Introduction
Heat stroke (HS), which results from exposure to a
high environmental temperature [classic heat stroke (CHS)] or
strenuous exercise [exertional heat stroke (EHS)], is characterized
by a core body temperature (Tc) >40°C and neurological
abnormalities. CHS and EHS can lead to multiple organ dysfunction
syndrome, which is associated with a systemic inflammatory response
and disseminated intravascular coagulation (DIC) (1–6). The
mortality rate in patients with HS is increasing, and ~30% of
survivors experience permanent deficits in neurological and
peripheral tissue function (3,7,8). A
previous study demonstrated that the majority of CHS reactions are
mimicked by exposing anesthetized rats to a high ambient
temperature (Ta) (9). In these
rats, arterial hypotension, hyperpyrexia, a hypercoagulable state,
activated inflammation and tissue injury occurred during HS
(10–12). Although more research has been
undertaken in recent years, specific and effective therapies for HS
are required.
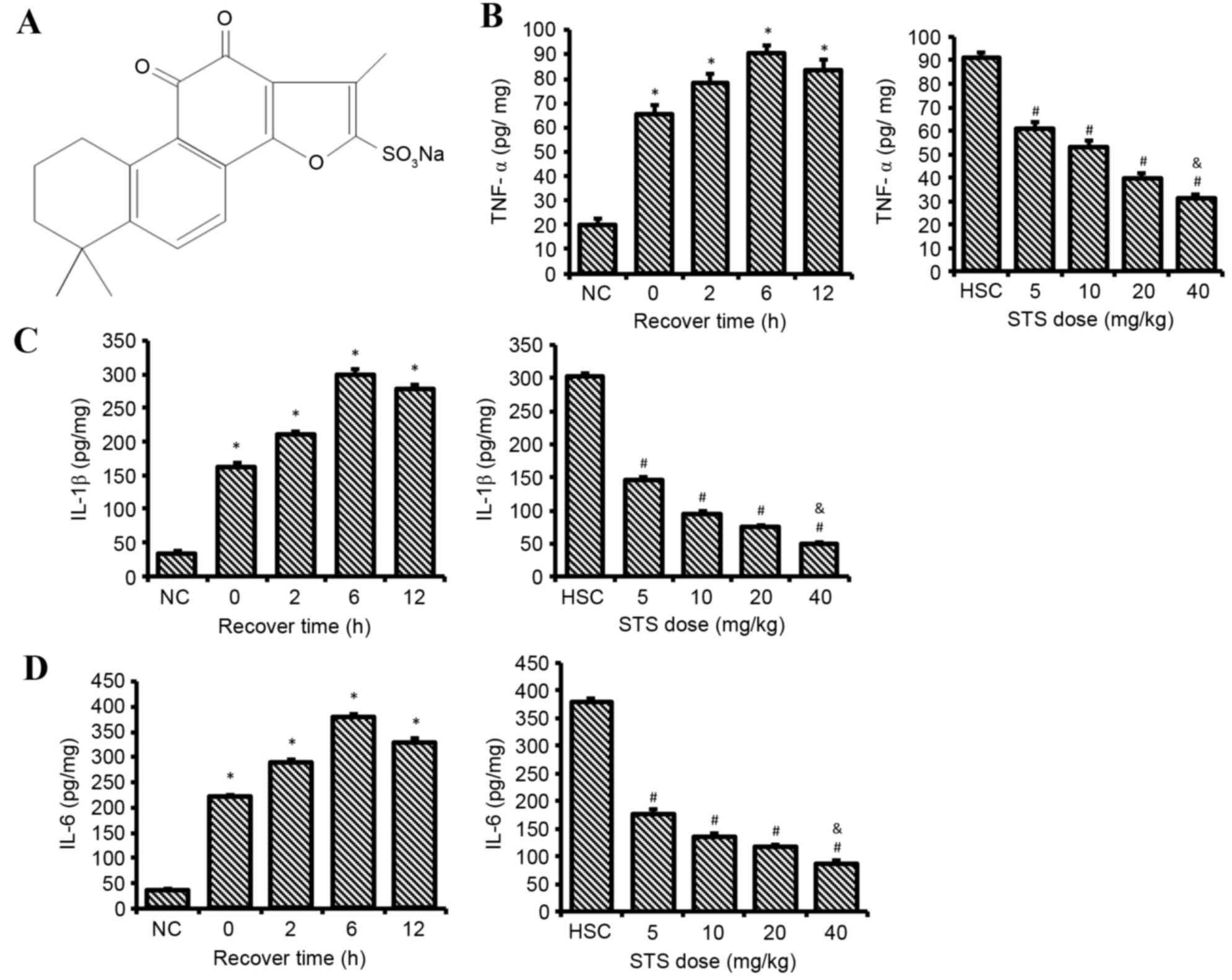
Sodium tanshinone IIA sulfonate (STS; Fig. 1A) is a water-soluble derivative of
tanshinone IIA. The latter is one of the primary lipophilic
components extracted from the dry root or rhizome of Salvia
miltiorrhiza Bge, termed ‘Danshen’ in traditional Chinese
medicine (13). Tanshinone IIA has
poor water solubility; therefore, STS was developed to increase the
bioavailability and has been successfully used to treat patients
with cardiovascular disorders. STS has been reported to have a wide
range of pharmacological activities, including anti-inflammatory
properties (14), antioxidant
capacity (15) and the ability to
inhibit apoptosis (16,17). Furthermore, STS has been
demonstrated to suppress cardiomyocyte hypertrophy (18) and protect human vascular
endothelial cells in vivo (19). Therefore, HS-induced inflammatory
and endothelial cell injuries may be reduced by STS treatment.
