Introduction
Pulmonary fibrosis is a fatal and progressive
disease characterized by injury to alveolar epithelial cells,
infiltration of inflammatory cells, enhanced proliferation of
fibroblasts, aberrant accumulation of the extracellular matrix and
abnormality in lung architectural remodeling (1,2).
Currently, glucocorticosteroids, which inhibit the expression of
pro-inflammatory cytokines, are the primary treatment option for
pulmonary fibrosis (3). However,
glucocorticosteroids cannot reverse or delay the course of the
disease (4). Therefore,
identification of novel and effective treatment options that have
the potential to improve the mortality rate are urgently
required.
Recent evidence suggested that oxidative stress, due
to the imbalance between the functions of oxidants and antioxidant
proteins, has an important role in the progression of lung
fibrosis, by activating redox-sensitive signaling pathways,
modification of immune cell function, and activation of fibroblasts
(5,6). Previous studies have additionally
suggested that reactive oxygen species (ROS) cause the activation
of growth-regulatory cytokines, including transforming growth
factor (TGF)-β, a critical pro-fibrosis cytokine (7). Felton et al (8) previously analyzed the effects of
N-acetylcysteine, an exogenous antioxidant, and demonstrated that
it dramatically decreases lung damage in the presence of TGF-β1 by
reducing intracellular ROS production. Additionally, the activity
of the intracellular antioxidant enzyme catalase (CAT) has been
linked with decreased lung fibrosis in a mouse model (9). Therefore, based on these studies, it
was hypothesized that a strategy to upregulate the expression of
endogenous multiple antioxidants maybe an efficient approach to
prevent or treat lung fibrosis (10).
The transcription factor nuclear factor
erythroid2-related factor-2 (Nrf2) has a primary role in regulating
cellular antioxidant responses (11). Upon exposure to oxidative burden,
the Nrf2-antioxidant signaling pathway is activated to stimulate
transcription of various antioxidant defense enzymes, including
NADPH quinone oxidoreductase (NQO1), heme oxygenase-1 (HO-1),
superoxide dismutase (SOD), and CAT (12). Upregulation of the expression of
these antioxidant enzymes promotes antioxidative response,
detoxification, and anti-inflammation. It has been reported that
Nrf2 and its downstream antioxidants enzymes serve critical roles
in a lung fibrosis model (13),
whereas deletion of the Nrf2 gene increases susceptibility to
bleomycin (BLM) due to reduced antioxidant activity (14). BLM, an anti-cancer medicine, can
induce oxidative stress and DNA damage, and can induce pulmonary
fibrosis in clinical treatment. Intratracheal instillation of BLM
has been used to establish pulmonary fibrotic animal models for
many years (15,16).
Sulforaphane (SFN), a dietary organosulfur compound,
is isolated from cruciferous vegetables and has an indirect
antioxidant activity. It upregulates the expression of endogenous
antioxidant proteins against oxidative stress and damage via Nrf2
activation (17). Previous studies
have demonstrated that the antitoxic and antioxidant properties of
SFN in experimental models most likely involves activation of the
Nrf2 gene (18,19). However, it is unclear if SFN
alleviates lung fibrosis, resulting in upregulation of the Nrf2
gene and its downstream antioxidant targets.
Therefore, the present study aimed to investigate if
SFN may prevent the development of BLM-induced lung fibrosis in a
mouse model, and to identify the potential mechanisms of
action.
Materials and methods
Animals
C57/BL6 male mice (age, 8–10 weeks; weight, ~20 g;
n=60) were purchased from the Animal Center of Jilin University
(Changchun, China) and housed in the animal testing center of the
Second Hospital of Jilin University. They were housed at a constant
temperature of 22°C in 50–60% humidity and a 12 h light/dark cycle,
and were fed standard rodent feed and tap water. The experimental
protocol was approved by the Animal Care and Use Committee of Jilin
University.
Mice were divided randomly into four groups
(n=15/group): Control, SFN, BLM and BLM/SFN. To generate a
pulmonary fibrosis model, experimental mice were administered a
single dose of 5 mg/kg body weight BLM (Nippon Kayaku, Co., Ltd.,
Tokyo, Japan) by intratracheal instillation, and control mice
received saline. The SFN and BLM/SFN group mice received a
subcutaneous injection of SFN (5 mg/kg body weight; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) each day. The dose of SFN used in
the present study was in accordance with a previously published
study (18). Mice in the control
group received an equal volume of vehicle (1% dimethyl sulfoxide in
PBS). Finally, mice from each group were sacrificed at days 7 or 28
after initial BLM or saline administration, and samples were
collected.
Histopathological examination
Following anesthesia, the thorax was opened and
lungs were isolated from all mice. The left lungs were fixed in 10%
buffered formalin, following which they were dehydrated in a series
of graded alcohol. Subsequently, they were washed with xylene,
embedded into paraffin blocks and sliced into 3-µm sections. To
examine basic structural alterations and collagen content, slices
were stained with hematoxylin & eosin (H&E) and Masson's
trichrome. Pulmonary inflammation and fibrosis was evaluated using
the Szapiel score (20).
Nrf2 expression was assessed by immunohistochemical
staining. Tissue sections were dewaxed and then incubated with
target retrieval solution (Dako; Agilent Technologies, Inc., Santa
Clara, CA, USA) in a microwave for 15 min at 98°C for antigen
retrieval, followed by treatment with 3% hydrogen peroxide for 15
min and then 5% bovine serum albumin (Sigma-Aldrich) for 2 h, at
room temperature. Sections were subsequently incubated with
anti-Nrf2 primary antibody (1:100; ab31163; Abcam, Cambridge, MA,
USA) overnight at 4°C and then incubated with a horseradish
peroxidase (HRP) conjugated goat anti-rabbit secondary antibody
(1:1,000; SA00001-2; Wuhan Sanying Biotechnology, Wuhan, China) for
1 h at room temperature. Sections were subsequently stained with
3,3-diaminobenzidine, and counterstained with hematoxylin. Staining
was observed under a Nikon Eclipse E600 microscope (Nikon
Corporation, Tokyo, Japan).
