Introduction
A dicentric Y chromosome has 2 centromeres and is a
common abnormal structural rearrangement of the Y chromosome that
is unstable during cell division. It is likely to generate various
cell lines and most affected patients that have been reported are
chromosomal mosaics, typically including the 45,X cell line
(1). If there are completely
symmetrical arms on the dicentric chromosome, it is considered an
isodicentric chromosome. Regardless of the proportion of the two
cell lines in peripheral blood, the phenotypic spectrum of
chromosomal mosaics of all ages may vary widely and includes
healthy infertile males, females with or without Turner syndrome,
individuals with ambiguous genitalia and mixed gonadal dysgenesis
(2–6). However, there are few reports
regarding patients with three different cell lines, particularly
the 46,XY normal karyotype.
Hypospadias is a common abnormality of the external
genitalia in males, and patients with hypospadias may exhibit
chromosomal abnormalities. Kojima et al (7) assessed 400 patients who underwent
surgery to repair hypospadias and identified chromosomal anomalies
in 22 (6%).
The present case report is of a hypospadiac male
infant with a 45,X/46,X,psu idic(Y)(p11.32)/46,XY karyotype. The
patient carried a pseudodicentric Y chromosome with the break point
located at pseudoautosomal region 1 (PAR1). To the best of our
knowledge, this is the first description of a mosaic karyotype
containing three cell lines. The proband manifested short stature
due to haploinsufficiency of short stature homeobox (SHOX),
confirmed through single nucleotide polymorphism (SNP)-array
comparative genomic hybridization (CGH) detection. The combination
of cytogenetic, fluorescence in situ hybridization (FISH)
and SNP-array CGH technologies was beneficial for diagnosing the
karyotype accurately, predicting the prognosis and preparing an
effective treatment plan.
Case report
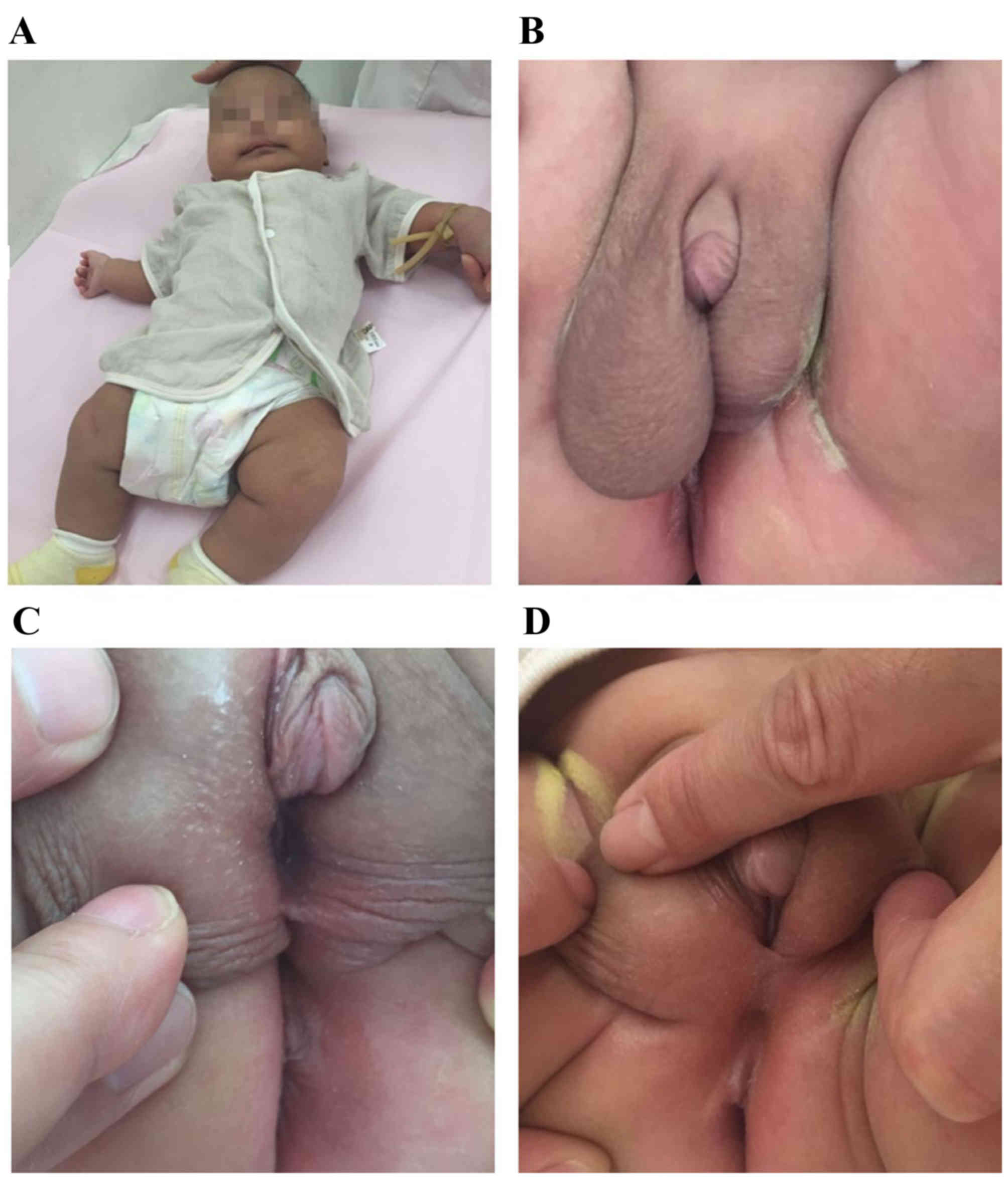
A newborn infant was investigated due to hypospadias
and differential testicular volumes (Fig. 1). Written informed consent was
obtained from the parents. The parents of the infant had not had
contact with hazardous substances and were healthy 28-year-olds.
