Introduction
Trauma-induced hemorrhage remains the leading cause
of mortality for people under the age of 45, and affects almost
every community (1,2). The pathophysiological process of
trauma and severe hemorrhage-induced shock (THS) is complex; it
involves a systemic inflammatory reaction and pathological
alterations, such as hypovolemia, hypoxemia, microcirculatory
disturbances and oxidative stress (3). Major complications of THS include
systemic inflammatory response syndrome, multiple organ dysfunction
syndrome and sepsis, which are the main causes of the high
mortality rate (4). Since the
inflammatory response is a key element in THS-induced injury
(5,6), the majority of studies have focused
on the regulation of proinflammatory mediators (7,8).
Bruton's tyrosine kinase (BTK) is a prototypical
member of the Tec family of protein tyrosine kinases. It serves an
essential role in B cell development, and mature B cell activation
and survival; BTK gene mutations result in B cell
deficiency-related X-linked agammaglobulinemia in humans and
X-linked immunodeficiency in mice (9,10).
Previous studies have demonstrated BTK to be a crucial effector for
B cell receptor-, immunoglobulin (Ig)E receptor-, Toll-like
receptor (TLR)- and cytokine receptor-dependent innate and adaptive
immunity systems (10–13). The activation (by tyrosine
phosphorylation) of BTK may stimulate the nuclear factor (NF)-κB
and mitogen-activated protein kinase (MAPK) signaling pathways, and
ultimately trigger a series of inflammatory reactions (14–18).
Previous studies have reported that BTK inhibition may be efficient
in controlling B cell malignancy and B cell-related autoimmune
disorders (19–23); however, whether BTK participates in
THS-induced lung injury remains to be elucidated.
NF-κB and MAPKs have been reported to be involved in
several proinflammatory signaling pathways (24,25).
The activation of NF-κB- or MAPK-mediated pathways have been
demonstrated to increase the levels of inflammatory mediators, such
as nitroc oxide (NO) and inducible NO synthase (iNOS), which serve
important roles in THS-induced organ injury (26). Various organs can be severely
affected by trauma-induced hemorrhage; however, THS-induced lung
injury is one of the main causes of post-traumatic mortality
(27). The present study aimed to
reveal the potential role of BTK in the progression of THS,
investigate the protective effects of BTK inhibition on THS-induced
lung injury in vivo, and explore the molecular mechanisms
underlying the actions of BTK by assessing the activation of NF-κB
and MAPK pathways.
Materials and methods
Animals
Male Sprague-Dawley rats (n=48; age, 10–14 weeks;
weight, 360–400 g) were purchased from Liaoning Changsheng
Biotechnology Co., Ltd. [permit no. SCXK (Liao) 2015–0001; Benxi,
China]. Rats were allowed to acclimate for 1 week in a controlled
environment: 22±1°C, 40–50% humidity, under a 12 h light-dark
cycle. Food pellets and tap water were available ad libitum
throughout the study. Animal care and handling procedures strictly
followed the National Institutes of Health Guide for the Care and
Use of Laboratory Animals (8th Edition, 2010) and were approved by
the Institutional Animal Care and Use Committee of the General
Hospital of Shenyang Military Area Command (Shenyang, China).
Development of the THS model
THS was induced in rats as previously described
(28). Briefly, rats were
anesthetized with an intraperitoneal injection of sodium
pentobarbital (50 mg/kg), bilateral groins were dissected and the
femoral arteries of both sides, and the femoral vein of one side,
were cannulated. Bilateral femur fractures were induced by
hemostatic forceps, one of the femoral arteries was connected to a
multichannel physiology recorder by catheter and the other artery
was induced to hemorrhage to a mean arterial pressure <50 mmHg
for 1.5 h. Rats were resuscitated with lactated Ringer's solution
(Hangzhou Empyrean Animal Health Co., Ltd., Hangzhou, China), at
four times the volume of shed blood, through the femoral vein and
were maintained under anesthesia for an additional 4.5 h.
Experimental groups
A total of 48 rats were randomly divided into four
groups (n=12/group): i) The Sham group, the femoral arteries and
veins were cannulated without induced fractures and bloodletting;
ii) the THS group, bilateral femur fractures and hemorrhage were
induced manually; iii) the THS + LFM-A13 group, rats received a
peritoneal injection of the BTK inhibitor LFM-A13 (25 mg/kg;
Shanghai Biochempartner Co., Ltd., Wuhan, China) and trauma was
immediately induced; and iv) the Sham + LFM-A13 group, rats
received a peritoneal injection of LFM-A13 (25 mg/kg) prior to
surgery but did not receive induced fractures and bloodletting.
Bronchoalveolar lavage fluid (BALF),
blood and lung tissue collection
A total of 6 h following the induction of trauma, 6
rats from each group received endotracheal intubation and the left
lungs were lavaged three times with 1.5 ml cold saline; the whole
BALF volume was collected. Then, the abdomen was opened and
peripheral blood was collected from inferior vena cava using a 10
ml syringe. Both lungs were removed by midline thoracolaparotomy.
The left lungs were snap-frozen in liquid nitrogen and stored at
−80°C, and the right lungs were fixed in 4% paraformaldehyde at 4°C
for 48 h for subsequent histological analysis. The remaining rats
were euthanized with anesthetic overdose (supplementary injection
with 50 mg/kg sodium pentobarbital), the lungs were removed and the
wet weight was measured. Subsequently, lungs were dried at 100°C in
a thermostabilized oven for 72 h, the dry weight was measured and
the wet/dry ratio was calculated. The BALF was divided into two
parts; one part was used for Giemsa staining, as follows: BALF
cells were collected by centrifugation at 300 × g for 10 min at 4°C
and stained with Giemsa solution (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). The total numbers of leukocytes and
eosinophils were counted under an optical microscope. The other
part of the BALF was centrifuged at 1,000 × g for 10 min at 4°C and
the protein concentration of the supernatant was determined using a
Bradford Protein Assay kit (Wanleibio, Shenyang, China).