Assay for hydroxyproline content
Pulmonary collagen content was analyzed with a
Hydroxyproline assay kit (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). Lung tissue was hydrolyzed by
incubating with NaOH at 95–100°C for 20 min. Following
neutralization with HCl, the tissue was diluted with distilled
water. Subsequently, hydroxyproline content in the tissue was
assessed by recording the absorbance at a wavelength of 550 nm. The
results were calculated as µg hydroxyproline/g wet lung tissue.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). RNA concentrations and purity were quantified using a
NanoDrop 2000™ Spectrophotometer (Thermo Fisher
Scientific, Inc.). A total of 3 µg RNA was used for cDNA synthesis
according to the manufacturer's protocol, using a Revert Aid First
Strand cDNA Synthesis kit (Gene Copoeia, Inc., Rockville, MD, USA).
Primers for Nrf2, NQO1, HO-1, SOD1, CAT and β-actin amplification
were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). The
primers were as follows: Nrf2 forward, TCC TAT GCG TGA ATC CCA AT,
and reverse, GCG GCT TGA ATG TTT GTC TT; NQO1 forward, GTT TCT GTG
GCT TCC AGG TC, and reverse, CGT TTC TTC CAT CCT TCC AG; HO-1
forward, GGG CTG TGA ACT CTG TCC AA, and reverse, GGT GAG GGA ACT
GTG TCA GG; SOD1 forward, TTC TCG TCT TGC TCT CTC TGG, and reverse,
GTT CAC CGC TTG CCT TCT; CAT forward, CCT CGT TCA GGA TGT GGT TT,
and reverse, CCT CGT TCA GGA TGT GGT TT; β-actin forward, GTG CTA
TGT TGC TCT AGA CTT CG, and reverse, ATG CCA CAG GAT TCC ATA CC.
qPCR was performed in a 20 µl reaction buffer containing 10 µl Fast
Start Universal SYBR®-Green Master mix (Roche
Diagnostics, Basel, Switzerland), 1 µl 10 mM primer pairs, 1 µl
cDNA and 8 µl ddH2O, using the Roche Light
Cycler® 480 Real-Time PCR system. The thermocycling
conditions were as follows: Initial step of 95°C for 5 min, then 40
cycles of 95°C for 30 sec, 58°C for 30 sec and 72°C for 60 sec. The
comparative cycle time (Cq) method was used to determine fold
differences between samples, values were normalized to the
endogenous reference (β-actin) using the 2ΔΔCq method
(21).
Western blot analysis
For preparation of total protein samples, lung
tissues were homogenized in ice-cold radioimmunoprecipitation assay
lysis buffer (Beijing Dingguo Changsheng Biotechnology Co., Ltd.,
Beijing, China), and protein concentrations were determined using a
bicinchoninic acid protein assay kit (Beijing Dingguo Changsheng
Biotechnology Co., Ltd.). Equal amounts (10 µg) of protein samples
were separated by 10% SDS-PAGE (15% SDS-PAGE was used for studies
of IL-1β) and transferred onto polyvinylidene difluoride membranes
(EMD Millipore, Billerica, MA, USA). Membranes were blocked with 5%
(w/v) skimmed milk for >1 hat room temperature and subsequently
incubated overnight at 4°C with the following primary antibodies:
Anti-Nrf2 (1:1,000; ab31163), anti-HO-1 (1:1,000; ab13243),
anti-NQO1 (1:1,000; ab34173), anti-SOD1 (1:2,000; ab16831),
anti-CAT (1:1,000; ab16731), anti-3-nitro-tyrosine (3-NT) (1:1,000;
ab110282), anti-4-hydroxynonenal (4-HNE) (1:1,000; ab46545),
anti-tumor necrosis factor (TNF)-α (1:200; ab6671), anti-TGF-β
(1:1,000; ab64715), anti-caspase-3 (1:1,000; #9665), anti-IL-1β
(1:300; sc-7884) and anti-β-actin (1:1,000; 66009-1-Ig). All
antibodies were purchased from Abcam, except for anti-caspase-3
(Cell Signaling Technology, Inc. Danvers, MA, USA), anti-IL-1β
(Santa Cruz Biotechnology, Inc. Dallas, TX, USA) and anti-β-actin
(Wuhan Sanying Biotechnology). After three washes with
Tris-buffered saline (pH 7.4) containing 0.1% Tween-20, membranes
were incubated with the appropriate HRP-conjugated secondary
antibodies for 1 h at room temperature. The secondary antibodies
were as follows: Goat anti-mouse (1:1,000; SA00001-1) and goat
anti-rabbit (1:1,000; SA00001-2) both from Wuhan Sanying
Biotechnology. Proteins were visualized using an Enhanced
Chemiluminescence kit (EMD Millipore), and the band density values
were normalized to β-actin expression. Densitometry was performed
using Quantity One 4.52 software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), and ≥6 blots were repeated to obtain
densitometric quantification.
Terminal deoxynucleotidyl transferase
UTP nick-end labeling (TUNEL) staining
TUNEL staining was performed on formalin-fixed,
paraffin-embedded lung sections using an In Situ Cell Death
Detection kit, POD (Roche Diagnostics) according to the
manufacturer's protocol. Positively stained apoptotic cells were
counted randomly in five microscopic fields from at least three
slides of each mouse, under a light microscope. The percentage of
TUNEL staining represented the number of TUNEL positive cells in a
field with a total of 100 nuclei.
