Introduction
Acute kidney injury (AKI) is a common and serious
complication characterized by a sharp decline in the glomerular
filtration rate (GFR) (1,2). AKI typically arises over a short
period of time and significantly increases morbidity and mortality
(1). In many cases, AKI results in
poor short- and long-term prognoses (2). Renal tubular epithelial cell (RTEC)
injury is the main cause and underlying mechanism of AKI induced by
ischemia-hypoxia (3). The
occurrence of cell death in RTEC injury may be regulated via
several mechanisms.
Recently, in addition to the three mechanisms of
cell death (apoptosis, autophagy and necrosis), a fourth mechanism
of cell death has been described: Necroptosis (4). Necroptosis differs from apoptosis and
autophagy in many ways (5).
Necroptosis is a type of necrosis that is promoted by death
receptor ligands when the apoptosis signalling pathway is
inhibited. It is dependent on the activity of receptor-interacting
protein kinase (RIP)1, and is a type of programmed cell death
(6). Generally, when receiving a
death signal, RIP1 activates caspase-8 and initiates apoptosis.
However, if the caspase-8 signalling pathway is inhibited, RIP1
activates NADPH oxidase, thus producing reactive oxygen species and
activating the necroptosis signalling pathway (7). Typically, apoptosis is a major cell
death pathway. However, when this pathway is inhibited, for example
following treatment with the caspase inhibitor
benzyloxycarbonyl-Val-Ala-Asp-fluoro-methylketone (zVAD-fmk),
necroptosis becomes dominant (8).
RIP1 is a critical factor in the necroptosis signaling pathway and
is regulated by a specific inhibitor, necrostatin-1 (Nec-1)
(9,10). Investigating these factors may
provide insight into novel treatments for AKI.
In addition, the type of cell death may be more
important than whether a cell dies. A necrotic cell may lead to the
leakage of inflammatory substances and cause severe tissue damage,
whereas an apoptotic cell does not. Thus, the type of cell death
underlying AKI requires further study.
Our previous study indicated that during the AKI
process induced by tumour necrosis factor (TNF)-α, the NRK52-E RTEC
line suffered injury that resulted in all types of cell death.
Additionally, Nec-1 may additionally protect RTECs from ischemia
injury-induced apoptosis via a dynamin-related protein
(DRP1)-dependent mechanism (11).
On the basis of these findings, additional factors were included
for further investigation (11),
such as the mitochondrial damage-related genes Drp-1, Cyclophilin
D, c-Jun N-terminal kinase, and factors of ischemia and hypoxia in
NRK52E cells.
TNF damage and ATP depletion are involved in
ischemia injury (12). Therefore,
the present study aimed to assess these mechanisms in AKI by
inducing damage to NRK-52E cells using TNF-α, followed by treatment
with antimycin A to inhibit mitochondrial respiration to stimulate
an ischemic and hypoxic environment (13). Following this, zVAD-fmk and Nec-1
were added to identify novel targets that may regulate the
underlying mechanism and quantity of cell death.
Materials and methods
Materials
The NRK-52E cell line was obtained from the American
Type Culture Collection (CRL-1571; ATCC; Manassas, VA, USA). A
vertical electrophoresis unit (BIO-RAD 300; Bio-Rad Laboratories,
Inc., Hercules, CA, USA), microplate reader (Multiskan MK3, Thermo
Fisher Scientific, Inc., Waltham, MA, USA), image analyser (Q500IW;
Leica Microsystems GmbH, Wetzlar, Germany), light microscope
(Olympus Corporation, Tokyo, Japan), and hypothermia high-speed
centrifuge (Beckman Coulter, Inc., Brea, CA, USA) were used in the
present study.
Recombinant human TNF-α (cat. no. 96-300-01A-10;
PeproTech, Inc., Rocky Hill, NJ, USA), zVAD-fmk, antimycin A
(ALX-260-020-M005; Enzo Life Sciences, Inc., Farmingdale, NY, USA),
Nec-1 (ALX-430-136; Enzo Life Sciences, Inc.) were used. The
following primary antibodies were used for the study:
microtubule-associated protein 1A/1B-light chain 3 (LC3)A/B (cat.
no. 4108; Cell Signalling Technology, Inc., Danvers, MA, USA),
anti-RIP1 (cat. no. sab3500420; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), anti-RIP3 (cat. no. prs2283; Sigma-Aldrich;
Merck KGaA) and anti-GAPDH (cat. no. G9545; Sigma-Aldrich; Merck
KGaA). The secondary antibody used was goat anti-rabbit
IgG-phycoerythrin (cat. no. sc-3739; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA). A BD FACSAria™ II flow cytometer
(BD Biosciences, Franklin Lakes, NJ, USA) was used for flow
cytometry.