Proinflammatory cytokines and endothelial cell injury have been
demonstrated to initiate coagulation disorders and ultimately lead
to DIC in HS (1,20). HS-induced DIC and multiple organ
damage may also be reduced by STS treatment.
To evaluate this hypothesis, the present study
assessed the therapeutic effects of STS treatment on inflammation,
aortic endothelial cell apoptosis, DIC and multiple organ damage in
a HS rat model.
Materials and methods
Animals
A total of 72 adult male Sprague-Dawley rats aged
42–47 days and weighing 200–260 g were obtained from the Animal
Resource Center of Hubei (Wuhan, China). The animals were housed
individually at a Ta of 25±1°C, a relative humidity of 50±10% and a
12-h light/dark cycle for 1 week prior to the start of the
experiments. Pelleted rat feed and tap water were provided ad
libitum. The experimental protocol was approved by the Animal
Resource Center of Hubei. Animal care and experiments were
conducted according to the National Institutes of Health Guidelines
for the Use of Laboratory Animals (21). The rats were anesthetized by
intraperitoneal injections of 50 mg/kg pentobarbital sodium
(Shanghai Haling Biotechnology Co., Ltd., Shanghai, China). The
anesthetic administration was completed when the corneal reflex and
pain reflexes induced by tail pinch were abolished in the rats.
Adequate anesthesia was maintained throughout the course of all
experiments.
Induction of HS and experimental
design
HS was induced by placing the rats in a warm blanket
in an animal temperature controller (ATC; SS-20-2, Huaibei Zhenghua
Biological Instrument Equipment Co., Ltd., Huaibei, China) preset
to 35°C. To avoid burns, a towel was placed between the animals and
the warm blanket.
The rats were randomly divided into the following
groups. In the normothermic control (NC) group, 8 rats were placed
into a temperature-controlled room (26°C) following the
administration of anesthesia throughout the entire experiment. In
the heat stroke (HS) groups, the Tc (represented by the rectal
temperature) of 32 rats were monitored every 5 min following the
administration of anesthesia until HS onset. When the Tc reached
43.5°C, which was considered the time of onset of HS, the rats were
removed from the ATC and allowed to recover at 26°C for 0, 2, 6 or
12 h (HS-0, HS-2, HS-6 and HS-12 groups, respectively). In the
STS-treated heat stroke (STS-HS) groups, 32 rats were exposed to
the same heat treatment described above. Immediately following the
onset of HS, the rats received intravenous injections of 5, 10, 20
or 40 mg/kg body weight STS (Shanghai No. 1
Biochemical-Pharmaceutical Co., Ltd, Shanghai, China; groups STS-5,
STS-10, STS-20 and STS-40, respectively) via the femoral vein and
were placed at a temperature of 26°C to recover for 6 h. After
recovering for the set time, each group of animals was subjected to
the following measurements: Serum levels of interleukin-1β (IL-1β),
tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6);
apoptotic endothelial cells in the aorta; plasma prothrombin time
(PT), activated partial thromboplastin time (aPTT), platelet count
and D-dimer; serum levels of blood urea nitrogen (BUN), creatinine
(Cr), alanine aminotransferase (ALT), aspartate aminotransferase
(AST), alkaline phosphatase (ALP) and lactate dehydrogenase (LDH);
and histological changes in the major organs.