Statistical analysis
Data were collected from >4 animals/group and are
presented as the mean ± standard deviation. Comparisons between
groups were assessed by one-way analysis of variance, followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
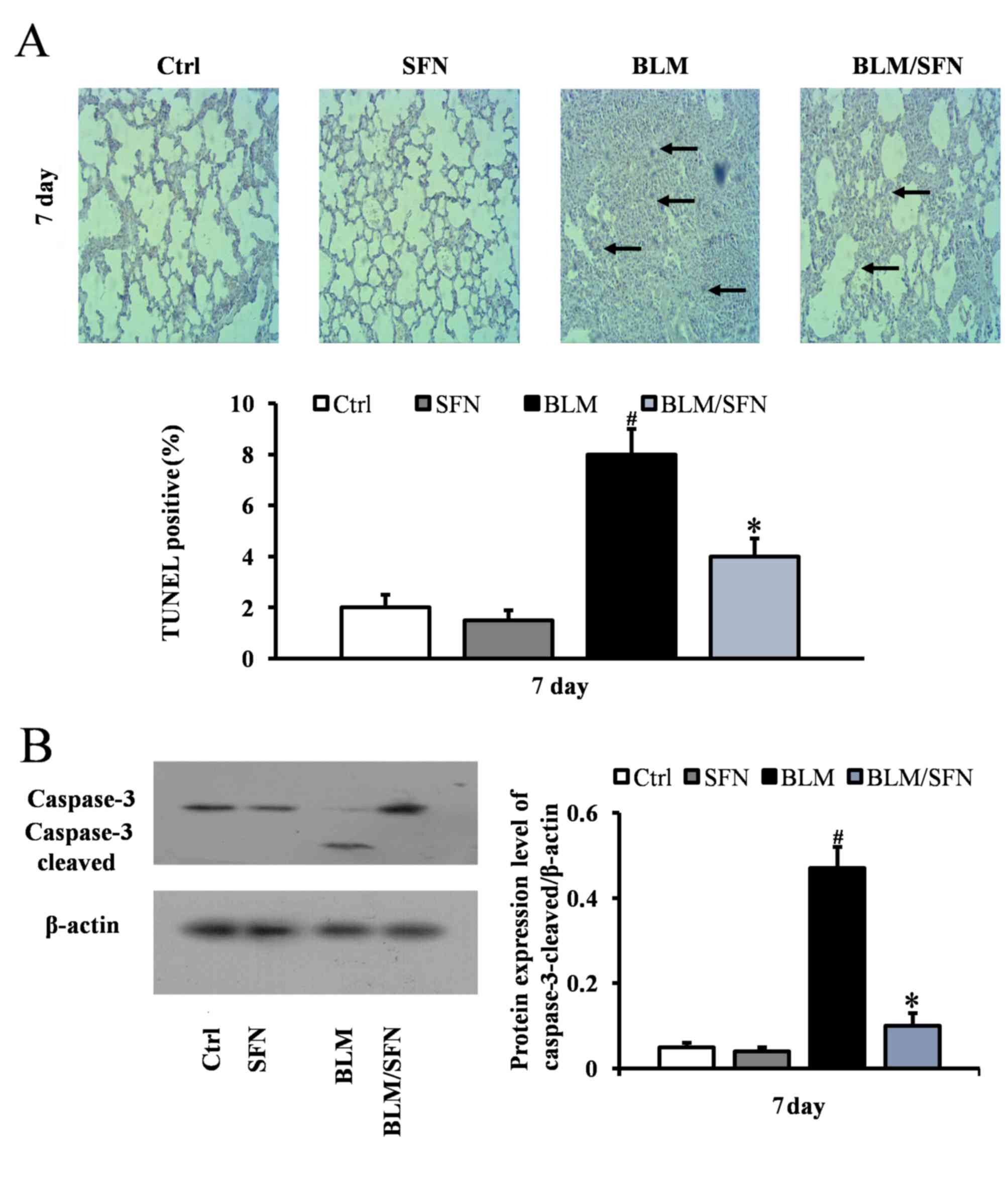
SFN alleviates BLM-induced alveolar
epithelial cell apoptosis
In the early stages of the pulmonary fibrosis
induced by BLM, an increase in alveolar epithelial cell apoptosis
was observed. At day 7, the BLM treatment mice had significantly
increased TUNEL-positive cells compared with control mice. However,
mice with combined BLM and SFN treatment exhibited a significant
decrease in epithelial cell apoptosis compared with BLM treatment
alone (P<0.05; Fig. 1A).
Consistent with TUNEL staining results, western blot analysis
additionally revealed a significant increase in the expression of
cleaved caspase-3 in the BLM group at day 7. However, the effect of
BLM treatment was neutralized by the presence of SFN (P<0.05;
Fig. 1B).