Cell culture and grouping
The NRK-52E rat proximal tubular epithelial cell
line (70–80% confluency; ATCC) was cultured in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum with high glucose. Cells were seeded
into 6-well cell culture plates at a density of 5×104/l
in serum-free media for 24 h to synchronize cell growth. Following
this, NRK-52E cells were treated with or without TNF-α, zVAD-fmk,
antimycin A, and Nec-1 based on the following principles. Cells
were divided into the following groups: Ctrl (Control), exposed to
nothing; T, exposed to 10 ng/ml TNF-α only; TA, exposed to 10 ng/ml
TNF-α + 10 µmol/l antimycin A; TAZ, exposed to 10 ng/ml TNF-α + 50
µmol/l zVAD-fmk + 10 µmol/l antimycin A.
Following modelling, each group was divided into two
subgroups: +N/A and +100 µmol/l Nec-1.
Cell morphological assessment
Cell morphology was observed using an inverted phase
contrast microscope. NRK52-E cells possess characteristics of both
proximal and distal tubular epithelial cells, and the cells
exhibited a single-layer, cobblestone-like arrangement.
Cell viability
Cell viability was evaluated using the Cell Counting
kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Cells (4 wells of each group/subgroup following modelling)
were used, and the supernatant was discarded. A total of 100 µl 10%
CCK-8 was added to the wells. After 2 h of incubation at 37°C and
5% CO2, the cells were assessed using a microplate
reader at a wavelength of 450 nm to assess cell viability using the
following calculation: Cell viability (%)=[(model group-blank)/(Ctl
group-blank)]100%.
Cell death analysis and necroptosis
blocking detection
Following modelling, all media were discarded and
the cells were washed with PBS for 3 min, twice. Subsequently, 4%
paraformaldehyde stationary liquid was added (30 min). The cells
were washed with PBS (3 min, three times) and 0.5 ml Hoechst 33258
dye (5 min) was added. Following staining, the cells were washed
with three times with PBS for 3 min and mounted with Fluoromount-G
mounting medium. Hoechst-stained cells were observed and imaged
using confocal laser scanning microscopy. Apoptotic cells were
quantified, and the apoptotic index (percentage of apoptotic cells)
was calculated (randomly, four high magnification fields were
observed; at least 1,000 cells were counted for each data point;
repeated in triplicate). The apoptotic index was calculated as:
Apoptotic index=(apoptotic cells/counted cells) × 100%.
For Annexin V/propidium iodide (PI) double staining,
a kit was used according to the manufacturer's instructions (cat.
no. KGA108; Nanjing KeyGen Biotech Co., Ltd., Nanjing, China).
Following modelling, the cells were centrifuged at a speed of 110 ×
g at 4°C for 3 min and collected following exposure to 0.25%
trypsin solution (EDTA-free). The medium was discarded, and cells
were washed twice with PBS for 3 min. Cells were subsequently
resuspended in 400 µl 1X Binding Buffer. Subsequently, 5 µl Annexin
V-fluorescein isothiocyanate was added. The solution was mixed and
incubated in the dark for 15 min (2–8°C). Following this, 5 µl
propidium iodide (PI) was added. The solution was mixed and
incubated in the dark for 5 min (2–8°C). The cells were assessed
within 1 h using a flow cytometer.
Western blotting
Cells treated under different experimental
conditions were lysed with radio-immunoprecipitation assay lysis
buffer (BioVision, Inc., Milpitas, CA, USA) The samples were
centrifuged (110 × g, 4°C, 4 min) and the supernatants were
collected as total cell extracts. The protein concentration was
determined using the Bicinchoninic Acid assay kit (cat. no. P0010;
Beyotime Institute of Biotechnology). Protein (40 µg) was separated
by 10% SDS-PAGE and transferred onto a polyvinylidene difluoride
membrane (Immobilon-P; EMD Millipore, Billerica, MA, USA) by
electroblotting. After blocking with non-fat milk for 2 h at 4°C,
the membranes were incubated overnight at 4°C with primary rabbit
monoclonal antibodies against RIP1, RIP3 and LC3-B (1:1,000
dilution) and with an anti-GAPDH antibody (1:2,000 dilution) as
loading control. Following washing the membranes, the secondary
antibody was added (1:500 dilution), and the membranes were
incubated for 1 h at room temperature. Protein bands were
visualized using Enhanced Chemiluminescence Plus Western Blotting
Detection reagents (cat. no. P0018; Beyotime Institute of
Biotechnology) and then exposed to X-ray film (Kodak, Rochester,
NY, USA). The bands of the resulting autoradiography were
densitometrically quantified using Bandscan software (ImageJ,
version 1.48; National Institutes of Health, Bethesda, MD, USA).
Protein expression was quantified as the ratio of the specific band
to GAPDH.