The karyotypes of the parents were normal male (46, XY) and normal
female (46, XX). There was no known parental consanguinity and
family history was negative for hypospadias and short adult
stature. During the pregnancy, there was no evidence of
intrauterine growth retardation except for short femur length at
23, 31, 32 and 33 weeks. Due to the low risk of Down's syndrome,
karyotype analysis of amniotic cells was not performed. At 6 weeks
pregnant, the infant's mother (gravida 1 para 1) had taken
progesterone for approximately one month due to low progesterone
levels. Due to hypothyroidism at 36 weeks pregnancy, levothyroxine
sodium tablets had been taken.
Cesarean delivery occurred at 38 weeks of gestation.
The birth weight of the infant was 3,150 g (−0.5 standard
deviation; 3,300 g being the average weight of a healthy male
Chinese newborn) and a length of 47 cm (−1.5 standard deviation;
49.9 cm being the average length of a healthy male Chinese
newborn). The Apgar score was 9/10/10. The infant was examined at a
pediatric day-surgery center (Department of Pediatric Surgery,
Beijing Children's Hospital, Capital Medical University, Beijing,
China) at 29 days. Inguinal ultrasonography revealed no uterus or
ovaries. Bilateral testes were both located in the scrotum, with a
left testicular size of 0.8×0.4 cm and a right testicular size of
1.2×0.7 cm. There was a liquid dark space of ~2.5×0.8 cm on the
testicular sheath membrane cavity. No abnormalities were detected
regarding bilateral testicular parenchyma, blood supply and
bilateral spermatic cords. An intra-abdominal investigation of
laparoscopy was not performed. At 56 days old, the weight of the
infant was 6,100 g (0.5 standard deviation; 5,600 g being the
average) and length was 56 cm (−1.5 standard deviation; 58.4 cm
being the average). Averages provided are according to the World
Health Organization standards of child growth.
The serum reproductive hormone levels of the patient
were detected at 56 days (Beijing Obstetrics and Gynecology
Hospital, Capital Medical University, Beijing, China) by
immunoassay, using a UniCel® DxI 800 Immunoassay system
(Beckman Coulter, Inc., Brea, CA, USA), and were normal for
luteinizing hormone (7.22 IU/l; normal range, 1.24–8.62 IU/l),
follicle-stimulating hormone (4.02 IU/l; normal range, 1.27–19.26
IU/l), estradiol (12.37 pg/ml; normal range, <47 pg/ml),
progesterone (0.70 ng/ml; normal range, 0.10–0.84 ng/ml) and
testosterone (1.13 ng/ml; normal range, 1.75–7.81 ng/ml). However,
prolactin levels were elevated (37.49 ng/ml; normal range,
2.64–13.13 ng/ml).
Chromosomal karyotype and FISH
analysis
Lymphocytes were obtained at 5 days after birth; 2
ml of peripheral blood was collected, and then 0.5 ml of peripheral
blood lymphocytes were cultured in lymphocyte culture medium
(Yishengjun; BaiDi Bio-Technology, Guangzhou, China) at 37°C for 72
h, followed by 50 µg/ml colchicine treatment (Yishengjun; BaiDi
Bio-Technology) 1 h before culture termination to arrest mitoses.
The lymphocytes were hypotonically treated in 0.075 M KCl and fixed
in methanol:acetic acid (3:1); then G-banding was performed.