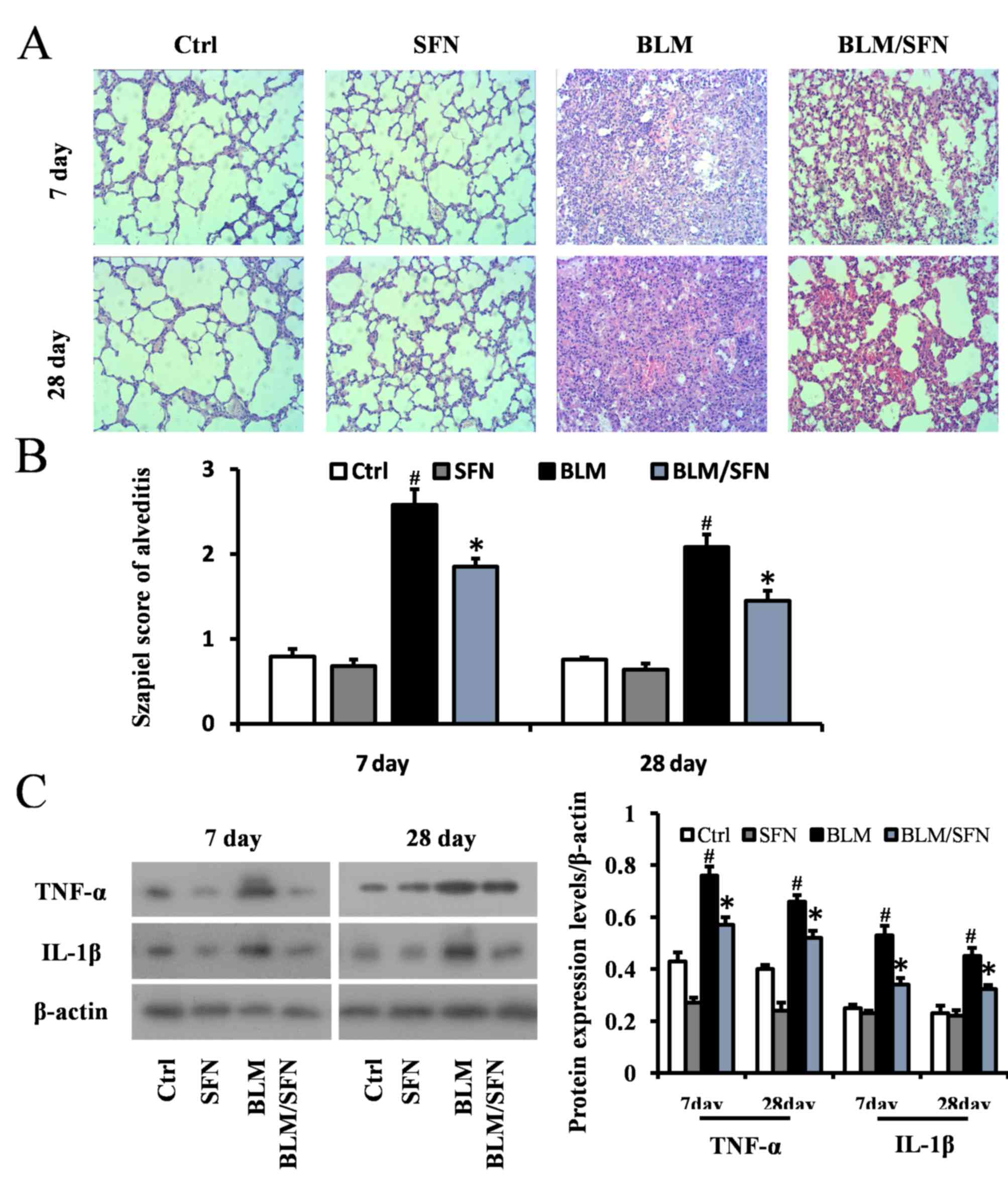
SFN attenuates BLM-induced alveolitis
and lung fibrosis
Histological examination using H&E staining of
mouse lungs from the BLM treated group revealed significant
progressive lesions in lung structure. This included excessive
infiltration of inflammatory cells, thickened pulmonary alveolar
septa and a large number of collapsed alveoli (Fig. 2A and B). However, combined SFN and
BLM markedly alleviated the degree of alveolitis at days 7 and 28.
In the cases of the control and SFN treated groups, the lungs had
healthy histology. Protein expression levels of the inflammatory
cytokines IL-1β and TNF-α were examined in lung tissues at days 7
and 28 by western blot analysis (Fig.
2C). In the fibrosis model (BLM treatment alone), the
expression levels of IL-1β and TNF-α were elevated compared with
the control group at days 7 and 28. However, SFN treatment in the
fibrosis model significantly downregulated the expression of TNF-α
and IL-1β compared with the BLM treatment alone at the two time
points (P<0.05).
Furthermore, lung fibrosis was examined by Masson's
staining, where blue staining represented collagen matrix. BLM
treatment predominantly induced collagen accumulation in the
interstitial areas at day 28 (Fig. 3A
and B), and its effects were significantly inhibited by SFN
treatment at day 28 (P<0.05). Simultaneously, hydroxyproline
content of lung samples on days 7 and 28 in the four groups were
examined. Hydroxyproline content in the lung fibrosis model (BLM
treatment alone) was increased compared with control mice; however,
combined BLM and SFN treatment significantly suppressed this effect
at day 28 (P<0.05; Fig. 3C). To
further examine the protective effect of SFN on pulmonary fibrosis,
protein expression levels of TGF-β, a critical pro-fibrotic
mediator, were assessed by western blot analysis. Expression levels
of TGF-β were markedly increased in the lungs of mice from the BLM
group compared with the control group. However, SFN treatment
significantly reduced this increase at days 7 and 28 (Fig. 3D).