Histological and immunohistochemical
analysis
Lung tissues (n=5 rats/group) fixed in
paraformaldehyde were embedded in paraffin and cross-sectioned (5
µm). Sections were stained with hematoxylin and eosin, and lung
morphology was observed under a light microscope with an Olympus
DP73 digital camera (Olympus Corporation, Tokyo, Japan).
For immunohistochemical analysis, the 5 µm-thick
lung sections were heated for 10 min in 0.01 mol/l citrate buffer
for antigen retrieval, followed by endogenous peroxidase
inactivation by incubation in 3% H2O2 for 15
min. Sections were blocked with normal goat serum (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) for 15 min at
room temperature, and incubated with rabbit primary antibodies
against phosphorylated (p)-BTK (1:200; cat no. bs-3055R; BIOSS,
Beijing, China) or inducible nitric oxide synthase (iNOS; 1:200;
cat no. BA0362; Wuhan Boster Biological Technology, Ltd., Wuhan,
China) at 4°C overnight. Following overnight incubation, slides
were washed with PBS and incubated with a secondary biotinylated
goat anti-rabbit IgG antibody (1:200; cat no. A0277; Beyotime
Institute of Biotechnology, Haimen, China) at 37°C for 30 min.
Specific proteins of interest were detected by horseradish
peroxidase (HRP)-conjugated streptavidin (cat no. A0303; Beyotime
Institute of Biotechnology) and visualized by 3,3′-Diaminobenzidine
solution (Beijing Solarbio Science & Technology Co. Ltd.);
sections were counterstained with hematoxylin and observed under an
optical microscope.
Inflammatory factors and nitric oxide
(NO) detection
Peripheral blood samples were centrifuged at 1,000 ×
g for 10 min at 4°C and the serum was collected. Serum protein
expression levels of tumor necrosis factor-α (TNF-α) (Rat TNF Alpha
PicoKine™ ELISA kit; cat no. EK0526), interleukin
(IL)-1β (Rat IL-1 Beta PicoKine™ ELISA kit; cat no.
EK0393), IL-6 (Rat IL-6 PicoKine™ ELISA kit; cat no.
EK0412) and monocyte chemotactic protein 1 (MCP-1; Rat MCP-1
PicoKine™ ELISA kit; cat no. EK0902) were determined
using commercially available ELISA kits purchased from Wuhan Boster
Biological Technology, Ltd., according to the manufacturers'
protocol.
Lung tissues were homogenized, freeze-thawed with
liquid nitrogen three times and centrifuged at 10,000 × g for 10
min at 4°C. Following centrifugation, supernatants were collected
and protein concentrations were measured using the Bicinchoninic
Acid Protein Assay kit (Wanleibio). Proteins were diluted to 2
µg/µl in PBS and NO concentrations were measured using the Total
Nitric Oxide Assay kit (cat no. S0023; Beyotime Institute of
Biotechnology).
Western blot analysis
Total and nuclear proteins were extracted from lung
tissue samples (n=5 rats/group) using the Nuclear and Cytoplasmic
Protein Extraction kit (Wanleibio) and quantified using a
bicinchoninic assay kit (Wanleibio). Equal amounts of extracted
protein samples (40 µg) were separated by 5–12% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
fat-free milk for 1 h at room temperature and probed with primary
antibodies against BTK (1:500; cat no. bs-2752R; BIOSS), p-BTK
(1:500; cat no. bs-3055R; BIOSS), iNOS (1:400; cat no. BA0362;
Wuhan Boster Biological Technology, Ltd.), inhibitor of NF-κB (IκB;
1:500; cat no. bs-1287R; BIOSS), p-IκB antibody (1:500; cat no.
bs-5515R; BIOSS), NF-κB (1:400; cat no. BA0610; Wuhan Boster
Biological Technology, Ltd.), extracellular signal-regulated kinase
(ERK; 1:500; cat no. bs-2637R; BIOSS), p-ERK (1:500; cat no.
bs-1522R; BIOSS), c-Jun N-terminal kinase (JNK; 1:500; cat no.
bs-10562R; BIOSS), p-JNK (1:500; cat no. bs-1640R; BIOSS), p38
(1:500; cat no. bs-0637R; BIOSS) or p-p38 (1:500; cat no. bs-5477R;
BIOSS) at 4°C overnight. Following overnight incubation, the
membranes were washed with PBS and the specific proteins of
interest were detected with secondary HRP-conjugated goat
anti-rabbit IgG (1:5,000; cat no. WLA023; Wanleibio) or goat
anti-mouse IgG (1:5,000; cat no. WLA024; Wanleibio) at 37°C for 45
min. Protein bands were visualized using an Enhanced
Chemiluminescence kit (Wanleibio). Densitometric analysis was
performed by Gel-Pro Analyzer version 3.0 (Media Cybernetics, Inc.,
Rockville, MD, USA), using β-actin (1:1,000; cat no. sc-47778;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and histone H3
(1:500; cat no. bs-17422R, BIOSS) as internal controls.
Electrophoretic mobility shift assay
(EMSA)
NF-κB DNA-binding activity was detected using an
NF-kB EMSA kit (cat no. BITF001; Viagene Biotech, Inc., Tampa, FL,
USA), according to the manufacturer's protocol. Nuclear proteins
were extracted and quantified as aforementioned. Proteins (25 µg)
were diluted in 5 µl PBS and incubated with 0.5 µl biotin-labeled
NF-κB specific probes (0.2 µmol/l; cat no. TF001BP; Viagene
Biotech, Inc.) at room temperature for 20 min. The NF-κB-specific
recognition sequence is: 5′-AGTTGAGGGGACTTTCCCAGGC-3′. The reaction
mixtures (10 µl) were electrophoresed on 6.5% non-denaturing
polyacrylamide gel at 180 V for 80 min. Protein-DNA complexes were
electrically transferred onto nylon membranes, cross-linked under
an ultraviolet lamp for 10 min and specific bands were detected by
HRP-conjugated streptavidin and visualized using the Enhanced
Chemiluminescence kit (Wanleibio).