Immunoassay was performed to detect the infant's serum reproductive
hormone levels. Chromosomal analysis of peripheral lymphocytes
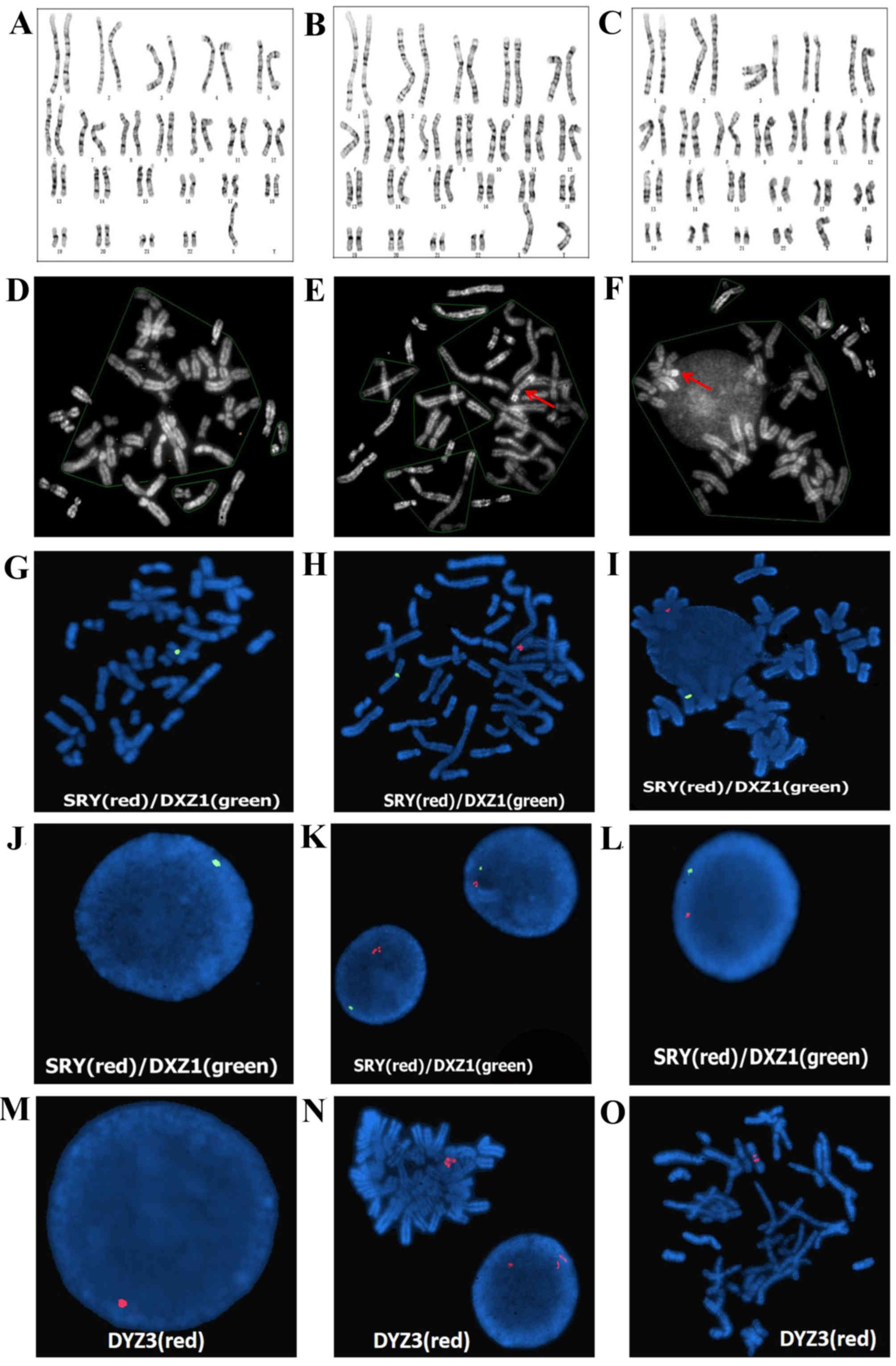
revealed the presence of 3 cell lines. In 23 of 50 (46%) analyzed
metaphases, a numerically abnormal karyotype was detected: 45,X
(Fig. 2A). In 24 of 50 (48%)
metaphases, a suspected isodicentric Y chromosome was detected:
46,X,?idic (Y)(p11.3) (Fig. 2B).
In 3 of 50 (6%) metaphases, a normal karyotype was detected: 46,XY
(Fig. 2C). Additional QFQ-banding
techniques revealed that the heterochromatic region of the long arm
of the Y chromosome was none, two copies and one copy in the above
three cell lines, respectively (Fig.
2D-F). The karyotype was designated as
45,X/46,X,?idic(Y)(p11.3)/46,XY.