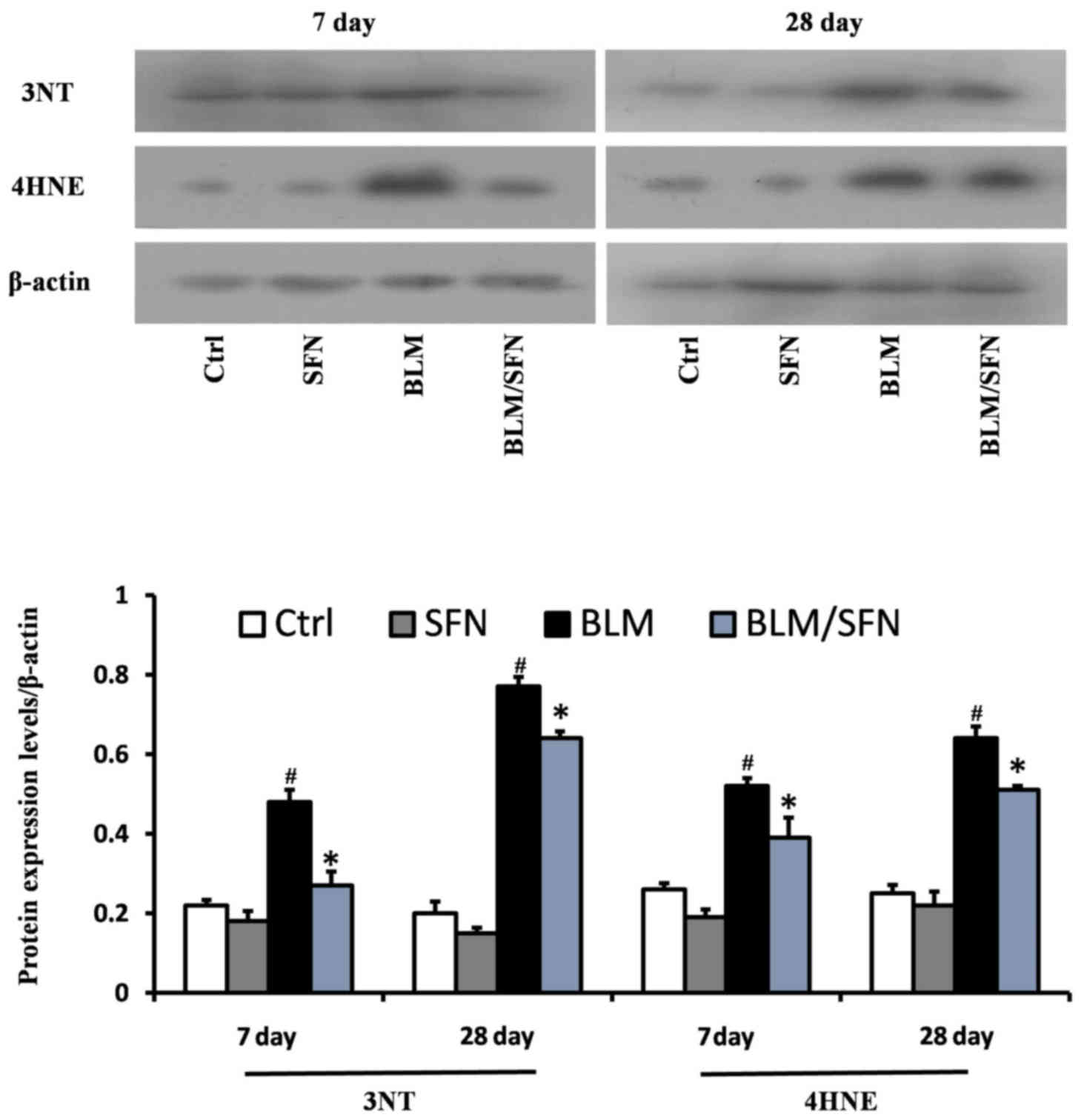
SFN protects against BLM-induced lung
oxidative stress
BLM treatment was demonstrated to significantly
increase lung oxidative stress, as detected by accumulation of 3-NT
and 4-HNE, which are indicators of nitration damage and lipid
peroxidation, respectively. Western blot analysis revealed that BLM
treatment significantly increased expression levels of 3-NT and
4-HNE at days 7 and 28. However, treatment with SFN partly
prevented oxidative damage at the two time-points compared with BLM
alone (P<0.05; Fig. 4).
SFN upregulates the expression of Nrf2
and downstream genes in the lungs
Based on the above results, it appeared that SFN may
protect against BLM-induced lung inflammation, fibrosis, apoptosis
and oxidative damage. Furthermore, as oxidative stress is
considered a pivotal mediator for pathological alterations in
pulmonary fibrosis, the underlying mechanisms of SFN action in
pulmonary fibrosis was investigated. Expression of the Nrf2 gene in
the lungs was assessed by immunochemical staining (Fig. 5A), RT-qPCR (Fig. 5B) and western blot analysis
(Fig. 5C). The results from all
these assays indicated that Nrf2 expression levels were increased
at days 7 and 28 in the lungs of BLM treated mice compared with
control group mice. Notably, an additional significant increase in
Nrf2 expression in the lungs of mice treated with BLM and SFN
simultaneously was observed compared with BLM treatment alone, at
the two time-points (P<0.05).