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least 5 independent experiments. The statistical significance
of the differences between groups was assessed by one-way analysis
of variance followed by a post hoc Bonferroni test for multiple
comparisons. Statistical analysis was performed using SPSS software
version 16.0 (SPSS, Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Pulmonary BTK is activated by THS
To investigate a potential role for BTK in
THS-induced pulmonary injury, the protein expression levels of
p-BTK were examined by immunohistochemistry. As shown in Fig. 1A, p-BTK was mainly localized to the
membrane of alveolar epithelial cells in the Sham group, and was
notably upregulated by THS-induced injury. Western blot analysis
also demonstrated that THS rats exhibited a significantly increased
expression of p-BTK in the lungs (P<0.01 vs. Sham group);
however, the protein expression levels of total BTK were unchanged
(Fig. 1B), suggesting that BTK was
activated in the lungs of rats with THS-induced injury.
Effects of BTK on pulmonary capillary
permeability and morphological alterations
To examine the role of BTK in pulmonary capillary
permeability, a specific inhibitor of BTK, LFM-A13, was
intraperitoneally injected prior to THS induction. Total protein
concentration in the BALF from rats with THS-induced injury was
significantly higher compared with the Sham group (P<0.01;
Fig. 2A), and this was
significantly reduced by LFM-A13 treatment (P<0.05); however,
protein concentrations in the Sham + LFM-A13 group remained
unaltered. Similarly, the pulmonary wet/dry ratio was significantly
increased in the THS group compared with rats in the Sham group
(P<0.01; Fig. 2B), and was
reduced in THS rats treated with LFM-A13 compared with untreated
THS rats (P<0.05). The wet/dry ratio was not altered in the Sham
+ LFM-A13 group. Histological analysis revealed that the lungs of
THS rats exhibited notable interstitial hyperplasia, edema and
neutrophil infiltration, which were reduced in LFM-A13-treated THS
rats. Converesely, LFM-A13 administration in Sham rats did not
produce significant histopathological alterations compared with the
Sham group (Fig. 2C).
Effects of BTK on the inflammatory
response in THS rats
The total number of leukocytes and eosinophils were
counted in BALF from each group to examine the effects of BTK on
pulmonary inflammatory cell infiltration. As shown in Figs. 3A and B, the number of leukocytes
and eosinophils were significantly increased in the BALF of THS
rats compared with rats in the Sham group, and were significantly