Statistical analysis
Each subgroup comprised at least 16 wells of cells,
which were randomly assigned to different conditions. Calculations
were performed using SPSS software (version 13.0; SPSS Inc.,
Chicago, IL, USA). Data are expressed as the mean ± standard
deviation. One-way analysis of variance followed by post hoc least
significant differences test or Dunnett's t-test was used to
compare the differences between groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Cell necroptosis hypoxic-ischemic
injury model in rat NRK52-E cells is successfully established by
using TNF-α, antimycin A and zVAD-fmk
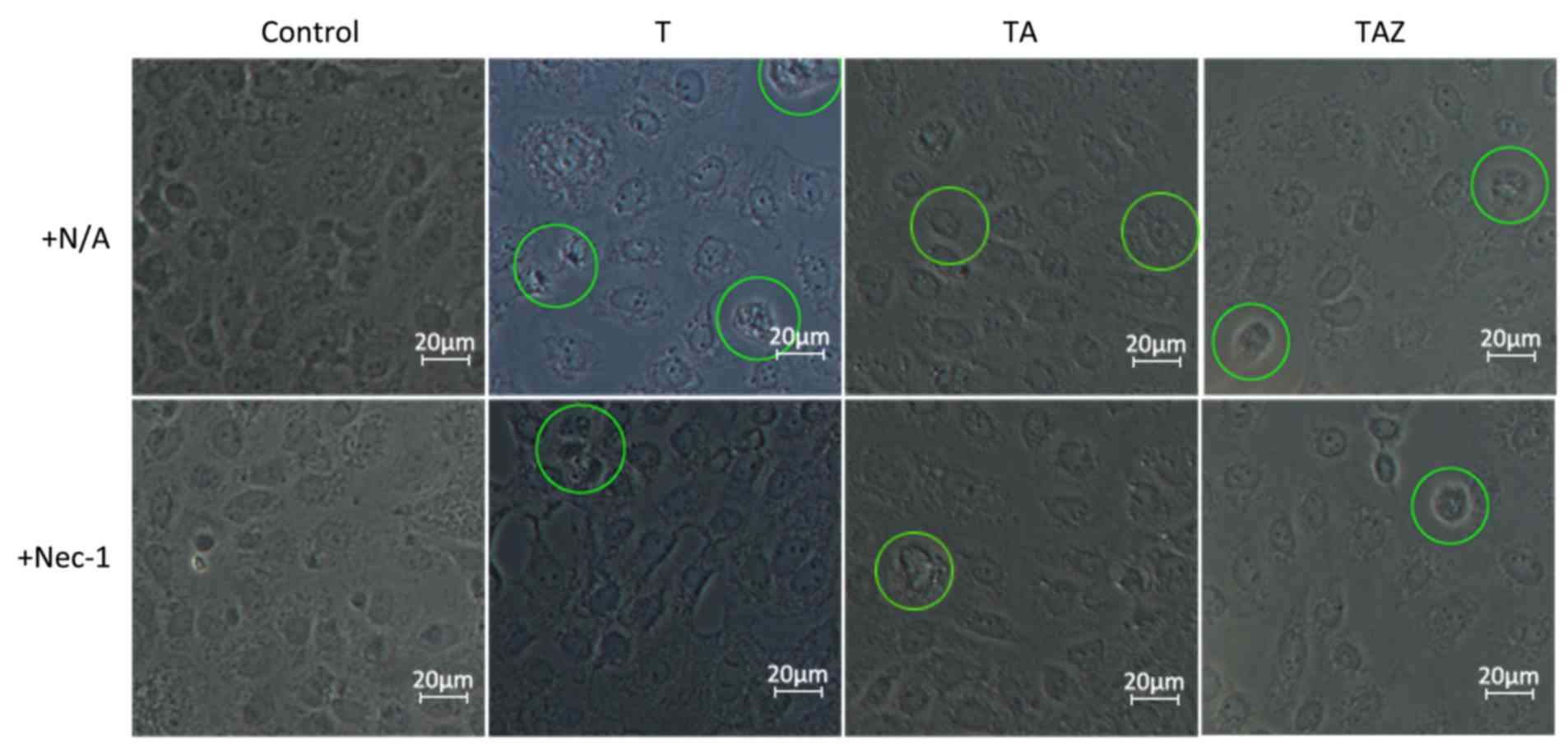
Inverted phase contrast microscopy revealed that the
Ctl group cells had an irregular or oval morphology with an oval
nucleus in the center. The cells were arranged on the bottom of the
culture flask, like paving stones, in the absence of injury
factors. Certain cells in the T and TA groups exhibited apoptotic
characteristics, became smaller or irregular in shape, and
exhibited cell fragmentation and cytoplasm condensation. Certain
cells demonstrated necrotic characteristics, including swelling and
rupturing of the plasma membrane. In the TAZ group, apoptosis was
rarely observed. Most of the cells in this group exhibited