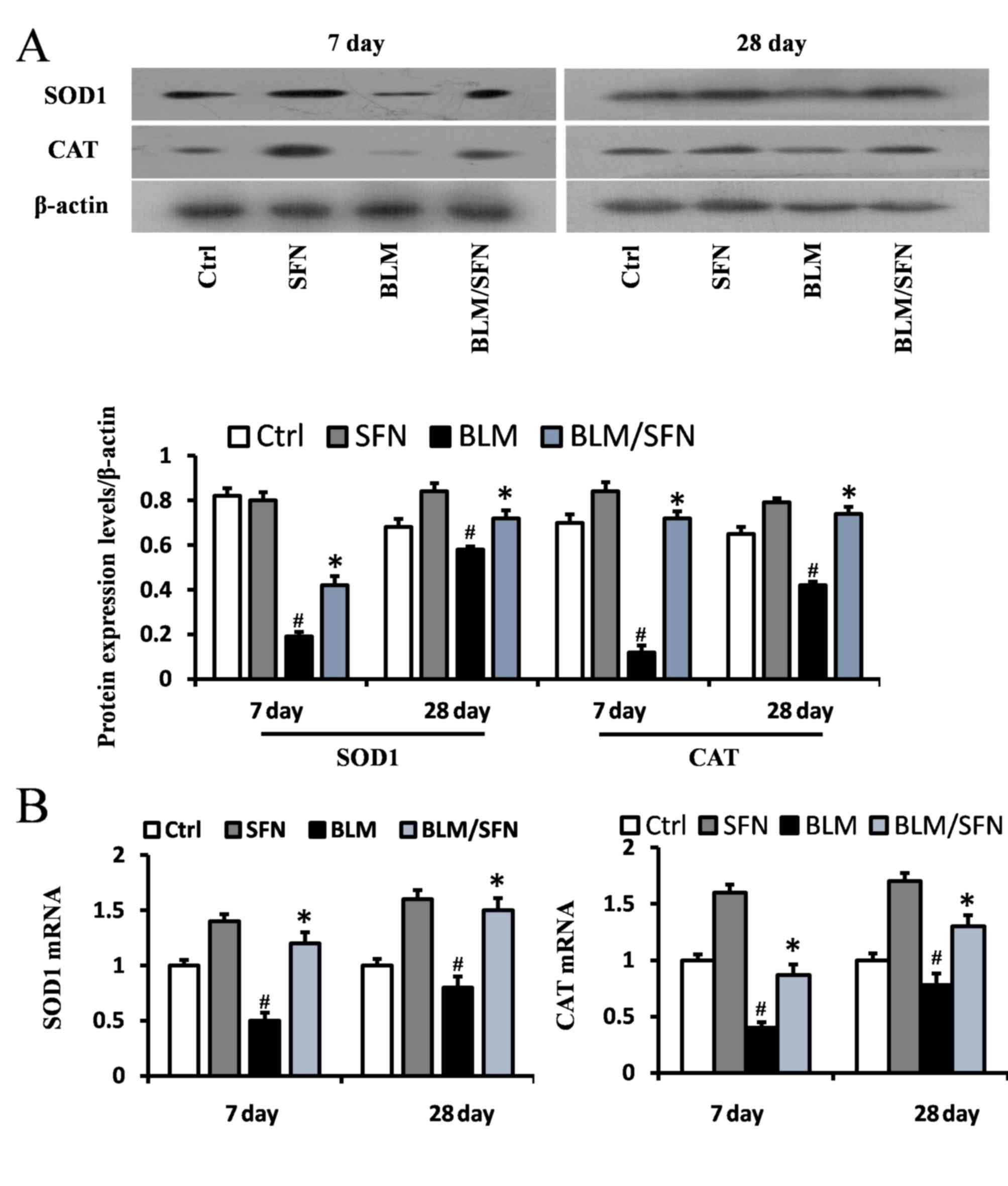
In addition, the transcriptional activity of Nrf2
was investigated by examining the mRNA and protein expression
levels of its downstream antioxidant target genes, including NQO1
and HO-1 (Fig. 6), and SOD1 and
CAT (Fig. 7). In the BLM group,
the protein expression levels of HO-1, NQO1, SOD1 and CAT were
significantly decreased in the lungs at days 7 and 28. However, the
mRNA expression levels of NQO1 and HO-1 were increased at day 7,
and the mRNA expression levels of SOD1 and CAT were decreased at
days 7 and day 28, compared with the control. Notably, the protein
levels and the mRNA levels of HO-1, NQO1, SOD1 and CAT were
enhanced following the addition treatment of SFN to BLM. Therefore,
it was hypothesized that SFN may inhibit pulmonary fibrosis by
activating the expression of Nrf2 and its downstream genes.
Discussion
Pulmonary fibrosis is a chronic disease of variable
etiology, and oxidative stress has been implicated as an important
cause in promoting its pathogenesis in humans and animals (22). Emerging evidence has indicated that
BLM induces lung injury as a result of its ability to generate ROS
and elevate oxidative burden by disturbing the antioxidant/oxidant
balance in the lungs (10).
Accordingly, in the present study, levels of oxidative stress and
antioxidant enzymes were investigated. It was demonstrated that
oxidative and nitrogen oxidative damage was increased and the
activities of the antioxidant enzymes SOD1 and CAT decreased
immediately following BLM injection compared with control group
mice. Of note, SFN treatment alleviated BLM-induced oxidative
burden and additionally increased the levels of antioxidant
enzymes. This data supported the hypothesis that upregulation of
antioxidative enzymes following SFN injection is responsible for
the alleviation of pulmonary fibrosis, at least in part by reducing
the oxidative damage. The present study demonstrated that
SFN-induced Nrf2 expression in the lungs protected against
BLM-mediated damage. This was supported by a significant reduction
of lung lesions, including lung epithelial cell apoptosis and
excessive infiltration of inflammatory cells, reduced BLM-induced
thickness of alveolar septa, and significant collagen deposition in
the interstitial areas. Furthermore, administration of SFN to BLM
treated mice attenuated the expression of IL-1β, TNF-α, TGF-β and
cleaved caspase-3.
A previous study demonstrated that Nrf2 expression
was increased in a lung fibrosis model (23). However, the precise role of Nrf2 in
a mouse fibrosis model was identified by Cho et al (24) using Nrf2 knockout mice. They used
BLM to induce fibrosis in Nrf2 knockout and wild-type mice and
demonstrated that compared to wild-type mice, Nrf2 knockout mice
exhibited an increased susceptibility, including increased lung
weight, inflammation, hydroxyproline content and fibrotic score
(24). Another study observed that
protein expression levels of the Nrf2 transcription factor was
involved in circadian regulation in the lungs of C57BL/6 mice. The
study observed a significantly higher fibrotic score when Nrf2
protein expression levels were reduced, even when BLM treatment was
decreased. The Nrf2 rhythmic activity affected tissue
susceptibility to BLM-induced lung fibrosis in a
time-of-day-dependent manner (13). Walters et al (25) identified elevated protein and mRNA
levels of Nrf2, and upregulation of NQO1 and HO-1 in a pulmonary
fibrosis model. Consistent with this, the present study observed
significantly increased mRNA and protein expression levels of Nrf2
at days 7 and 28 in a lung fibrosis model. However, the mRNA
expression levels of the Nrf2-associated downstream target genes
NQO1 and HO-1 were increased at day 7 following BLM administration.