decreased by LFM-A13 treatment. The expression levels of the
proteins involved in the inflammatory response were also determined
in samples of peripheral blood collected from rats in each group.
The results demonstrated that the levels of TNF-α, IL-1β, IL-6 and
MCP-1 in THS rats were significantly upregulated (P<0.01 vs.
Sham), as expected; LFM-A13 treatment effectively reduced the
levels of these inflammatory cytokines in THS rats (P<0.05 vs.
untreated THS rats; Fig. 3C-F).
Notably, treatment of Sham rats with LFM-A13 did not affect
inflammatory cell numbers or the levels of inflammatory
factors.
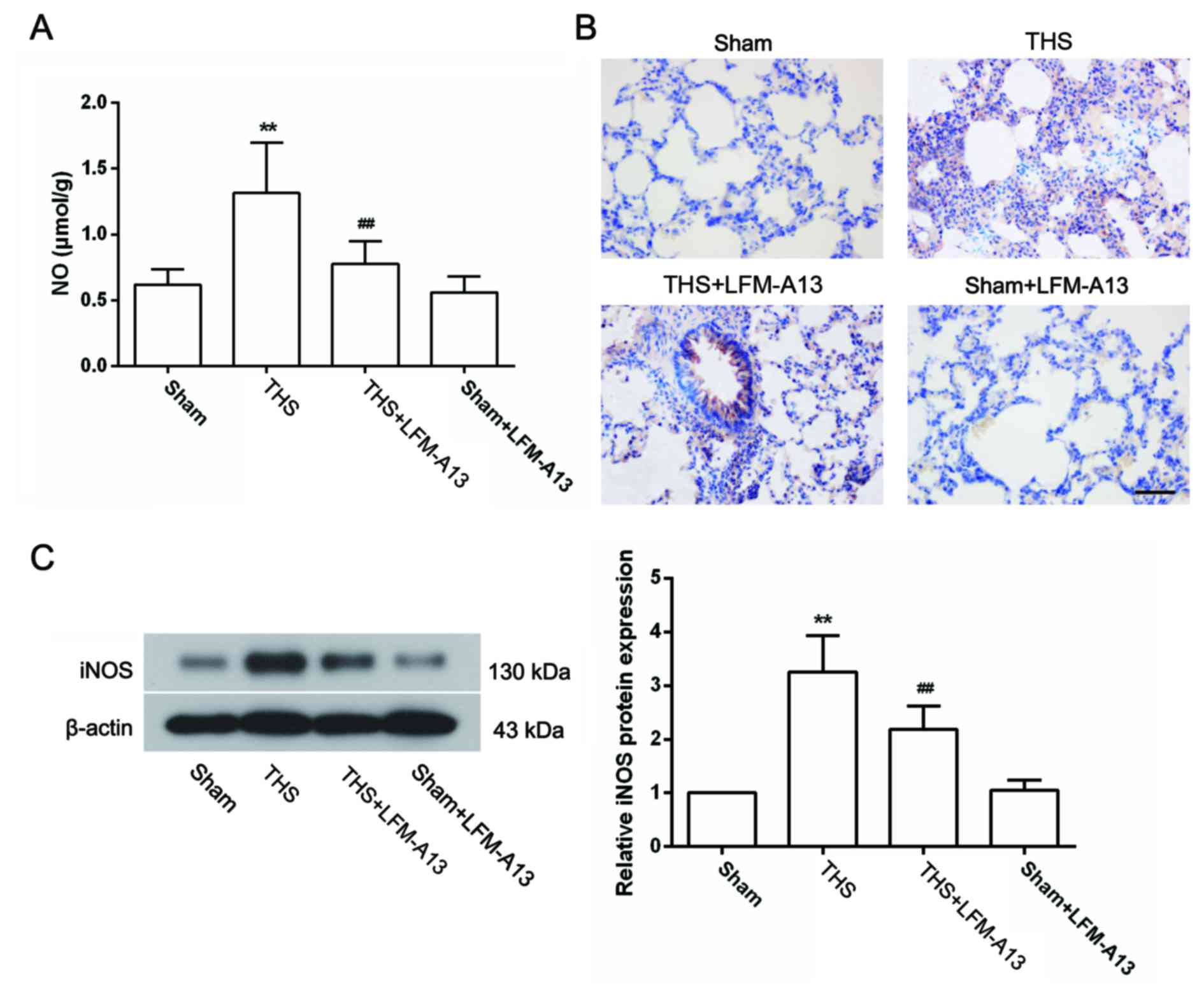
Effects of BTK on the expression of NO
and iNOS in THS rats
The excessive production of NO by iNOS has been
reported to be involved in the pathogenesis of THS-induced lung
injury (29); therefore, the
concentration of NO and the expression of iNOS in the lungs were
measured. As shown in Fig. 4A, the
concentration of NO in the lungs of THS rats was significantly
increased compared with rats in the Sham group (P<0.01), and was
significantly decreased by LFM-A13 treatment (P<0.01 vs.
untreated THS rats). Treatment with LFM-A13 did not affect NO
concentration in Sham rats (P>0.05 compared with the Sham
group). Immunohistochemical analysis demonstrated that very little
iNOS protein expression was detected in the lungs of rats in the
Sham group, whereas iNOS was widely expressed in lung tissues of
THS rats (Fig. 4B). The protein
expression levels of iNOS were notably reduced by LFM-A13 treatment
(Fig. 4B). These results were
confirmed by western blot analysis (Fig. 4C), which demonstrated that the
significant increase in iNOS protein expression induced by THS
(P<0.01 vs. Sham) was significantly inhibited by LFM-A13
treatment (Fig. 4C; P<0.01 vs.
untreated THS rats). In addition, immunohistochemical and western
blot analysis revealed that iNOS expression levels in rats from the
Sham + LFM-A13 group were not significantly different compared with
rats in the Sham group.
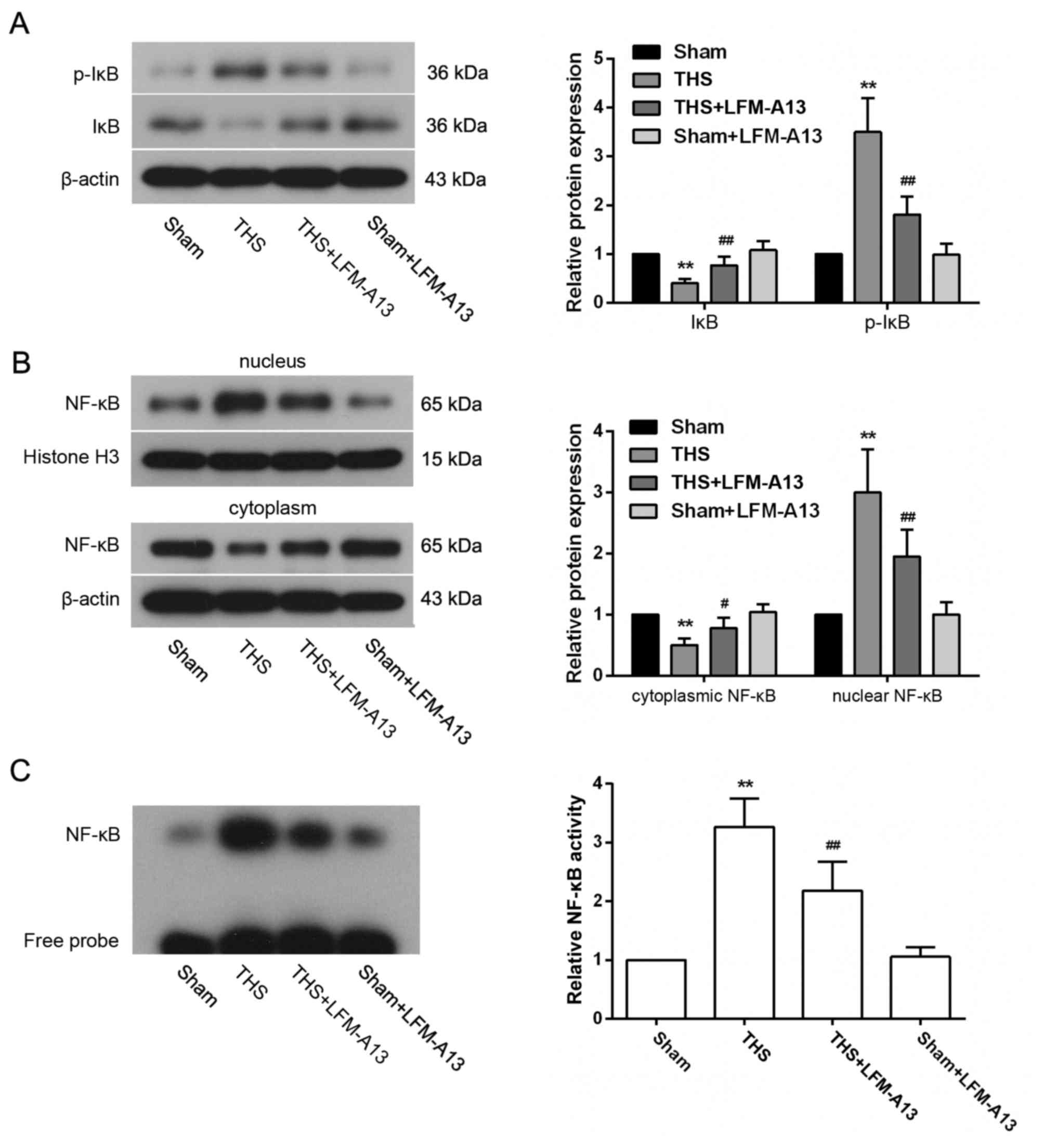
Effects of BTK on NF-κB activity
To investigate the role of NF-κB signaling in the
observed protective effects of LFM-A13 on THS-induced lung injury,
the activation of NF-κB expression in lungs post-THS induction was
examined. p-IκB expression was significantly increased and the
level of IκB was significantly decreased in the THS group compared
with rats in the Sham group (P<0.01; Fig. 5A); conversely, the cytoplasmic
expression of NF-κB was reduced and the nuclear expression levels
of NF-κB were increased following THS induction (P<0.05 vs.