FISH was performed using a sex-determining region Y
(SRY)/Vysis CEP X (DXZ1) probe and a Vysis CEP Y (DYZ3) probe on
the SRY region of Yq11.3, the centromeric region of the X
chromosome and Y chromosome (Vysis; Abbott Molecular, Inc., Des
Plaines, IL, USA). The probes were denatured for 2 min at 73°C. The
hybridization mixture (1 µl of each probe, 1 µl H2O and
7 µl of hybridization solution) was applied to each slide and
covered with a coverslip 20×20 mm. The hybridization mixture was a
70% solution of dextran sulphate and formamide in saline-sodium
citrate (SSC) buffer (pH 7). Each slide was then sealed with rubber
cement before hybridization was carried out overnight in a moist
chamber at 37°C. After hybirdization, the slides were washed for 3
min in a solution of X0.4 SSC at 73°C and a second time for 30 sec
in a solution of X2 SSC/0.1% Nonidet P40. Following the final wash,
slides were air dried in the dark. The slides were counterstained
with a solution of 4′, 6-Diamidine-2′-phenylindole dihydrochloride
(DAPI II; Vysis Inc., Downers Grove, IL, USA) diluted in an
antifade mounting medium. The SRY/DXZ1 probe was successfully
hybridized to metaphase cells (Fig.
2G-I) and interphase cell nuclei (Fig. 2J-L). Fig. 2J-L demonstrates that there was no
SRY signal in Fig. 2J, but there
were two signals and one signal on the SRY region of Yq11.3 in
(Fig. 2K and L, respectively). In
addition, there was one signal in the centromeric regions of the X
chromosome on metaphase cells (Fig.
2G-I) and one signal in interphase cell nuclei, respectively
(Fig. 2J-L). One red signal
revealed the active Y centromere on interphase cell nuclei (46,XY;
Fig. 2M). Two red signals revealed
the inactive Y centromere and one red signal revealed the active Y
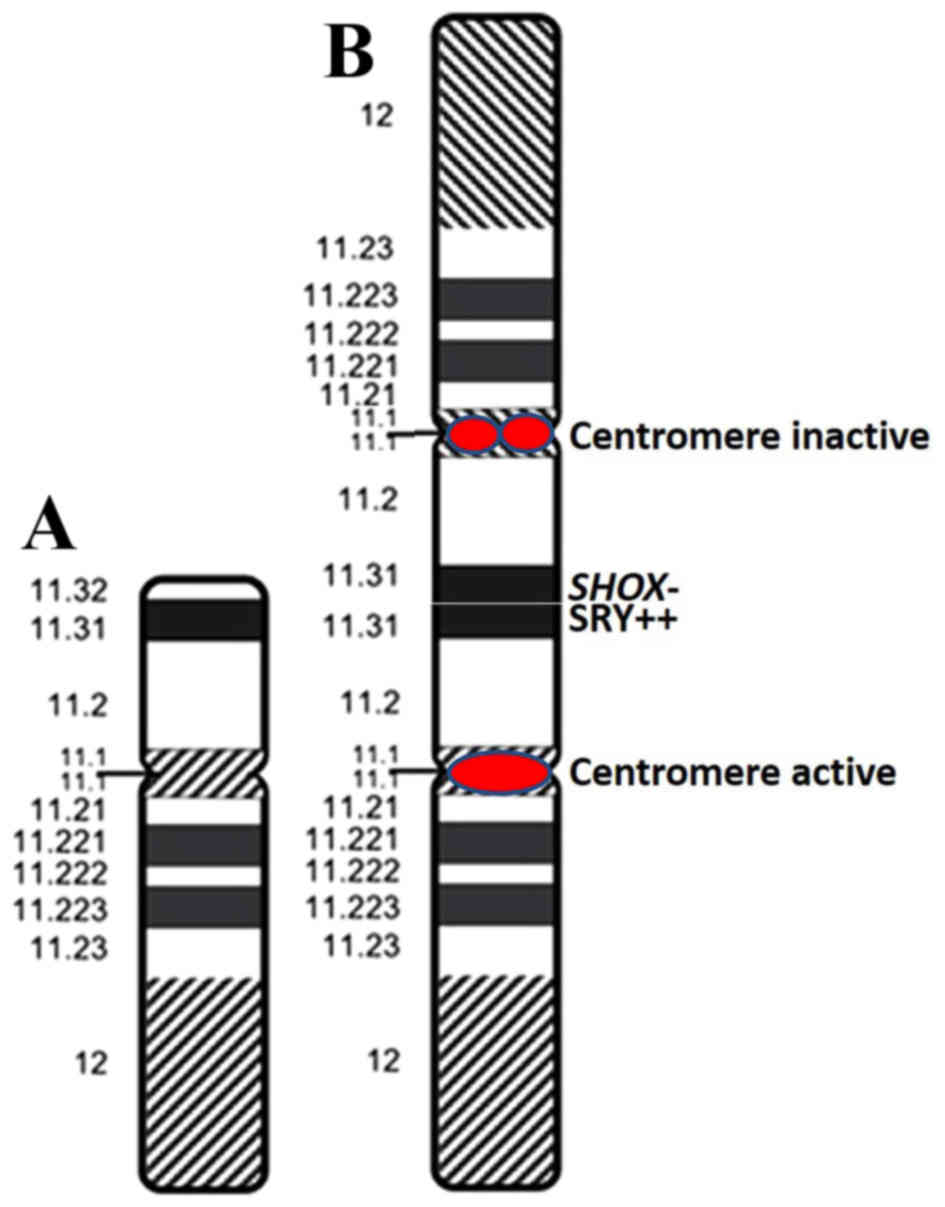
centromere on a metaphase cell [46,X,psu idic (Y)(p11.3); Fig. 2N-O]. According to these results,
the abnormal Y chromosome was identified as a dicentric derivate of
the Y chromosome with psuedoinactivation of one of the two
centromeres (Fig. 3). The
karyotype of the infant was designated as 45,X[20]/46,X,?idic
(Y)(p11.3).ish psu idic(Y)(p11.3) (SRY++, DYZ3++)[26]/46, X,.ish Y
(SRY+, DYZ3+)[4].