necrotic/necroptotic characteristics, including cell membrane
destruction, cell and organelle swelling, a dissolved nucleolus and
reduced refraction. A subgroup of TAZ cells exposed to Nec-1
exhibited an enhanced morphological status, and more cells appeared
normal, indicating that Nec-1 provided a protective effect
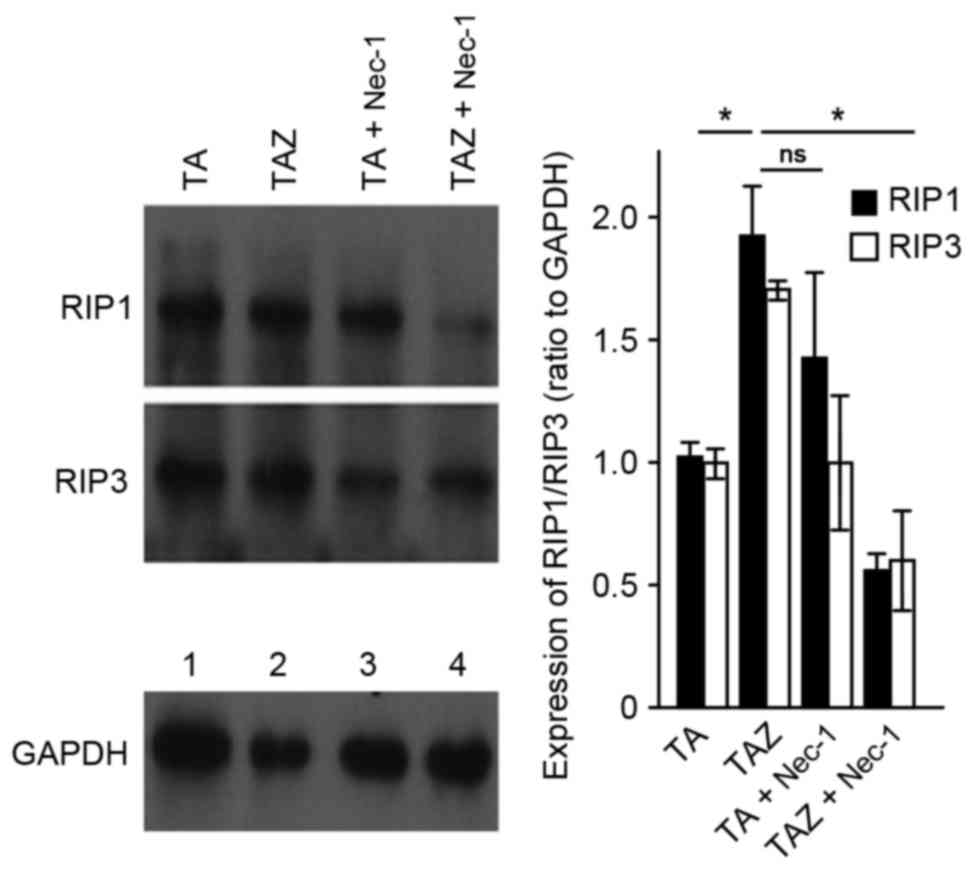
(Fig. 1). In the TAZ group,
apoptosis was inhibited by the caspase inhibitor, zVAD-fmk. These
results indicated that RIP1 and RIP3 expression increased during
key switches of cell death from apoptosis to necroptosis (14). In addition, in the TAZ + Nec-1
group, the expression of these proteins was inhibited by Nec-1
(Fig. 2).
Nec-1 exhibited a protective effect in
the rat NRK52-E cell injury model
Compared with the Ctl group, all of the modelling
groups exhibited reduced cell viability. The TAZ group demonstrated
the lowest cell viability among all of the modelling groups. With
all subgroups exposed to Nec-1, only the TAZ + Nec-1 group
demonstrated significant differences of increased cell viability
compared with cells not treated with Nec-1 (P<0.05; Fig. 3). No significant differences in
cell viability were noted in the Ctl group with or without exposure
to Nec-1 (P>0.05; Fig. 3).