Sham; Fig. 5B). However, these
changes in IκB, p-IκB and NF-κB expression levels were dampened by
LFM-A13 treatment in rats with THS-induced injury. p-IkB, IκB and
NF-κB levels in rats from the Sham + LFM-A13 group were not
significantly different compared with in rats in the Sham group
(P>0.05). Similarly, EMSA experiments demonstrated an increase
in the binding activity of NF-κB in the THS group, which was
strongly reduced by LFM-A13 treatment (P<0.01 vs. Sham or
untreated THS rats, respectively; Fig.
5C); treatment with LFM-A13 in Sham rats did not affect the
binding activity of NF-κB when compared to the Sham group
(P>0.05).
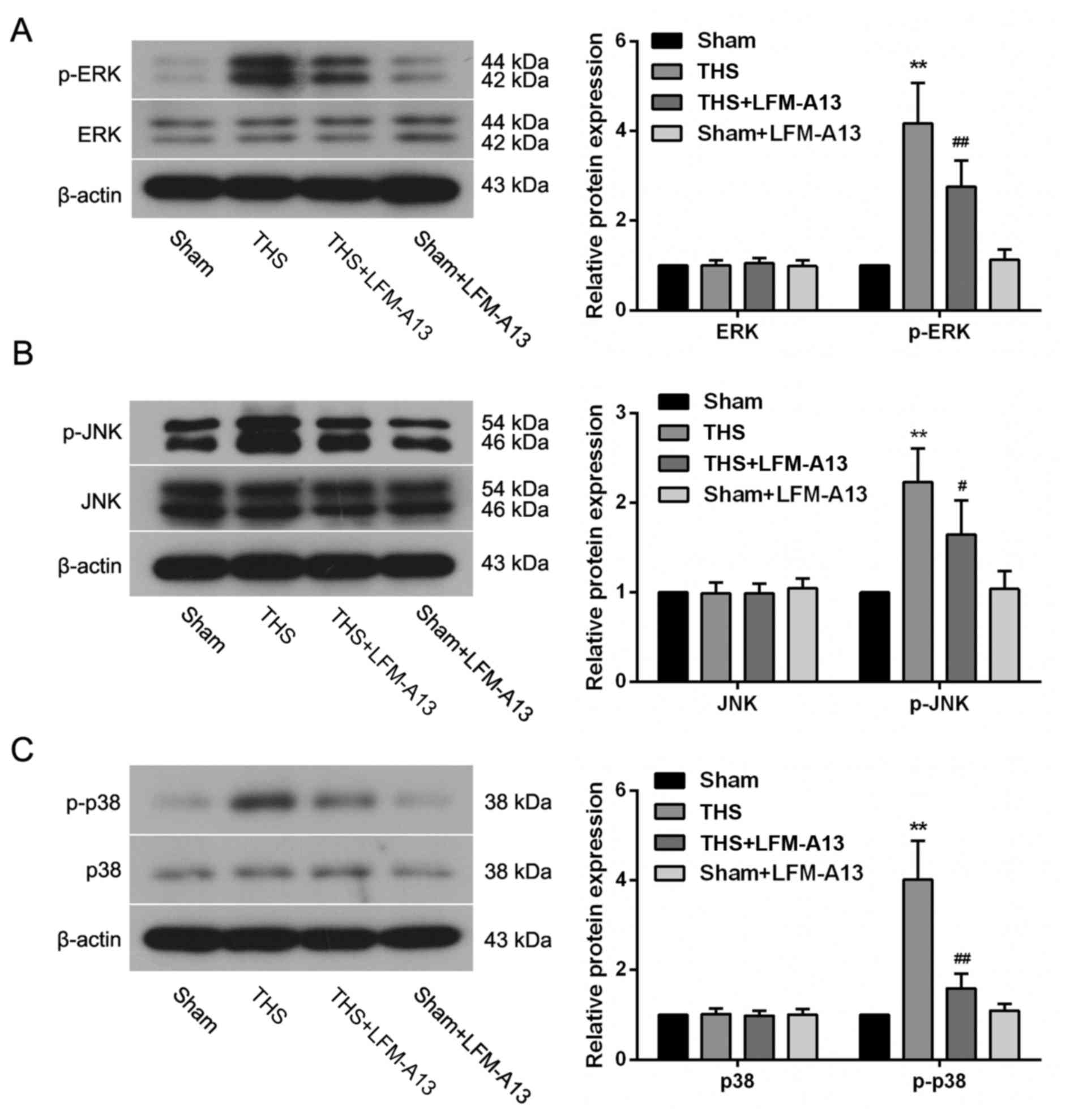
Effects of BTK on MAPK pathways
To further investigate the mechanism by which the
inhibition of BTK protected lungs from THS-induced injury,
components of the MAPK signaling pathway were examined. THS-induced
injury resulted in a pronounced upregulation in the levels of
p-ERK, p-JNK and p-p38 expression (P<0.01; Fig. 6A-C); however, treatment with
LFM-A13 significantly reversed these changes in THS rats,
indicating that inhibition of BTK may be able to suppress the
THS-induced activation of MAPK pathways. The activity of
MAPK-associated pathways in rats from the Sham + LFM-A13 group was
similar compared with in rats from the Sham group (P>0.05),.