SNP-array CGH analysis
Peripheral blood (1 ml) containing 2.25 mg/ml EDTA
was sent to Be Creative Lab Co. Ltd (Beijing, China) for processing
and an Affymetrix CytoScan® 750K (Affymetrix, Inc.,
Santa Clara, CA, USA) gene chip was used to determine the SNPs and
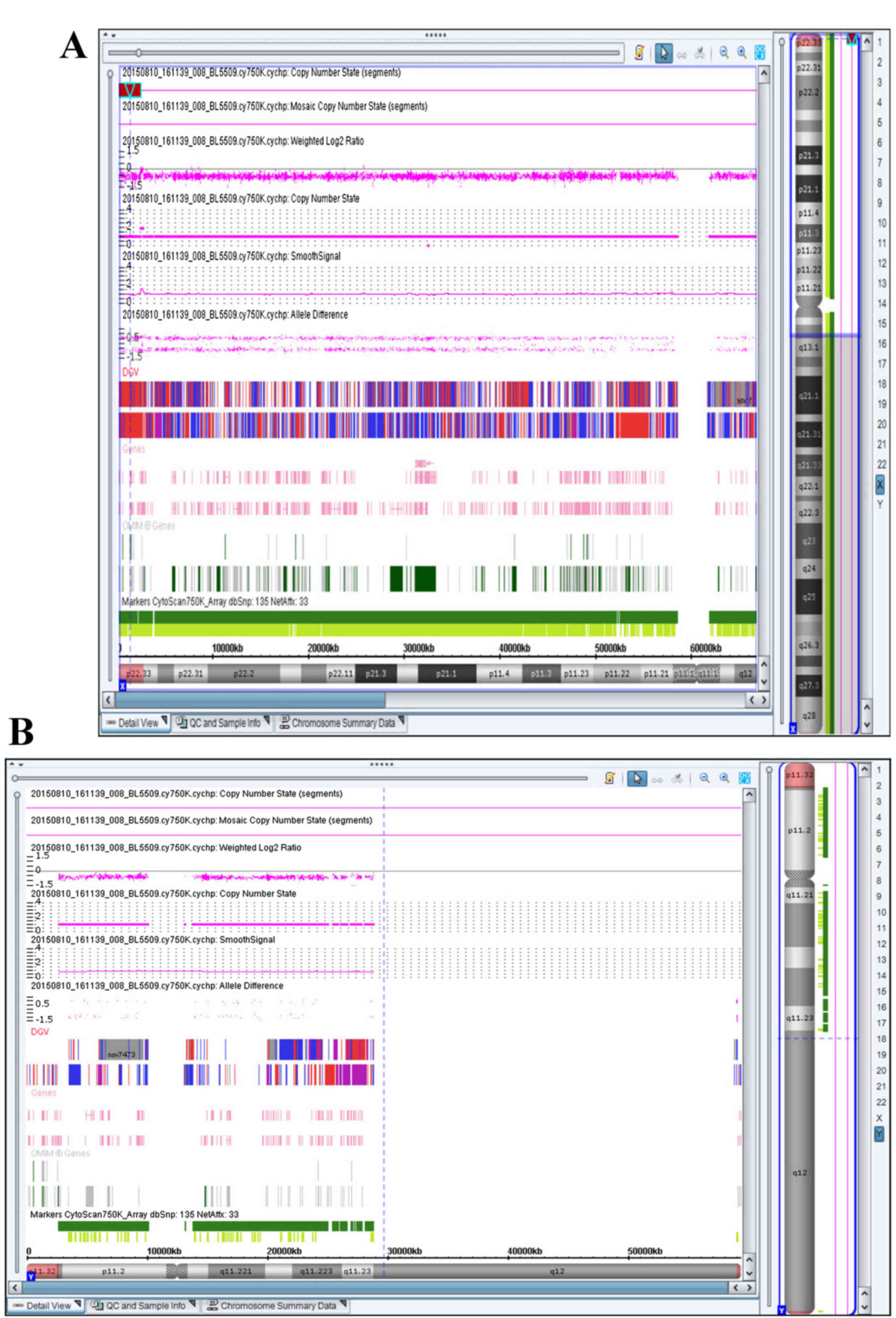
copy number variations (CNVs). SNP-array CGH detected a deletion on
the pseudoautosomal region of the Y chromosome (Yp11.32) that
encompassed ~2.2 Mb (Fig. 4).