Flow control effect of cell death is
potentially the mechanism underlying the protective effect of
Nec-1
All three mechanisms of cell death (necrosis,
apoptosis and necroptosis) occur when cells are exposed to
injurious factors. More cells undergo apoptosis when exposed to
antimycin A, whereas zVAD-fmk inhibits apoptosis and forces the
cells to die in an apoptosis-independent manner. When Nec-1
interferes with the necroptosis signalling pathway, it appears to
regulate flow control behaviour. Nec-1 inhibited the necroptosis
signalling pathway and induced cells to undergo necrosis and
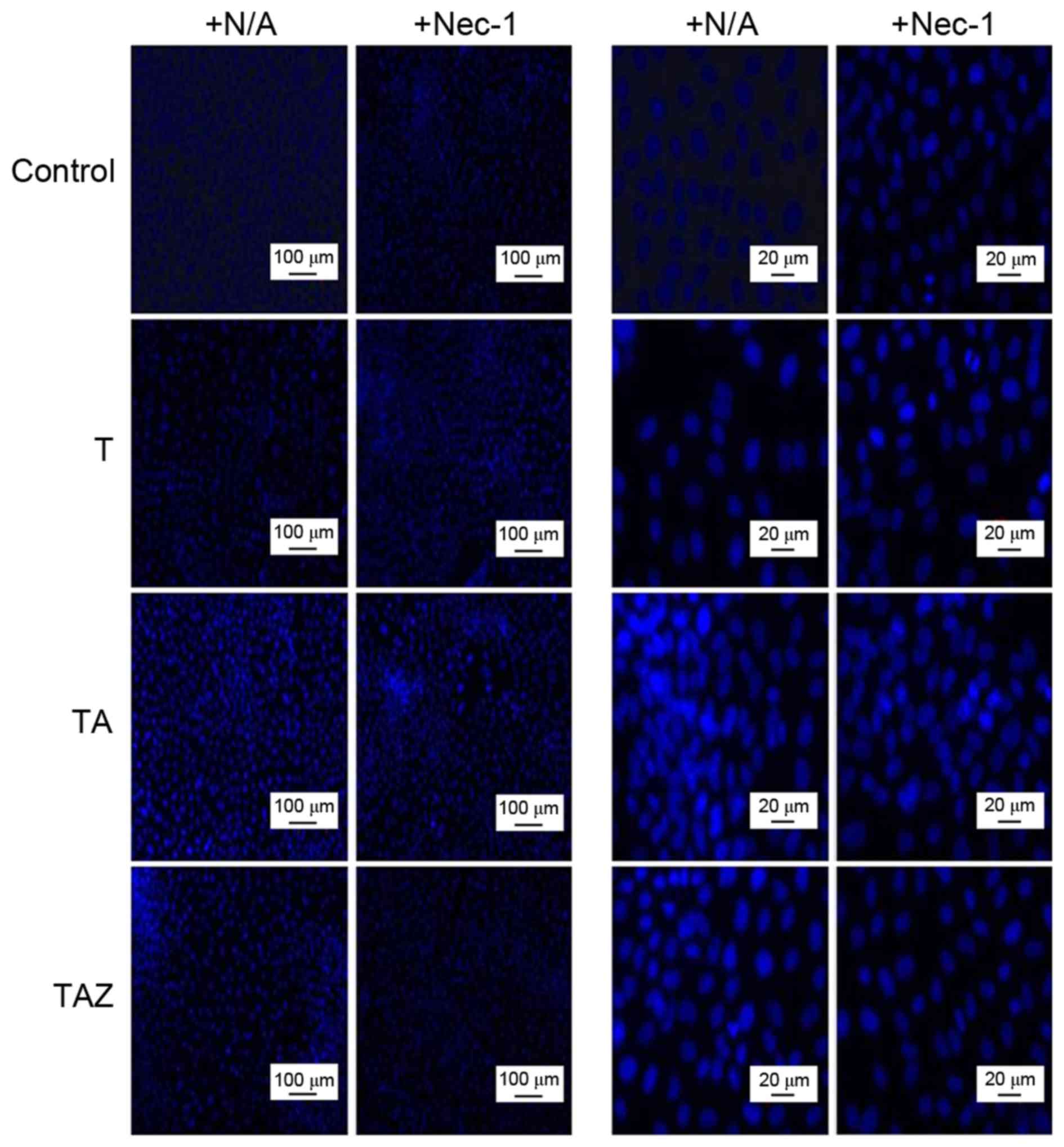
apoptosis. Hoechst staining indicated that more cells committed to
the apoptosis signalling pathways in the TA group. In the presence
of Nec-1, more cells died via apoptosis compared with the +N/A