Discussion
The present study demonstrated that the activation
of BTK was significantly increased in the lungs of rats following
THS-induced injury. The results revealed that treatment with the
BTK-specific inhibitor LFM-A13 appeared to protect pulmonary
capillary permeability, suppress inflammatory cell infiltration,
inhibit the inflammatory response and alleviate pathological
damage. In addition, LFM-A13 treatment suppressed NO production,
iNOS expression and the activation of NF-κB and MAPK signaling in
rats with THS-induced injury, suggesting that these pathways may be
a part of the mechanisms responsible for the pulmonary protective
effects of BTK inhibition in THS rats.
Systemic inflammation is a major cause of mortality
in patients with THS. A previous study reported that during
hemorrhagic shock an overabundance of inflammatory cytokines were
produced and severe visceral injury occurred (4). BTK is expressed in all hematopoietic
cells, with the exception of plasma cells and T lymphocytes
(30), and it is essential for
lipopolysaccharide (LPS)-induced TNF-α production in mononuclear
cells (31). Additional studies
have demonstrated that the downregulation of BTK expression by
small interfering RNA conferred strong protective effects against
sepsis-induced acute lung injury (32,33).
However, whether BTK is involved in THS-induced lung injury remains
unknown. The present study revealed that BTK was highly activated
in the lungs following THS-induced injury, indicating an essential
role for BTK in THS-related pulmonary damage.
Hemorrhage-induced ischemia and subsequent
reperfusion may result in the release of toxic mediators, resulting
in systemic inflammatory reactions (34). Lung tissues are particularly
vulnerable to injury caused by ischemia-reperfusion (I/R) (35). Inflammatory molecules, such as
TNF-α, IL-1β, IL-6 and NO, and the infiltration of neutrophils may
lead to an increase in microvascular permeability and pulmonary
edema (36–38). Results from the present study
demonstrated that BTK inhibition via LFM-A13 treatment attenuated
pulmonary capillary permeability, reduced the inflammatory response
and alleviated pulmonary pathological damage in THS rats. These
findings were consistent with a previous study demonstrating that
following BTK knockdown, the levels of inflammatory cytokines, as
well as the lung pathological scores, were reduced in mice
following cecal ligation and puncture-induced sepsis (32).
Low concentrations of NO were reported to be
essential for microvascular perfusion and in maintaining organ
function during the early phase of hypovolemic shock (39). However, the levels of iNOS
expression were revealed to be upregulated following hemorrhage
(40), and the resulting
overproduction of NO enhanced inflammatory reactions during
hemorrhagic shock-induced I/R, and further aggravated lung and
liver injury (41). The inhibition
of iNOS expression significantly reduced the strength of the
inflammatory response and lung injury in hemorrhagic shock model
mice (29). In the present study,
LFM-A13 treatment significantly reduced the levels of NO
concentration and iNOS expression in the lungs of THS rats,
suggesting that the inhibition of BTK may protect the lungs from
THS-induced injury through the suppression of NO production.
NF-κB and MAPK signaling are important regulatory
pathways that have been previously reported to participate in the
recruitment of neutrophils and the release of inflammatory
cytokines (42,43). Additional reports demonstrated that
hemorrhagic shock induced abnormal activation of the NF-κB and MAPK
pathways, whereas the suppression of these pathways was revealed to
aid in the protection of shock-induced organ damage (44,45).
BTK may directly bind to TLR4 and mediate the expression of its
downstream targets, such as p38 MAPK and NF-κB (11), and the inhibition of BTK expression
significantly weakened LPS-induced NF-κB activation (46). In the absence of BTK,
TLR3-triggered activation of MAPK and NF-κB signaling was abrogated
(47). In line with these studies,
results from the present study demonstrated that BTK inhibition
significantly suppressed the THS-induced activation of NF-κB and
MAPK signaling pathways. Therefore, the inhibition of BTK may
protect lungs from THS-induced damage, in part by suppressing NF-κB
and MAPK signaling.
In conclusion, the present study demonstrated that
BTK was activated in the lungs of THS model rats and that the
inhibition of BTK significantly attenuated pulmonary capillary
permeability, reduced inflammatory reactions, improved lung
pathological injury, and decreased NO and iNOS levels in THS model
rats. The pulmonary protective effects of BTK inhibition appear to
be at least partly due to the suppression of NF-κB and MAPK
signaling. These data suggested that the inhibition of BTK may be a
potential therapeutic method to protect lungs from THS-induced
damage.
References
|
1
|
Kauvar DS, Lefering R and Wade CE: Impact
of hemorrhage on trauma outcome: An overview of epidemiology,
clinical presentations, and therapeutic considerations. J Trauma.