Thus, the SNP-array CGH results of the patient were arr[hg19]
Yp11.32(118,551–2,393,500)x0. When the results of chromosome
karyotype analysis, FISH and SNP-array CGH were combined, it was
possible to identify the precise breakage of the abnormal Y
chromosome on Yp11.32. The results were analyzed using the Online
Mendelian Inheritance in Man (OMIM) database (omim.org/), Gene Review (Reviews of genetic
disorders/syndromes and lab testing; www.ncbi.nlm.nih.gov/gtr/#genereviews), the
International Standards for Cytogenomic Arrays (ISCA) and the
Consortium Clinical CNV Database (www.ncbi.nlm.nih.gov/dbvar). Data analysis revealed
that the 2.2 Mb deletion encompassed ~20 OMIM genes, including
SHOX. No clinically significant microdeletions or
microduplications on other chromosomes were observed.
Discussion
The phenotypic spectrum of 45, X/46, XY, dic(Y)
mosaicism is broad and variable. Table
I presents previous studies regarding patients with differing
sex, age, proportions of cell lines and break points. It has been
suggested that the diverse clinical phenotypes are dependent on the
proportions of the different cell lines, and the variable sites of
breakage and fusion on Y chromosomes.
 | Table I.Genotype-phenotype correlations in
reported patients with dic(Y) chromosomes. |
Table I.
Genotype-phenotype correlations in
reported patients with dic(Y) chromosomes.
| Author, year | Peripheral
karyotype | Sex | Age, years | Phenotype | Sex Determining
Region Y, copies | (Refs.) |
|---|
| Cui et al,
2015 | 46,X,idic(Y)
(p11.32) | Male | 32 | Short stature, severe
oligozoospermia | 2 | (13) |
| Yoshida et al,
1997 | 45,X[7]/46,X,psu
dic(q11.2)[33] | Male | 28 | Azoospermia |
| (14) |
| Hes et al,
2009 | 45,X[22]/46,X,psu
dic(Y)(pter→q11.21:: p11.31→p11.2::q11.21→pter)[12] | Male | 50 | Mental retardation
and hypogonadism | 3 | (17) |
| Batstone et
al, 1991 | 45,X/46,X dic
(Y) | Male | 14 | Noonan's
syndrome | / | (18) |
| Fernandez et
al, 2002; Yoshida et al, 1997 |
45,X/46,X,dic(Y)(pter→q11::q11→pter) | Female | 24 | Widely spaced
nipples, Pterygium colli, coarctation of aorta, a small uterus,
rudimentary gonads, deficient intrauterine growth, low weight at
birth, psychomotor and mental delay, lumbar scoliosis,
strabismus | 2 different | (3,14) |
| Gole et al,
2008 |
45,X[10]/46,X,idic(Y)(q11.2)[90] | Female | 2.7 | Clitoromegaly, short
stature | 2 | (19) |
| Smith et al,
1996 |
45,X[70%]/46,X,dic(q11.2)[30%] | Female | 66 | Clitoromegaly,
primary amenorrhea, no breast development, a large right inguinal
hernia | / | (6) |
| Shimoda et al,
1998 |
45,X[13]/46,X,dic(q11.2)[17] | Female | 29 | Ambiguous genitalia
with clitoromegaly | / | (20) |
| Kaprova-Pleskacova
et al, 2013 | 45,X[92.2%]/46,X,psu
dic(Y)(p12)[7.8%] | / | Infant | Congenital ambiguous
genitalia | 2 | (4) |
| Reddy et al,
1996 | 45,X[92%]/46,X,psu
dic(q11.2)[8%] | / | Infant | Mixed gonadal
dysgenesis | / | (5) |
| Bittmann et
al, 2005 | 45,X(13)/46,X,dic (Y) (del(Y) (q?-qter) | Male | Infant | Right-sided inguinal
hernia, ambiguously differentiated gonad | / | (21) |
| The present
study | 45,X[23]/46,X,psu
dic(Y)(p11.3)[24]/46,XY[3] | Male | Infant | Hypospadias, short
stature | 2 | – |
The present study reported a patient with
hypospadias and a mosaicism karyotype of 3 cell lines
45,X[23]/46,X,?idic(Y)(p11.32)[24]/46,XY[3]. The dic(Y) chromosome
of the male infant was a result of meiosis I exchange between
sister chromatids at the pseudoautosomal region, followed by
centromere misdivision at meiosis II during spermatogenesis in the
father. Another cause probably occurred during the first division
following fertilization (8). The
most important reason for chromosomal breakage and reunion is
exposure to hazardous substances including radiation, medication,
chemical and biological factors. Although the parents of the
present infant denied contact with the above factors, the presence
or absence of hazardous material tends to be unpredictable in daily
life.