subgroup. Among all of the modelling groups outside the control
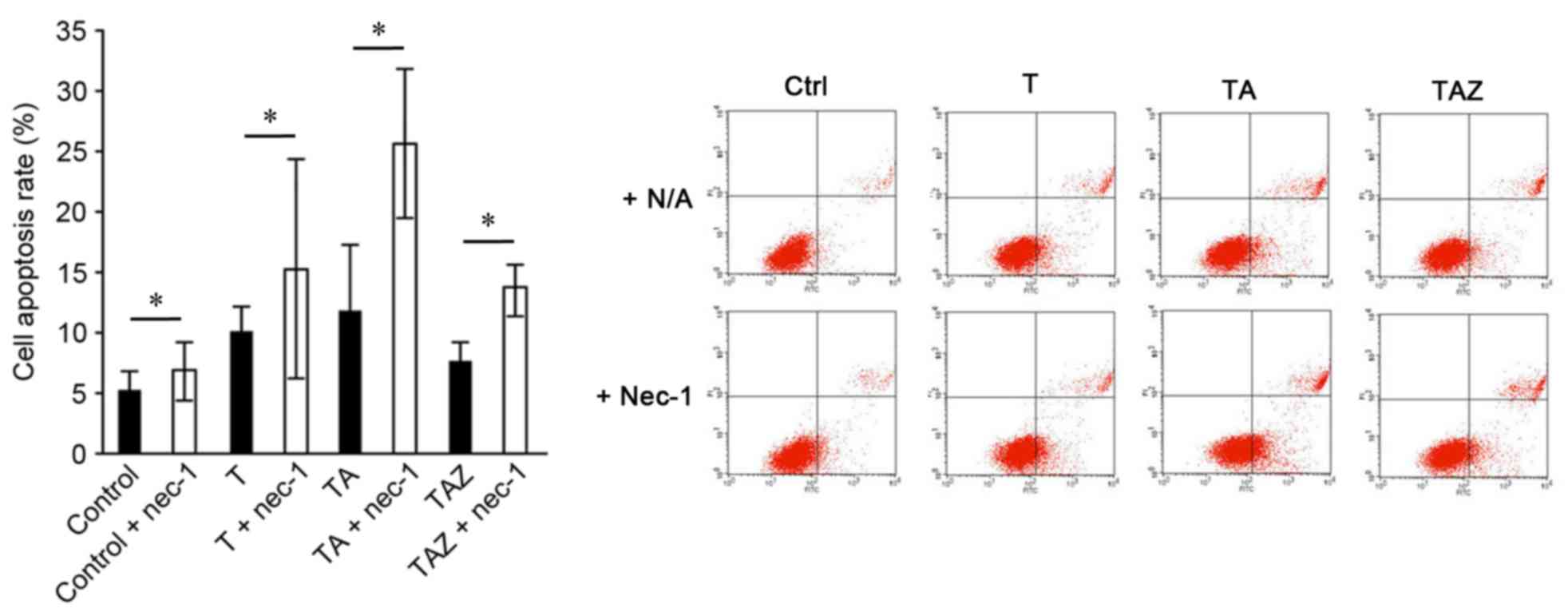
group, the TAZ group exhibited the lowest apoptosis rate (Fig. 4). In addition, Annexin V/PI
staining demonstrated that the apoptosis rate increased in addition
to other subjoining injury factors. However, the rate decreased
when zVAD-fmk (a caspase inhibitor) was added. All subgroups with
the addition of Nec-1 exhibited an increased apoptosis rate, and
the TA + Nec-1 group exhibited the highest apoptosis rate
(25.50±6.15%; Fig. 5).
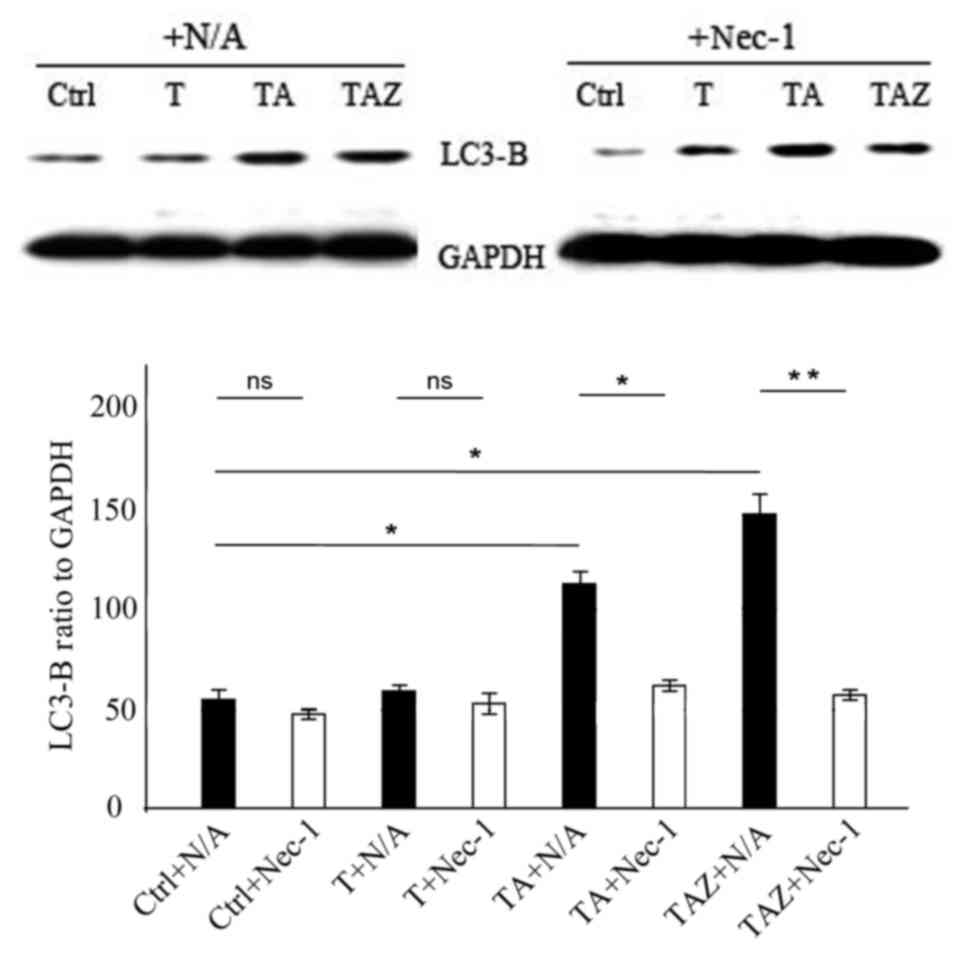
Nec-1 combined with zVAD-fmk
downregulates autophagy marker LC3-B
In vitro western blotting revealed that Nec-1
addition had no effect on LC3-B protein expression levels in the
control and T groups (P>0.05; Fig.