60:(6 Suppl). S3–S11. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kauvar DS and Wade CE: The epidemiology
and modern management of traumatic hemorrhage: US and international
perspectives. Crit Care. 9:(Suppl 5). S1–S9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Angele MK, Schneider CP and Chaudry IH:
Bench-to-bedside review: Latest results in hemorrhagic shock. Crit
Care. 12:2182008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cai B, Deitch EA and Ulloa L: Novel
insights for systemic inflammation in sepsis and hemorrhage.
Mediators Inflamm. 2010:6424622010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee CC, Chang IJ, Yen ZS, Hsu CY, Chen SY,
Su CP, Chiang WC, Chen SC and Chen WJ: Delayed fluid resuscitation
in hemorrhagic shock induces proinflammatory cytokine response. Ann
Emerg Med. 49:37–44. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Claridge JA, Schulman AM and Young JS:
Improved resuscitation minimizes respiratory dysfunction and blunts
interleukin-6 and nuclear factor-kappa B activation after traumatic
hemorrhage. Crit Care Med. 30:1815–1819. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang H, Huang Y, Xu H, Hu R and Li QF:
Inhibition of hypoxia inducible factor-1α ameliorates lung injury
induced by trauma and hemorrhagic shock in rats. Acta Pharmacol
Sin. 33:635–643. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koscsó B, Trepakov A, Csóka B, Németh ZH,
Pacher P, Eltzschig HK and Haskó G: Stimulation of A2B adenosine
receptors protects against trauma-hemorrhagic shock-induced lung
injury. Purinergic Signal. 9:427–432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang WC, Collette Y, Nunès JA and Olive D:
Tec kinases: A family with multiple roles in immunity. Immunity.
12:373–382. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mohamed AJ, Yu L, Bäckesjö CM, Vargas L,
Faryal R, Aints A, Christensson B, Berglöf A, Vihinen M, Nore BF
and Smith CI: Bruton's tyrosine kinase (Btk): Function, regulation,
and transformation with special emphasis on the PH domain. Immunol
Rev. 228:58–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jefferies CA and O'Neill LA: Bruton's
tyrosine kinase (Btk)-the critical tyrosine kinase in LPS
signalling? Immunol Lett. 92:15–22. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fluckiger AC, Li Z, Kato RM, Wahl MI, Ochs
HD, Longnecker R, Kinet JP, Witte ON, Scharenberg AM and Rawlings
DJ: Btk/Tec kinases regulate sustained increases in intracellular
Ca2+ following B-cell receptor activation. EMBO J. 17:1973–1985.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Turner H and Kinet JP: Signalling through
the high-affinity IgE receptor Fc epsilonRI. Nature. 402:(6760
Suppl). B24–B30. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bajpai UD, Zhang K, Teutsch M, Sen R and
Wortis HH: Bruton's tyrosine kinase links the B cell receptor to
nuclear factor kappaB activation. J Exp Med. 191:1735–1744. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiu Y and Kung HJ: Signaling network of
the Btk family kinases. Oncogene. 19:5651–5661. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lindvall J and Islam TC: Interaction of
Btk and Akt in B cell signaling. Biochem Biophys Res Commun.
293:1319–1326. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mueller H, Stadtmann A, Van Aken H, Hirsch
E, Wang D, Ley K and Zarbock A: Tyrosine kinase Btk regulates
E-selectin-mediated integrin activation and neutrophil recruitment
by controlling phospholipase C (PLC) gamma2 and PI3Kgamma pathways.
Blood. 115:3118–3127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakuma C, Sato M, Takenouchi T, Chiba J
and Kitani H: Critical roles of the WASP N-terminal domain and Btk
in LPS-induced inflammatory response in macrophages. PLoS One.
7:e303512012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kutsch N, Marks R, Ratei R, Held TK and
Schmidt-Hieber M: Role of tyrosine kinase inhibitors in indolent
and other mature B-Cell neoplasms. Biomark Insights. 10:(Suppl 3).
S15–S23. 2015.
|
|
20
|
de Rooij MF, Kuil A, Geest CR, Eldering E,
Chang BY, Buggy JJ, Pals ST and Spaargaren M: The clinically active
BTK inhibitor PCI-32765 targets B-cell receptor- and
chemokine-controlled adhesion and migration in chronic lymphocytic
leukemia. Blood. 119:2590–2594. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Di Paolo JA, Huang T, Balazs M, Barbosa J,
Barck KH, Bravo BJ, Carano RA, Darrow J, Davies DR, DeForge LE, et
al: Specific Btk inhibition suppresses B cell- and myeloid
cell-mediated arthritis. Nat Chem Biol. 7:41–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akinleye A, Chen Y, Mukhi N, Song Y and
Liu D: Ibrutinib and novel BTK inhibitors in clinical development.
J Hematol Oncol. 6:592013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Honigberg LA, Smith AM, Sirisawad M,
Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA and
Buggy JJ: The Bruton tyrosine kinase inhibitor PCI-32765 blocks
B-cell activation and is efficacious in models of autoimmune
disease and B-cell malignancy. Proc Natl Acad Sci USA.
107:13075–13080. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lai EW, Toledo-Pereyra LH, Walsh J,
Lopez-Neblina F and Anaya-Prado R: The role of MAP kinases in
trauma and ischemia-reperfusion. J Invest Surg. 17:45–53. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jarrar D, Chaudry IH and Wang P: Organ
dysfunction following hemorrhage and sepsis: Mechanisms and
therapeutic approaches (Review). Int J Mol Med. 4:575–583.