The SRY gene, located at the tip of the Y short arm
(Yp11.3), is a critical switch that results in testis development
(9). To confirm whether the break
point on the Y chromosome involved SRY and sex differentiation,
FISH was performed on metaphase cells and interphase cell nuclei
using a SRY/DXZ1 probe. The results revealed that there were two
signals on dic(Y), which suggested that the breakage was on Yp11.3.
A DYZ3 probe was additionally used to verify the activity of the
two Y chromosomal centromeres on dic(Y). The rearranged Y
chromosome was of dicentric structure in which only one centromere
was active, meaning that the aberrant Y chromosome was stable in
mitotic cell division.
SHOX, located in the PAR1 on the tip of the
short arms of the X and Y sex chromosomes (Xp22.33 and Yp11.32), is
comprised of ~2.6 Mb (10). To
evaluate the development of the patient and to confirm the
deficiency of SHOX gene on the terminal of the short arm of
the Y chromosome, SNP-array CGH was used for genome-wide sequencing
and to identify CNVs. Only one copy of SHOX was detected on
Xp22.33. However, the copy on Yp11.32 was deletion. This revealed
that the break point on dic(Y) chromosome was Yp11.23, and it had
lost the pseudoautosomal region. The deficiency of SHOX
causes Leri-Weill Syndrome, which is characterized by short stature
and abnormal limbs (11). This
indicates that the short femur length during pregnancy and short
stature following birth in the infant described in the present
study were due to the deletion of the short arm end of the Y
chromosome and the haploinsufficiency of the SHOX gene.
Previous evidence regarding the use of recombinant growth hormone
in patients with SHOX deficiency has indicated the
beneficial effect of this treatment, which improved growth speed
and final height (12).
It is likely that this infant will suffer
infertility upon maturation. Multiple previous reports have
observed that male patients with 45,X/46,X,dic(Y) exhibit
azoospermia or oligozoospermia (2,13–15).
The cause of spermatogenic failure may be explained by the presence
of an abnormal Y chromosome that may not form a sex vesicle, which
appears to be necessary for the completion of the meiosis process
and the formation of sperm, or the presence of the 45,X cell line
(16).
In conclusion, the present study reports the case of
an 8-week-old hypospadiac male infant with 45,X/46,X,psu
idic(Y)(p11.32)/46,XY mosaicism and haploinsufficiency of
SHOX. Doctors and genetic consultants must pay greater
attention to pregnant women with low risk of prenatal screening and
ultrasonic structural abnormalities. The combination of
cytogenetic, FISH and SNP-array CGH technologies was beneficial for
diagnosing the karyotype accurately, predicting the prognosis, and
preparing an effective treatment plan for the patient.
Acknowledgements
The authors thank the patient and his family for
participating in the present study, and all staff of the Be
Creative Lab Co., Ltd. (Beijing, China). The present study was
supported by the Basic-Clinical Scientific Research Cooperation
Fund, Capital Medical University (grant no. 15JL76) and the Beijing
Obstetrics and Gynecology Hospital, Capital Medical University
(grant no. fcyy201534).
References
|
1
|
Hsu LY: Prenatal diagnosis of 45,X/46,XY
mosaicism-a review and update. Prenat Diagn. 9:31–48. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Codina-Pascual M, Oliver-Bonet M, Navarro
J, Starke H, Liehr T, Gutierrez-Mateo C, Sánchez-García JF, Arango
O, Egozcue J and Benet J: FISH characterization of a dicentric Yq
(p11.32) isochromosome in an azoospermic male. Am J Med Genet A.