6). LC3-B expression levels were higher in the TA and TAZ
groups compared with control (P<0.05; Fig. 6), and when Nec-1 was added, this
effect was reversed, with LC3-B expression being significantly
downregulated in the TZ (P<0.05) and TAZ (P<0.01) groups
(Fig. 6).
Discussion
AKI is a type of functional and structural disorder
or sign of renal damage. AKI is mainly caused by a decrease in
renal or intra-renal perfusion and in certain cases is fatal, with
a mortality rate of >60% (15,16).
Ischemia-reperfusion injury is a major cause of AKI, and this type
of injury is particularly harmful to RTECs. The key to controlling
the prognosis of AKI involves reducing the death rate of RTECs
(17). RTECs may die via various
mechanisms: Apoptosis, autophagy, necrosis or necroptosis.
Apoptosis induces less damage to the surrounding tissue because the
cell contents do not leak out during the process (4). However, the leakage of inflammatory
substances may lead to serious injury to the surrounding cells when
RTECs die via the necrosis pathway (18).
Our previous study revealed that TNF-α induces
apoptosis and necroptosis (11).
However, as demonstrated in the present study, necroptosis did not
occur when TNF-α was combined with zVAD-fmk. The current study
identified that antimycin A plus TNF-α combined with zVAD-fmk
generates a nearly apoptosis-free model with a cell apoptosis rate
of 7.03±0.40% vs. the control group (4.07±0.20%). In addition,
Annexin V/PI staining strengthened this conclusion, demonstrating a
cell apoptosis rate of 7.49±1.65 vs. 4.77±1.96% in the control
group. Using this model, further research is required in order to
fully elucidate the effects of Nec-1 in AKI.
In current study, cell morphology and CCK-8 cell
viability results revealed significant differences in the TAZ group
treated with and without Nec-1, and cell viability increased from
30.77±1.66 to 69.36±1.69%. Furthermore, Nec-1 exhibited minimal
toxic effects in response to NRK52-E. In the control group, Nec-1
exposure resulted in a cell viability rate of 95.56±6.05%.
Therefore, Nec-1 clearly provides a protective effect to the TAZ
group. However, no differences were observed between the T and TA
groups, and the mechanism may be explained as follows.
When a cell confronts overwhelming injury factors,
including a high concentration of TNF, antimycin A, and severe
hypoxic-ischemic injury similar to that simulated in in
vitro models, the cell may die via four mechanisms: Necrosis,
apoptosis, autophagic death or necroptosis. Without regulation, all
of the above pathways exist, but only a small number of cells
undergo necroptosis. Therefore, Nec-1 cannot provide a protective
effect to the T and TA groups. In the presence of zVAD-fmk, which
inhibits pan-caspase pathways, the apoptosis rate decreased and
cell viability did not increase. These results indicated that many
cells undergo other cell death pathways, including necrosis and
necroptosis. Furthermore, the addition of Nec-1 protected a large
number of cells from dying and directly led to an ~40% increase in
cell viability. Furthermore, certain cells sustained significant
injury from damaging factors to the extent that could not avoid
dying due to Nec-1 inhibition of the necroptosis pathway. All
subgroups exposed to Nec-1 exhibited an observable increase in
apoptosis, which was confirmed by Hoechst staining and Annexin V/PI
staining. The study attempted to control this and inhibit the
apoptosis and necroptosis pathways. Subsequently, certain cells
could not be saved, but the largest alternative pathway was blocked
which increased cell viability by 38.59%, thereby reducing cell
death by 44.26%. This is a notable finding.
