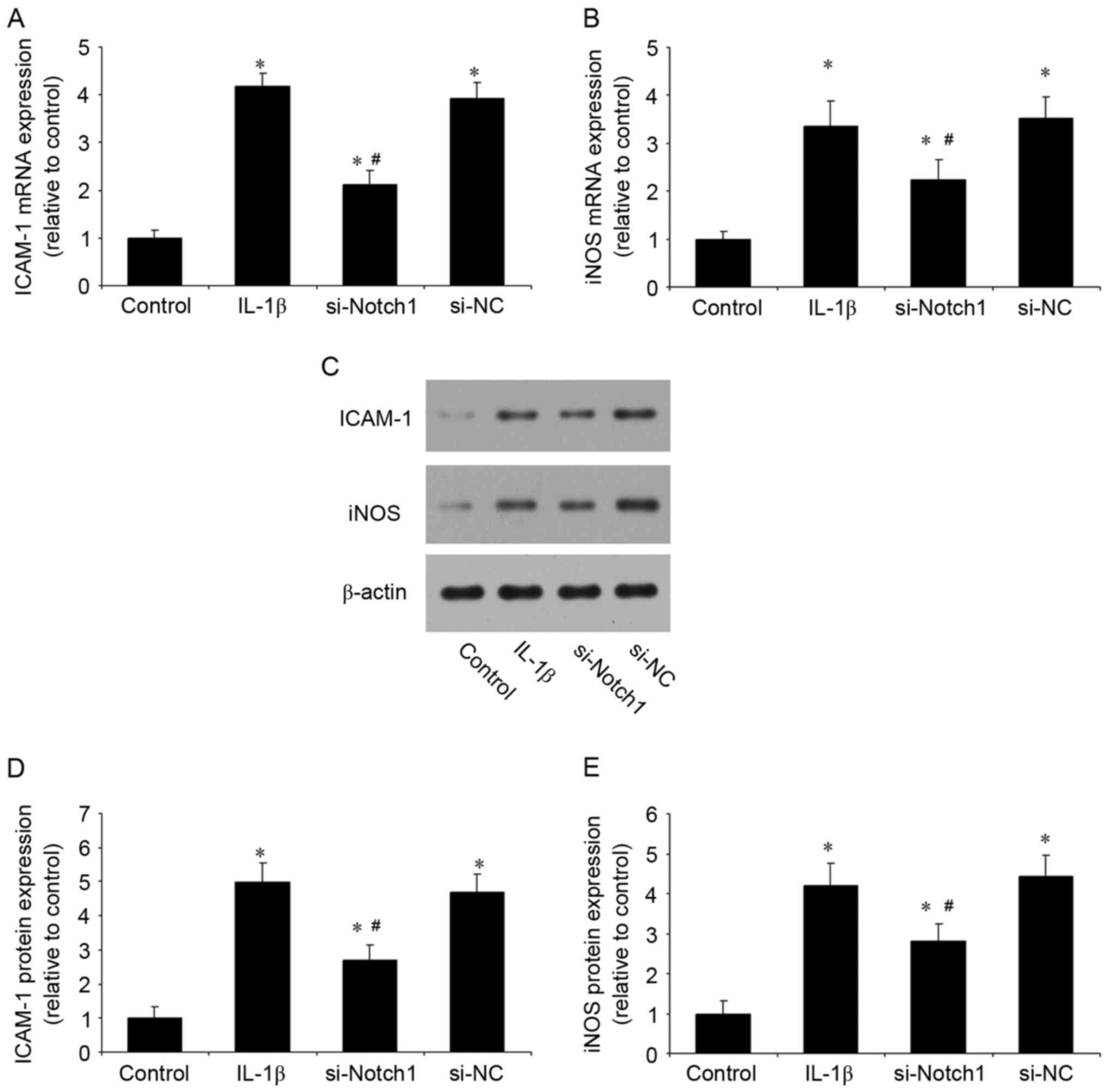
reduced these levels and control siRNA had no effect (Fig. 3A and B). In addition, the
inhibition of Notch1 significantly reduced the IL-1β-induced mRNA
(Fig. 4A and B) and protein
(Fig. 4C-E) expression levels of
ICAM-1 and iNOS (Fig. 4). These
results suggested that the inhibition of Notch1 suppressed the
IL-1β-induced inflammatory response in chondrocytes.