127A:302–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fernandez R, Marchal JA, Sanchez A and
Pasaro E: A point mutation, R59G, within the HMG-SRY box in a
female 45,X/46,X, psu dic(Y) (pter->q11::q11->pter). Hum
Genet. 111:242–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaprova-Pleskacova J, Snajderova M, Stoop
J, Koudova M, Kocarek E, Novotna D, Drop SL, Obermannova B, Lebl J,
Oosterhuis JW and Looijenga LH: 45,X/46,X,psu dic(Y) gonadal
dysgenesis: Influence of the two cell lines on the clinical
phenotype, including gonadal histology. Sex Dev. 7:282–288. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reddy KS, Sulcova V, Ho CK, Conner ED and
Khurana A: An infant with a mosaic 45,X/46,X,psu dic(Y) (pter->
q11.2::q11.2->pter) karyotype and mixed gonadal dysgenesis
studied for extent of mosaicism in the gonads. Am J Med Genet.
66:441–444. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith YR, Stetten G, Charity L, Isacson C,
Gearhart JP and Namnoum AB: Ambiguous genitalia in an elderly woman
with a mosaic 45,X/46,X,dic(Y)(Q11.2) karyotype. Urology.
47:259–262. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kojima Y, Hayashi Y, Yanai Y, Tozawa K,
Sasaki S and Kohri K: Molecular analysis of hypospadias in a boy
with dicentric Y chromosome. J Urol. 165:1244–1245. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fernandez R and Pasaro E: Molecular
analysis of an idic(Y)(qter ->p11.32::p11.32->qter)
chromosome from a female patient with a complex karyotype. Genet
Mol Res. 5:399–406. 2006.PubMed/NCBI
|
|
9
|
Sinclair AH, Berta P, Palmer MS, Hawkins
JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R
and Goodfellow PN: A gene from the human sex-determining region
encodes a protein with homology to a conserved DNA-binding motif.
Nature. 346:240–244. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Binder G: Short stature due to SHOX
deficiency: Genotype, phenotype, and therapy. Horm Res Paediatr.
75:81–89. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shears DJ, Vassal HJ, Goodman FR, Palmer
RW, Reardon W, Superti-Furga A, Scambler PJ and Winter RM: Mutation
and deletion of the pseudoautosomal gene SHOX cause Leri-Weill
dyschondrosteosis. Nat Genet. 19:70–73. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blum WF, Ross JL, Zimmermann AG, Quigley
CA, Child CJ, Kalifa G, Deal C, Drop SL, Rappold G and Cutler GB
Jr: GH treatment to final height produces similar height gains in
patients with SHOX deficiency and Turner syndrome: Results of a
multicenter trial. J Clin Endocrinol Metab. 98:E1383–E1392. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cui YX, Wang WP, Li TF, Li WW, Wu QY, Li
N, Zhang C, Yao Q, Hu YA and Xia XY: Clinical and cytogenomic
studies in a case of infertility associated with a nonmosaic
dicentric Y chromosome. Andrologia. 47:477–481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshida A, Nakahori Y, Kuroki Y, Motoyama
M, Araki Y, Miura K and Shirai M: Dicentric Y chromosome in an
azoospermic male. Mol Hum Reprod. 3:709–712. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sasagawa I, Ishigooka M, Kato T, Hayami S,
Hashimoto T and Nakada T: Dicentric Y chromosome without evidence
of mosaicism in an azoospermic male. Scand J Urol Nephrol.
30:75–76. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hamerton JL, Canning N, Ray M and Smith S:
A cytogenetic survey of 14,069 newborn infants. I. Incidence of
chromosome abnormalities. Clin Genet. 8:223–243. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hes FJ, Madan K, Rombout-Liem IS, Szuhai
K, Sørensen H, van Amstel HK, Bakker E, Visser TJ, Smit JW and
Hansson K: Multiple genomic aberrations in a patient with mental
retardation and hypogonadism: 45,X/46,X,psu dic(Y) karyotype,
thyroid hormone receptor beta (THRB) mutation and heterozygosity
for Wilson disease. Am J Med Genet A. 149A:2231–2235. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Batstone PJ, Faed MJ, Jung RT and Gosden
J: 45,X/46,X dic (Y) mosaicism in a phenotypic male. Arch Dis
Child. 66:252–253. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gole LA, Lim J, Crolla JA and Loke KY:
Gonadal mosaicism 45,X/46,X,psu dic(Y)(q11.2) resulting in a turner
phenotype with mixed gonadal dysgenesis. Singapore Med J.
49:349–351. 2008.PubMed/NCBI
|
|
20
|
Shimoda N, Sato K, Satoh S, Ogawa O, Ito S
and Kato T: Atypical true hermaphroditism with a mosaic
45,X/46,X,dic(Y)(q11.2) karyotype. J Urol. 160:1434–1435. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bittmann S, Wieczorek D, Stallmach T and
Ulus H: 45,X/46,X,dic(Y)-Mosaicism in the newborn. Klin Padiatr.
217:300–303. 2005. View Article : Google Scholar : PubMed/NCBI
|