LC3-B is associated with the formation of
autophagosomes, and an increase in LC3-B is a main biochemical
parameter of autophagy (19). In
the current research, the expression of LC3-B in the TAZ group
increased, indicating that autophagic death was present and served
an important role in cell death (20–22).
However, the expression of LC3-B decreased and was almost close to
normal levels upon exposure to Nec-1 in the TAZ group, suggesting
that Nec-1 combined with zVAD-fmk reduced autophagy and provided a
protective effect to hypoxic-ischemic injury in RTECs.
In conclusion, the present study established a novel
method to generate a necroptosis cell line model, and demonstrated
that Nec-1 has a protective effect on RTECs via a flow-control-like
effect.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81570609) and the Natural
Science Foundation of Guangdong Province (grant no. 2014A030313545)
and the National Clinical Key Specialty Construction Preparatory
Projects.
References
|
1
|
Ricci Z and Ronco C: New insights in acute
kidney failure in the critically ill. Swiss Med Wkly.
142:w136622012.PubMed/NCBI
|
|
2
|
Bagshaw SM: The long-term outcome after
acute renal failure. Curr Opin Crit Care. 12:561–566. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heyman SN, Evans RG, Rosen S and
Rosenberger C: Cellular adaptive changes in AKI: Mitigating renal
hypoxic injury. Nephrol Dial Transplant. 27:1721–1728. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tharaux PL and Huber TB: How many ways can
a podocyte die? Semin Nephrol. 32:394–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Muppidi J, Porter M and Siegel RM:
Measurement of apoptosis and other forms of cell death. Curr Protoc
Immunol Chapter. 3:Unit 3.17. 2004. View Article : Google Scholar
|
|
6
|
Fulda S: The mechanism of necroptosis in
normal and cancer cells. Cancer Biol Ther. 14:999–1004. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Webster KA: Mitochondrial membrane
permeabilization and cell death during myocardial infarction: Roles
of calcium and reactive oxygen species. Future Cardiol. 8:863–884.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Linkermann A, Hackl MJ, Kunzendorf U,
Walczak H, Krautwald S and Jevnikar AM: Necroptosis in immunity and
ischemia-reperfusion injury. Am J Transplant. 13:2797–2804. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moriwaki K and Chan FK: RIP3: A molecular
switch for necrosis and inflammation. Genes Dev. 27:1640–1649.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Christofferson DE and Yuan J: Necroptosis
as an alternative form of programmed cell death. Curr Opin Cell
Biol. 22:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Jiang F, Chen Y, Luo J, Liu S,
Zhang B, Ye Z, Wang W, Liang X and Shi W: Necrostatin-1 attenuates
ischemia injury induced cell death in Rat tubular cell line NRK-52E
through decreased Drp1 expression. Int J Mol Sci. 14:24742–24754.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vandenabeele P, Galluzzi L, Vanden Berghe
T and Kroemer G: Molecular mechanisms of necroptosis: An ordered
cellular explosion. Nat Rev Mol Cell Biol. 11:700–714. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plaisance I, Duthe F, Sarrouilhe D and
Hervé JC: The metabolic inhibitor antimycin A can disrupt
cell-to-cell communication by an ATP- and Ca(2+)-independent
mechanism. Pflugers Arch. 447:181–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Coca SG, Yusuf B, Shlipak MG, Garg AX and
Parikh CR: Long-term risk of mortality and other adverse outcomes
after acute kidney injury: A systematic review and meta-analysis.
Am J Kidney Dis. 53:961–973. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li PK, Burdmann EA and Mehta RL: World
Kidney Day Steering Committee 2013: Acute kidney injury: Global
health alert. Transplantation. 95:653–657. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang JR, Yao FH, Zhang JG, Ji ZY, Li K,
Zhan J, Tong YN, Lin LR and He YN: Ischemia-reperfusion induces
renal tubule pyroptosis via the CHOP-Caspase-11 pathway. Am J
Physiol Renal Physiol. 306:F75–F84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
González-Guerrero C, Ocaña-Salceda C,
Berzal S, Carrasco S, Fernández-Fernández B, Cannata-Ortiz P, Egido
J, Ortiz A and Ramos AM: Calcineurin inhibitors recruit protein
kinases JAK2 and JNK, TLR signaling and the UPR to activate
NF-κB-mediated inflammatory responses in kidney tubular cells.
Toxicol Appl Pharmacol. 272:825–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanaka A and Youle RJ: A chemical
inhibitor of DRP1 uncouples mitochondrial fission and apoptosis.
Mol Cell. 29:409–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Chen Z, Zhang Y, Zhang M, Zhu X,
Fan Y, Shi S, Zen K and Liu Z: Rhein protects pancreatic β-cells
from dynamin-related protein-1-mediated mitochondrial fission and
cell apoptosis under hyperglycemia. Diabetes. 62:3927–3935. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamin-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001. View Article : Google Scholar : PubMed/NCBI
|