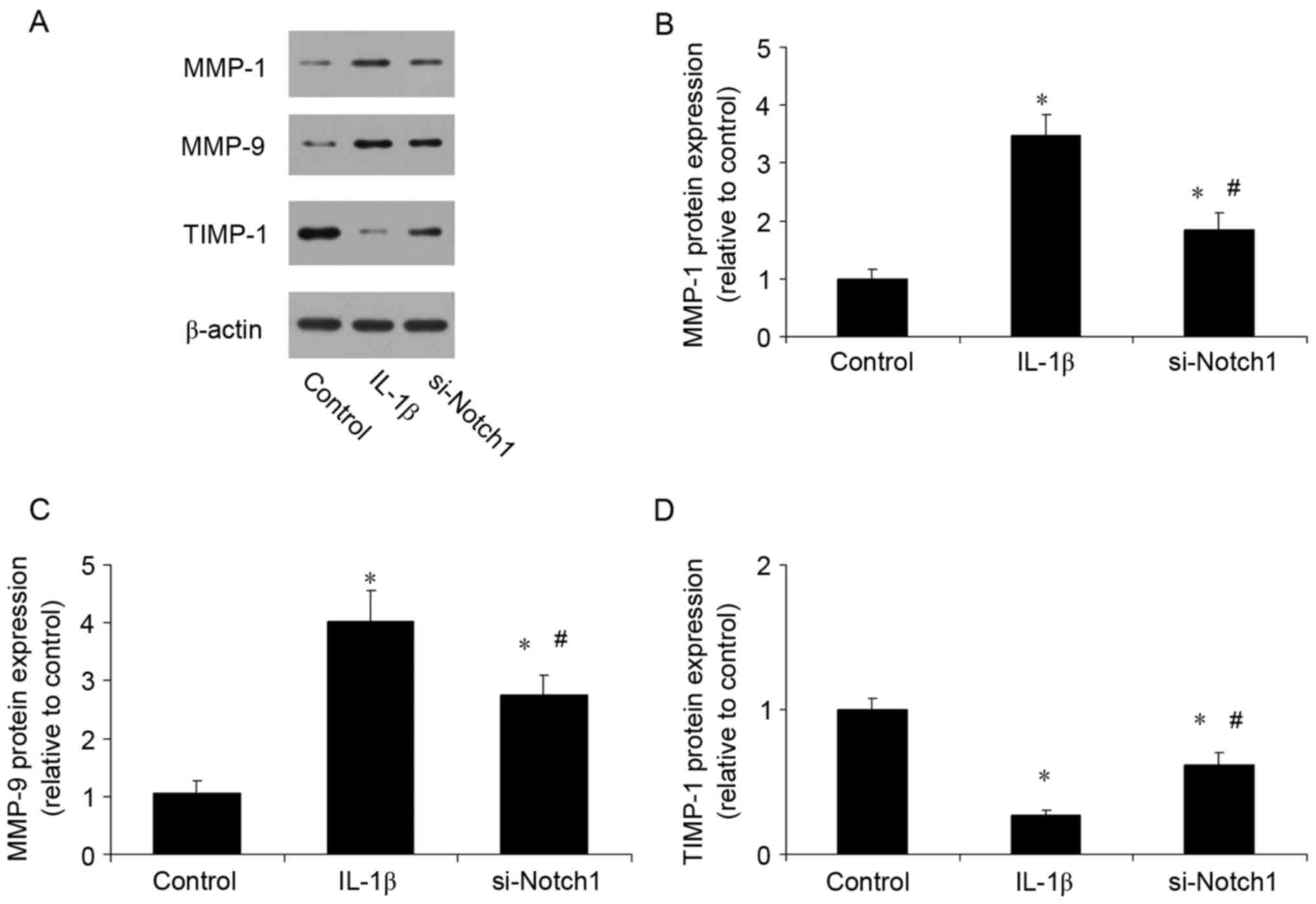
Notch1 inhibition reduces the
expression of MMPs and increases the expression of TIMP-1
An imbalance between MMPs and TIMPs has been
confirmed to be important in the progression of TMD. The present
study investigated the effect of inhibiting Notch1 on the
expression levels of MMPs and TIMP-1. Compared with the control,
IL-1β stimulation increased the expression levels of MMP-1 and
MMP-9 (Fig. 5A-C), and reduced the
expression of TIMP-1 (Fig. 5D).