This high level of Nrf2 expression in the mouse fibrosis model
appeared to be protective, because Nrf2 expression was rapidly
upregulated in tissues in response to oxidative stress at an early
stage; however, gradually decreased following chronic oxidative
stress. The protective role of Nrf2 was additionally demonstrated
in previous studies investigating other disease models (26,27).
For instance, Nrf2 expression in the renal tissue of multiple
low-dose streptozotocin-induced type1 diabetic mice was
significantly increased at 3 months; however, not at 6 months
(18).
A study by Artaud-Macari et al (28) used SNF to treat primary lung
fibroblasts cultured from healthy and patients with idiopathic
pulmonary fibrosis (IPF). They observed that Nrf2 activation
increased antioxidant defenses and reversed the myofibroblastic
differentiation in IPF patient-derived fibroblasts. Furthermore,
SFN treatment was demonstrated to increase the expression of Nrf2
and antioxidants proteins, and decrease ROS levels and
myofibroblastic dedifferentiation in cultured fibroblasts from
healthy and IPF patients. In addition, SFN treatment inhibited
TGF-β1-mediated pro-fibrotic deleterious effects in IPF and healthy
fibroblasts; however, not in Nrf2 ablated fibroblasts. Another
study demonstrated that pretreatment of mouse fibroblasts with SFN
increased their resistance to subsequent treatment with paraquat or
hydrogen peroxide in an Nrf2-dependent manner (29). SFN has additionally been reported
to protect against diabetes-induced aorta damage, cardiomyopathy
and nephropathy (18,30,31),
and attenuate hepatic fibrosis (32), via upregulation and activation of
Nrf2. In the present study, the data is consistent with the
observations that SFN may act as a protective agent against
BLM-induced oxidative stress by alleviating pulmonary lesions.
In conclusion, SFN may protect against BLM-induced
pulmonary fibrosis in mice. Fibrosis-induced and age-matched
control mice were administered with a daily dose of 0.5 mg/kg SFN,
which prevented the progression of lung damage. Lung damage
prevention was accompanied by significant upregulation of Nrf2
expression and its downstream target antioxidant genes NQO1, HO-1,
SOD1 and CAT. These results suggested that lung fibrosis maybe
prevented by SFN treatment, most likely via upregulation of
Nrf2-mediated protein expression. Thus, the results from this study
may facilitate the development of therapies for BLM-toxicity and
pulmonary fibrosis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81370162).
References
|
1
|
Todd NW, Luzina IG and Atamas SP:
Molecular and cellular mechanisms of pulmonary fibrosis.
Fibrogenesis Tissue Repair. 5:112012. View Article : Google Scholar :
|
|
2
|
Fernandez IE and Eickelberg O: New
cellular and molecular mechanisms of lung injury and fibrosis in
idiopathic pulmonary fibrosis. Lancet. 380:680–688. 2012.
View Article : Google Scholar
|
|
3
|
Shi K, Jiang J, Ma T, Xie J, Duan L, Chen
R, Song P, Yu Z, Liu C, Zhu Q and Zheng J: Dexamethasone attenuates
bleomycin-induced lung fibrosis in mice through TGF-β, Smad3 and
JAK-STAT pathway. Int J Clin Exp Med. 7:2645–2650. 2014.
|
|
4
|
Barratt S and Millar A: Vascular
remodelling in the pathogenesis of idiopathic pulmonary fibrosis.
QJM. 107:515–519. 2014. View Article : Google Scholar :
|
|
5
|
Bocchino M, Agnese S, Fagone E, Svegliati
S, Grieco D, Vancheri C, Gabrielli A, Sanduzzi A and Avvedimento
EV: Reactive oxygen species are required for maintenance and
differentiation of primary lung fibroblasts in idiopathic pulmonary
fibrosis. PLoS One. 5:e140032010. View Article : Google Scholar :
|
|
6
|
Kinnula VL and Myllärniemi M:
Oxidant-antioxidant imbalance as a potential contributor to the
progression of human pulmonary fibrosis. Antioxid Redox Signal.
10:727–738. 2008. View Article : Google Scholar
|
|
7
|
Liu RM, Vayalil PK, Ballinger C, Dickinson
DA, Huang WT, Wang S, Kavanagh TJ, Matthews QL and Postlethwait EM:
Transforming growth factor β suppresses glutamate-cysteine ligase
gene expression and induces oxidative stress in a lung fibrosis
model. Free Radic Biol Med. 53:554–563. 2012. View Article : Google Scholar :
|
|
8
|
Felton VM, Borok Z and Willis BC:
N-acetylcysteine inhibits alveolar epithelial-mesenchymal
transition. Am J Physiol Lung Cell Mol Physiol. 297:L805–L812.
2009. View Article : Google Scholar :
|
|
9
|
Odajima N, Betsuyaku T, Nagai K, Moriyama
C, Wang DH, Takigawa T, Ogino K and Nishimura M: The role of
catalase in pulmonary fibrosis. Respir Res. 11:1832010. View Article : Google Scholar :
|
|
10
|
Kinnula VL, Fattman CL, Tan RJ and Oury
TD: Oxidative stress in pulmonary fibrosis: A possible role for
redox modulatory therapy. Am J Respir Crit Care Med. 172:417–422.
2005. View Article : Google Scholar :
|
|
11
|
Sykiotis GP, Habeos IG, Samuelson AV and
Bohmann D: The role of the antioxidant and longevity-promoting Nrf2
pathway in metabolic regulation. Curr Opin Clin Nutr Metab Care.
14:41–48. 2011. View Article : Google Scholar :
|
|
12
|
Nguyen T, Nioi P and Pickett CB: The
Nrf2-antioxidant response element signaling pathway and its
activation by oxidative stress. J Biol Chem. 284:13291–13295. 2009.