1999.PubMed/NCBI
|
|
26
|
Kiang JG, Agravante NG, Smith JT and
Bowman PD: 17-DMAG diminishes hemorrhage-induced small intestine
injury by elevating Bcl-2 protein and inhibiting iNOS pathway,
TNF-α increase, and caspase-3 activation. Cell Biosci. 1:212011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levy G, Fishman JE, Xu DZ, Dong W, Palange
D, Vida G, Mohr A, Ulloa L and Deitch EA: Vagal nerve stimulation
modulates gut injury and lung permeability in trauma-hemorrhagic
shock. J Trauma Acute Care Surg. 73:338–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Menzel CL, Sun Q, Loughran PA, Pape HC,
Billiar TR and Scott MJ: Caspase-1 is hepatoprotective during
trauma and hemorrhagic shock by reducing liver injury and
inflammation. Mol Med. 17:1031–1038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hierholzer C, Harbrecht B, Menezes JM,
Kane J, MacMicking J, Nathan CF, Peitzman AB, Billiar TR and
Tweardy DJ: Essential role of induced nitric oxide in the
initiation of the inflammatory response after hemorrhagic shock. J
Exp Med. 187:917–928. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Desiderio S: Role of Btk in B cell
development and signaling. Curr Opin Immunol. 9:534–540. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Horwood NJ, Mahon T, McDaid JP, Campbell
J, Mano H, Brennan FM, Webster D and Foxwell BM: Bruton's tyrosine
kinase is required for lipopolysaccharide-induced tumor necrosis
factor alpha production. J Exp Med. 197:1603–1611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou P, Ma B, Xu S, Zhang S, Tang H, Zhu
S, Xiao S, Ben D and Xia Z: Knockdown of Burton's tyrosine kinase
confers potent protection against sepsis-induced acute lung injury.
Cell Biochem Biophys. 70:1265–1275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krupa A, Fol M, Rahman M, Stokes KY,
Florence JM, Leskov IL, Khoretonenko MV, Matthay MA, Liu KD, Calfee
CS, et al: Silencing Bruton's tyrosine kinase in alveolar
neutrophils protects mice from LPS/immune complex-induced acute
lung injury. Am J Physiol Lung Cell Mol Physiol. 307:L435–L448.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Olanders K, Sun Z, Börjesson A, Dib M,
Andersson E, Lasson A, Ohlsson T and Andersson R: The effect of
intestinal ischemia and reperfusion injury on ICAM-1 expression,
endothelial barrier function, neutrophil tissue influx, and
protease inhibitor levels in rats. Shock. 18:86–92. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kao MC, Yang CH, Sheu JR and Huang CJ:
Cepharanthine mitigates pro-inflammatory cytokine response in lung
injury induced by hemorrhagic shock/resuscitation in rats.
Cytokine. 76:442–448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding R, Han J, Tian Y, Guo R and Ma X:
Sphingosine-1-phosphate attenuates lung injury induced by
intestinal ischemia/reperfusion in mice: Role of inducible
nitric-oxide synthase. Inflammation. 35:158–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hierholzer C, Harbrecht BG, Billiar TR and
Tweardy DJ: Hypoxia-inducible factor-1 activation and
cyclo-oxygenase-2 induction are early reperfusion-independent
inflammatory events in hemorrhagic shock. Arch Orthop Trauma Surg.
121:219–222. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishii H, Ishibashi M, Takayama M, Nishida
T and Yoshida M: The role of cytokine-induced neutrophil
chemoattractant-1 in neutrophil-mediated remote lung injury after
intestinal ischaemia/reperfusion in rats. Respirology. 5:325–331.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cabrales P, Tsai AG and Intaglietta M:
Exogenous nitric oxide induces protection during hemorrhagic shock.
Resuscitation. 80:707–712. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Szabo C and Billiar TR: Novel roles of
nitric oxide in hemorrhagic shock. Shock. 12:1–9. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Collins JL, Vodovotz Y, Hierholzer C,
Villavicencio RT, Liu S, Alber S, Gallo D, Stolz DB, Watkins SC,
Godfrey A, et al: Characterization of the expression of inducible
nitric oxide synthase in rat and human liver during hemorrhagic
shock. Shock. 19:117–122. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Partrick DA, Moore FA, Moore EE, Barnett
CC Jr and Silliman CC: Neutrophil priming and activation in the
pathogenesis of postinjury multiple organ failure. New Horiz.
4:194–210. 1996.PubMed/NCBI
|
|
43
|
Botha AJ, Moore FA, Moore EE, Kim FJ,
Banerjee A and Peterson VM: Postinjury neutrophil priming and
activation: An early vulnerable window. Surgery. 118:358–365. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jeong KY, Suh GJ, Kwon WY, Kim KS, Jung YS
and Kye YC: The therapeutic effect and mechanism of niacin on acute
lung injury in a rat model of hemorrhagic shock: Down-regulation of
the reactive oxygen species-dependent nuclear factor κB pathway. J
Trauma Acute Care Surg. 79:247–255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kochanek AR, Fukudome EY, Li Y, Smith EJ,
Liu B, Velmahos GC, deMoya M, King D and Alam HB: Histone
deacetylase inhibitor treatment attenuates MAP kinase pathway
activation and pulmonary inflammation following hemorrhagic shock
in a rodent model. J Surg Res. 176:185–194. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Jefferies CA, Doyle S, Brunner C, Dunne A,
Brint E, Wietek C, Walch E, Wirth T and O'Neill LA: Bruton's
tyrosine kinase is a Toll/interleukin-1 receptor domain-binding
protein that participates in nuclear factor kappaB activation by
Toll-like receptor 4. J Biol Chem. 278:26258–26264. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee KG, Xu S, Kang ZH, Huo J, Huang M, Liu
D, Takeuchi O, Akira S and Lam KP: Bruton's tyrosine kinase
phosphorylates Toll-like receptor 3 to initiate antiviral response.
Proc Natl Acad Sci USA. 109:5791–5796. 2012. View Article : Google Scholar : PubMed/NCBI
|