However, the inhibition of Notch1 had the opposite effect, reducing
the expression levels of MMPs and increasing the expression of
TIMP-1.
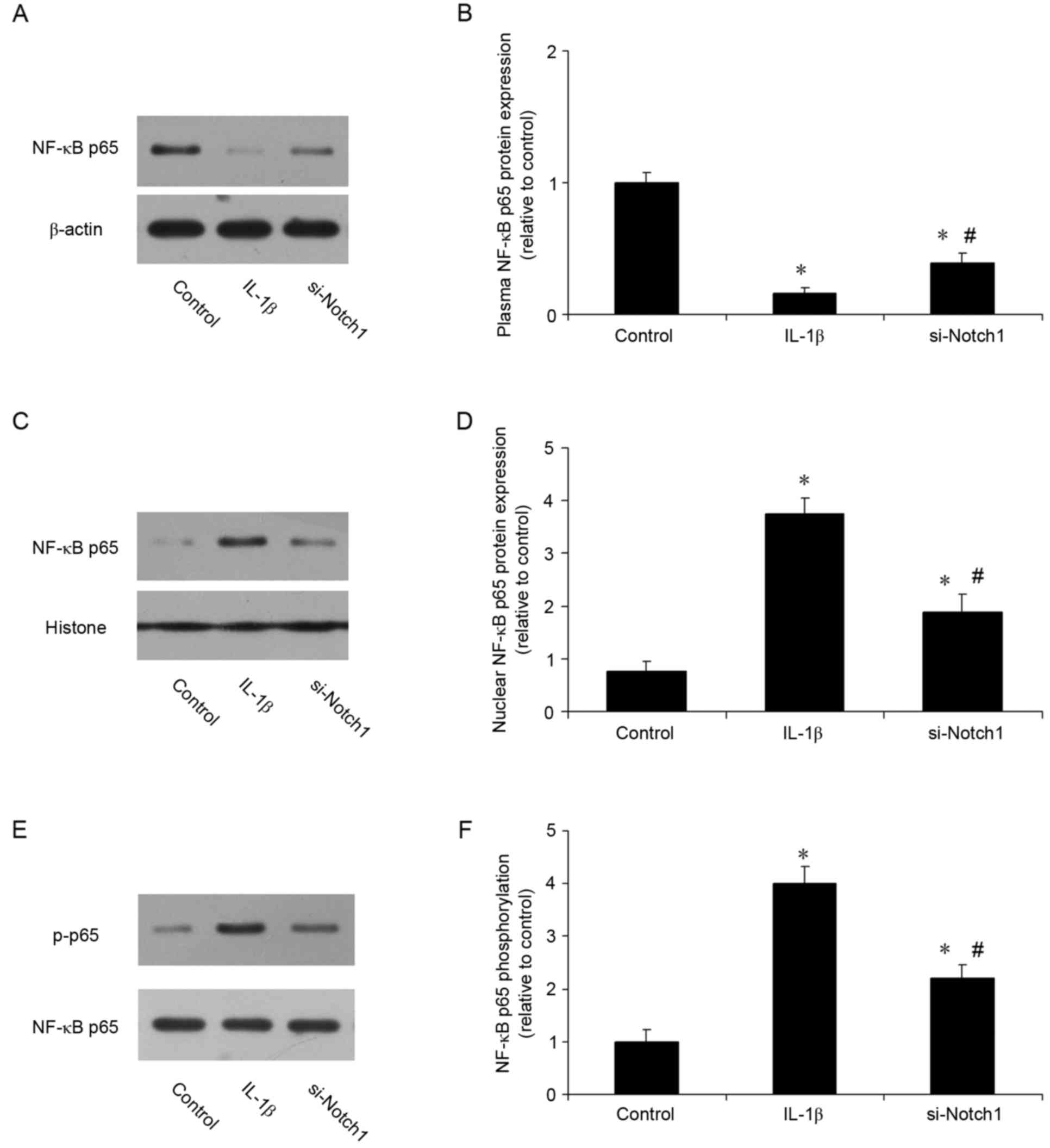
Notch1 inhibition suppresses IL-1β
induced activation of NF-кB in chondrocytes
The present study also investigated whether the
protective effects of inhibiting Notch1 involved the NF-кB
signaling pathway. The nuclear translocation of NF-κB p65 was
analyzed using western blot analysis. As shown in Fig. 6A-D, the localization of NF-κB p65
was predominantly cytoplasmic prior to IL-1β stimulation, however,
it was translocated to the nucleus when cells were stimulated with
IL-1β. The inhibition of Notch1 suppressed this IL-1β-induced p65
nuclear translocation. The expression of p-p65 was also measured.