View Article : Google Scholar :
|
|
13
|
Pekovic-Vaughan V, Gibbs J, Yoshitane H,
Yang N, Pathiranage D, Guo B, Sagami A, Taguchi K, Bechtold D,
Loudon A, et al: The circadian clock regulates rhythmic activation
of the NRF2/glutathione-mediated antioxidant defense pathway to
modulate pulmonary fibrosis. Genes Dev. 28:548–560. 2014.
View Article : Google Scholar :
|
|
14
|
Kikuchi N, Ishii Y, Morishima Y, Yageta Y,
Haraguchi N, Itoh K, Yamamoto M and Hizawa N: Nrf2 protects against
pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2
balance. Respir Res. 11:312010. View Article : Google Scholar :
|
|
15
|
Kandhare AD, Bodhankar SL, Mohan V and
Thakurdesai PA: Effect of glycosides based standardized fenugreek
seed extract in bleomycin-induced pulmonary fibrosis in rats:
Decisive role of Bax, Nrf2, NF-κB, Muc5ac, TNF-α and IL-1β. Chem
Biol Interact. 237:151–165. 2015. View Article : Google Scholar
|
|
16
|
Mouratis MA and Aidinis V: Modeling
pulmonary fibrosis with bleomycin. Curr Opin Pulm Med. 17:355–361.
2011. View Article : Google Scholar
|
|
17
|
Fahey JW and Talalay P: Antioxidant
functions of sulforaphane: A potent inducer of Phase II
detoxication enzymes. Food Chem Toxicol. 37:973–979. 1999.
View Article : Google Scholar
|
|
18
|
Cui W, Bai Y, Miao X, Luo P, Chen Q, Tan
Y, Rane MJ, Miao L and Cai L: Prevention of diabetic nephropathy by
sulforaphane: Possible role of Nrf2 upregulation and activation.
Oxid Med Cell Longev. 2012:8219362012. View Article : Google Scholar :
|
|
19
|
Noh JR, Kim YH, Hwang JH, Choi DH, Kim KS,
Oh WK and Lee CH: Sulforaphane protects against
acetaminophen-induced hepatotoxicity. Food Chem Toxicol.
80:193–200. 2015. View Article : Google Scholar
|
|
20
|
Szapiel SV, Elson NA, Fulmer JD,
Hunninghake GW and Crystal RG: Bleomycin-induced interstitial
pulmonary disease in the nude, athymic mouse. Am Rev Respir Dis.
120:893–899. 1979.
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Cheresh P, Kim SJ, Tulasiram S and Kamp
DW: Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta.
1832:1028–1040. 2013. View Article : Google Scholar
|
|
23
|
Walters DM and Kleeberger SR: Mouse models
of bleomycin-induced pulmonary fibrosis. Curr Protoc Pharmacol
Chapter. 5:Unit 5 462008.
|
|
24
|
Cho HY, Reddy SP, Yamamoto M and
Kleeberger SR: The transcription factor NRF2 protects against
pulmonary fibrosis. FASEB J. 18:1258–1260. 2004.
|
|
25
|
Walters DM, Cho HY and Kleeberger SR:
Oxidative stress and antioxidants in the pathogenesis of pulmonary
fibrosis: A potential role for Nrf2. Antioxid Redox Signal.
10:321–332. 2008. View Article : Google Scholar
|
|
26
|
Jiang X, Bai Y, Zhang Z, Xin Y and Cai L:
Protection by sulforaphane from type 1 diabetes-induced testicular
apoptosis is associated with the up-regulation of Nrf2 expression
and function. Toxicol Appl Pharmacol. 279:198–210. 2014. View Article : Google Scholar
|
|
27
|
Wang Y, Zhang Z, Sun W, Tan Y, Liu Y,
Zheng Y, Liu Q, Cai L and Sun J: Sulforaphane attenuation of type 2
diabetes-induced aortic damage was associated with the upregulation
of Nrf2 expression and function. Oxid Med Cell Longev.
2014:1239632014. View Article : Google Scholar :
|
|
28
|
Artaud-Macari E, Goven D, Brayer S, Hamimi
A, Besnard V, Marchal-Somme J, Ali ZE, Crestani B, Kerdine-Römer S,
Boutten A and Bonay M: Nuclear factor erythroid 2-related factor 2
nuclear translocation induces myofibroblastic dedifferentiation in
idiopathic pulmonary fibrosis. Antioxid Redox Signal. 18:66–79.
2013. View Article : Google Scholar
|
|
29
|
Higgins LG, Kelleher MO, Eggleston IM,
Itoh K, Yamamoto M and Hayes JD: Transcription factor Nrf2 mediates
an adaptive response to sulforaphane that protects fibroblasts in
vitro against the cytotoxic effects of electrophiles, peroxides and
redox-cycling agents. Toxicol Appl Pharmacol. 237:267–280. 2009.
View Article : Google Scholar
|
|
30
|
Miao X, Bai Y, Sun W, Cui W, Xin Y, Wang
Y, Tan Y, Miao L, Fu Y, Su G and Cai L: Sulforaphane prevention of
diabetes-induced aortic damage was associated with the
up-regulation of Nrf2 and its down-stream antioxidants. Nutr Metab
(Lond). 9:842012. View Article : Google Scholar :
|
|
31
|
Bai Y, Cui W, Xin Y, Miao X, Barati MT,
Zhang C, Chen Q, Tan Y, Cui T, Zheng Y and Cai L: Prevention by
sulforaphane of diabetic cardiomyopathy is associated with
up-regulation of Nrf2 expression and transcription activation. J
Mol Cell Cardiol. 57:82–95. 2013. View Article : Google Scholar
|
|
32
|
Oh CJ, Kim JY, Min AK, Park KG, Harris RA,
Kim HJ and Lee IK: Sulforaphane attenuates hepatic fibrosis via
NF-E2-related factor 2-mediated inhibition of transforming growth
factor-β/Smad signaling. Free Radic Biol Med. 52:671–682. 2012.
View Article : Google Scholar
|