Compared with the control, IL-1β increased the expression of p-p65
3-fold, whereas inhibition of Notch1 reduced the IL-1β-induced
expression of p-p65 (Fig. 6E and
F). These results suggested that the protective effect of
Notch1 inhibition occurred, at least in part, through the NF-кB
signaling pathway.
Discussion
TMD is an assorted set of clinical conditions
characterized by pain and dysfunction of the masticatory muscles in
the temporomandibular joint (TMJ) (21). The prevalence of TMD varies between
5 and 86% depending on different epidemiological studies,
diagnostic criteria and methods of assessment (22). According to the latest
understanding, inflammatory cytokines are the most important
pathogenic factors involved in the pathogenesis of TMD (23), responsible for the loss of
metabolic homeostasis by promoting catabolic and destructive
processes. Among the numerous representatives of these inflammatory
cytokines, the highest importance is attributed to IL-1β, TNF-α and
IL-6. In the present study, temporomandibular chondrocytes were
stimulated with IL-1β. Compared with the control group, IL-1β
significantly induced the inflammatory response in vitro,
including the secretion of TNF-α and IL-6, and mRNA and protein
expression of ICAM-1 and iNOS. These results were accordance with
previous findings in vivo (24).
MMPs are a family of >20 metalloenzymes, which
are crucial in tissue remodeling and cartilage degradation due to
their ability to degrade extracellular matrix (25). TIMPs are considered to be specific
inhibitors of MMPs. An imbalance between the activity of MMPs and
their inhibitors is considered to underlie cartilage destruction.
The expression of MMP-2 and MMP-9 has been reported to increase in
the rat trigeminal ganglion during the development of TMJ
inflammation (26). In the present
study, it was found that IL-1β stimulation increased the expression
of MMP-1 and MMP-9, and reduced the expression of TIMP-1, whereas
the inhibition of Notch1 by siRNA reduced the expression of MMPs
and increased the expression of TIMP-1. Therefore, maintaining the
balance of MMPs/TIMP-1 is the precise mechanism by which the
inhibition of Notch1 prevents cartilage matrix degradation. This
prompted investigation of the probable cell signaling
transduction.
NF-κB is pivotal in regulating
inflammation-associated genes (27). Various stimuli, including
pro-inflammatory cytokines, lead to the nuclear translocation of
NF-κB p65 and subsequently to the upregulated transcription of
various proinflammatory cytokines, including IL-1, TNFα, IL-6 and
IL-8 (28). NF-κB remains inactive
in the cytoplasm through interaction with the inhibitory protein,
inhibitor κB (IκB). The activation of NF-κB requires the
phosphorylation and subsequent degradation of IκBα (29). It has been demonstrated that the
phosphorylation of p65 is critical for binding to its target sites
on DNA (30). Considering NF-κB is
a pivotal regulator of the inflammatory response, the cross-talk
between IL-1β and NF-κB in chondrocytes may be one of the major
causes of TMJ inflammation. As expected, stimulation with IL-1β in
the present study induced the nuclear translocation and
phosphorylation of NF-κB p65, with an increased expression of
inflammatory cytokines, including TNFα, IL-6, ICMA-1 and iNOS.
In conclusion, the results of the present study
provide novel insights into the regulatory mechanisms of Notch1 in
temporomandibular chondrocytes during TMD. IL-1β stimulation
induced Notch1, and subsequent variation was associated with NF-κB
pathway, which may be an important pathogenetic factor in the
development of TMD. The present study, to the best of our
knowledge, is the first to provide evidence that the inhibition of
Notch1 effective treated TMD. However, further in vivo
experiments are required to identify novel and potent agents for
the prevention and treatment of TMD through the inhibition of
Notch1.
Acknowledgements
This study was supported by the Science and
Technology Development Plans of Shandong Province (grant no.
2014GSF118027) and the Specialized Research Fund for the Doctoral
Program of Higher Education of China (grant no.
20120131110074).
References
|
1
|
Reissmann DR, John MT, Schierz O and
Wassell RW: Functional and psychosocial impact related to specific
temporomandibular disorder diagnoses. J Dent. 35:643–650. 2007.
View Article : Google Scholar
|
|
2
|
Kiener HP and Karonitsch T: The synovium
as a privileged site in rheumatoid arthritis: Cadherin-11 as a
dominant player in synovial pathology. Best Pract Res Clin
Rheumatol. 25:767–777. 2011. View Article : Google Scholar
|
|
3
|
Wu M, Xu T, Zhou Y, Lu H and Gu Z:
Pressure and inflammatory stimulation induced increase of
cadherin-11 is mediated by PI3K/Akt pathway in synovial fibroblasts
from temporomandibular joint. Osteoarthritis Cartilage.
21:1605–1612. 2013. View Article : Google Scholar
|
|
4
|
Slade GD, Conrad MS, Diatchenko L, Rashid
NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W
and Nackley AG: Cytokine biomarkers and chronic pain: Association
of genes, transcription, and circulating proteins with
temporomandibular disorders and widespread palpation tenderness.
Pain. 152:2802–2812. 2011. View Article : Google Scholar :
|
|
5
|
Dai SM, Shan ZZ, Nakamura H, Masuko-Hongo
K, Kato T, Nishioka K and Yudoh K: Catabolic stress induces
features of chondrocyte senescence through overexpression of
caveolin 1: Possible involvement of caveolin 1-induced
down-regulation of articular chondrocytes in the pathogenesis of
osteoarthritis. Arthritis Rheum. 54:818–831. 2006. View Article : Google Scholar
|
|
6
|
Okamoto K, Kiga N, Shinohara Y, Tojyo I
and Fujita S: Effect of interleukin-1beta and
dehydroepiandrosterone on the expression of lumican and
fibromodulin in fibroblast-like synovial cells of the human
temporomandibular joint. Eur J Histochem. 59:24402015. View Article : Google Scholar :
|
|
7
|
Dinarello CA: A clinical perspective of
Il-1β as the gatekeeper of inflammation. Eur J Immunol.
41:1203–1217. 2011. View Article : Google Scholar
|
|
8
|
Korherr C, Hofmeister R, Wesche H and Falk
W: A critical role for interleukin-1 receptor accessory protein in
interleukin-1 signaling. Eur J Immunol. 27:262–267. 1997.
View Article : Google Scholar
|
|
9
|
Yudoh K, Nguyen vT, Nakamura H,
Hongo-Masuko K, Kato T and Nishioka K: Potential involvement of
oxidative stress in cartilage senescence and development of
osteoarthritis: Oxidative stress induces chondrocyte telomere
instability and downregulation of chondrocyte function. Arthritis
Res Ther. 7:R380–R391. 2005. View
Article : Google Scholar :
|
|
10
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster
JC: Activating mutations of notch1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004. View Article : Google Scholar
|
|
11
|
Weng AP and Aster JC: Multiple niches for
notch in cancer: Context is everything. Curr Opin Genet Dev.
14:48–54. 2004. View Article : Google Scholar
|
|
12
|
Selkoe D and Kopan R: Notch and
presenilin: Regulated intramembrane proteolysis links development
and degeneration. Annu Rev Neurosci. 26:565–597. 2003. View Article : Google Scholar
|
|
13
|
Bray SJ: Notch signalling: A simple
pathway becomes complex. Nat Rev Mol Cell Biol. 7:678–689. 2006.
View Article : Google Scholar
|
|
14
|
Eagar TN, Tang Q, Wolfe M, He Y, Pear WS
and Bluestone JA: Notch 1 signaling regulates peripheral T cell
activation. Immunity. 20:407–415. 2004. View Article : Google Scholar
|
|
15
|
Palaga T, Miele L, Golde TE and Osborne
BA: TCR-mediated notch signaling regulates proliferation and
IFN-gamma production in peripheral T cells. J Immunol.
171:3019–3024. 2003. View Article : Google Scholar
|
|
16
|
Ando K, Kanazawa S, Tetsuka T, Ohta S,
Jiang X, Tada T, Kobayashi M, Matsui N and Okamoto T: Induction of
notch signaling by tumor necrosis factor in rheumatoid synovial
fibroblasts. Oncogene. 22:7796–7803. 2003. View Article : Google Scholar
|
|
17
|
Monsalve E, Pérez MA, Rubio A,
Ruiz-Hidalgo MJ, Baladrón V, Garcia-Ramirez JJ, Gómez JC, Laborda J
and Diaz-Guerra MJ: Notch-1 up-regulation and signaling following
macrophage activation modulates gene expression patterns known to
affect antigen-presenting capacity and cytotoxic activity. J
Immunol. 176:5362–5373. 2006. View Article : Google Scholar
|
|
18
|
Palaga T, Buranaruk C, Rengpipat S, Fauq
AH, Golde TE, Kaufmann SH and Osborne BA: Notch signaling is
activated by TLR stimulation and regulates macrophage functions.
Eur J Immunol. 38:174–183. 2008. View Article : Google Scholar
|
|
19
|
Pastrana JL, Sha X, Virtue A, Mai J, Cueto
R, Lee IA, Wang H and Yang XF: Regulatory T cells and
atherosclerosis. J Clin Exp Cardiolog. 2012 Suppl 12:S22012.
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Kou XX, Wu YW, Ding Y, Hao T, Bi RY, Gan
YH and Ma X: 17β-estradiol aggravates temporomandibular joint
inflammation through the NF-κB pathway in ovariectomized rats.
Arthritis Rheum. 63:1888–1897. 2011. View Article : Google Scholar
|
|
22
|
Aliko A, Ciancaglini R, Alushi A, Tafaj A
and Ruci D: Temporomandibular joint involvement in rheumatoid
arthritis, systemic lupus erythematosus and systemic sclerosis. Int
J Oral Maxillofac Surg. 40:704–709. 2011. View Article : Google Scholar
|
|
23
|
Wojdasiewicz P, Poniatowski ŁA and
Szukiewicz D: The role of inflammatory and anti-inflammatory
cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm.
2014:5614592014. View Article : Google Scholar :
|
|
24
|
Kaneyama K, Segami N, Yoshimura H, Honjo M
and Demura N: Increased levels of soluble cytokine receptors in the
synovial fluid of temporomandibular joint disorders in relation to
joint effusion on magnetic resonance images. J Oral Maxillofac
Surg. 68:1088–1093. 2010. View Article : Google Scholar
|
|
25
|
Parks WC, Wilson CL and López-Boado YS:
Matrix metalloproteinases as modulators of inflammation and innate
immunity. Nat Rev Immunol. 4:617–629. 2004. View Article : Google Scholar
|
|
26
|
Nascimento GC, Rizzi E, Gerlach RF and
Leite-Panissi CR: Expression of MMP-2 and MMP-9 in the rat
trigeminal ganglion during the development of temporomandibular
joint inflammation. Braz J Med Biol Res. 46:956–967. 2013.
View Article : Google Scholar :
|
|
27
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: New discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar
|
|
28
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar
|
|
29
|
Ogata N, Yamamoto H, Kugiyama K, Yasue H
and Miyamoto E: Involvement of protein kinase C in superoxide
anion-induced activation of nuclear factor-kappa B in human
endothelial cells. Cardiovasc Res. 45:513–521. 2000. View Article : Google Scholar
|
|
30
|
Li Q and Verma IM: Nf-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar
|