Cytokine measurements
The serum levels of TNF-α, IL-6, and IL-1β were
measured using commercially available ELISA kits (catalog no. RTA00
for TNF-α; HS600B for IL-6; RLB00 for IL-1β; R&D Systems, Inc.,
Minneapolis, MN, USA) according to the manufacturer's protocol.
Biochemical analysis
Whole blood was obtained from the abdominal aorta.
The serum levels of Cr, BUN, ALT, AST, ALP, and LDH were determined
with an automatic biochemical analyzer (DXC-800; Beckman Coulter,
Inc., Brea, CA, USA). The plasma PT and aPTT, and D-dimer
concentration were measured using an automated coagulation
instrument (ACL-TOP; Beckman Coulter, Inc.). The platelet count was
determined using an automated blood cell counting instrument
(XT-4000i; Sysmex Corporation, Kobe, Japan).
Histological examination
Tissues specimens from livers, kidneys, adrenal
glands, small intestines, spleens, and lungs were fixed in 4%
paraformaldehyde, embedded in paraffin, sectioned at 4-µm
thickness, stained with hematoxylin and eosin, and subsequently
imaged under a light microscope with digital camera system (BX51;
Olympus Corporation, Tokyo, Japan).
Combined terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL)/CD31
immunofluorescence assay
For the detection and quantification of apoptotic
endothelial cells in aortic sections, TUNEL technology (In
Situ Cell Death Detection kit; Roche Applied Science, Madison,
WI, USA) was used in combination with immunofluorescence for CD31.
The paraffin-embedded sections were stained with a primary rabbit
polyclonal anti-CD31 antibody (1:50 dilution; catalog no. ab28364;
Abcam, Cambridge, UK) followed by incubation with a Texas
Red-conjugated goat anti-rabbit IgG secondary antibody (1:100
dilution; catalog no. BA1032; Wuhan Boster Biological Technology,
Ltd., Wuhan, China) to visualize the endothelial cell layer.
Briefly, sections were blocked with normal goat serum (Wuhan Boster
Biological Technology, Ltd.) for 30 min at room temperature prior
to immunofluorescence staining. The sections were incubated with
anti-CD31 antibody overnight at 4°C. Subsequently, after washing
with PBS containing 0.05% Tween-20, the secondary antibody was
applied to the sections for 1 h at 20–37°C. The aortic segments
embedded in paraffin were then used to detect TUNEL-positive
apoptotic cells, according to the manufacturer's protocol. The
nuclei were counterstained with 4,6-diamidino-2-phenylindole
(DAPI). The percentage of apoptotic endothelial cells was
calculated as the proportion of TUNEL/DAPI/CD31-positive cells out
of total DAPI/CD31-positive cells.
Statistical analysis
Data are expressed as mean ± standard deviation and
were analyzed by one-way analysis of variance followed by least
significant difference post hoc test (equal variances) or Dunnett's
T3 post hoc test (unequal variances). All analyses were performed
using SPSS software version 19.0 (IBM SPSS, Armonk, NY, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
STS reduces the levels of TNF-α, IL-1β
and IL-6 in HS
Clear differences were observed in the serum levels
of TNF-α (Fig. 1B), IL-1β
(Fig. 1C) and IL-6 (Fig. 1D) between the NC, HS and STS-HS
groups. The serum levels of TNF-α, IL-1β and IL-6 in all the HS-0,
HS-2, HS-6 and HS-12 groups were significantly increased compared
with the NC group. The serum levels of TNF-α, IL-1β and IL-6 were
greatest in the HS-6 group compared with the other HS groups.
Compared to rats that received no STS, treatment with 5–40 mg/kg
STS significantly attenuated the increased serum levels of IL-1β,
TNF-α and IL-6 after HS following recovery for 6 h (P<0.05).
Furthermore, the serum levels of IL-1β, TNF-α and IL-6 were
maintained at an extremely low level in the rats treated with STS
at 40 mg/kg compared with the 5–20 mg/kg groups (P<0.05).
STS reduces the number of apoptotic
aortic endothelial cells in HS
Significant differences were observed in the number
of apoptotic aortic endothelial cells between the NC, HS and STS-HS
groups. The number of apoptotic cells in the HS-0, HS-2, HS-6 and
HS-12 groups were significantly increased compared with the NC
group (P<0.05). Numbers in the HS-6 group were increased
compared with the other HS groups. Treatment with 5–40 mg/kg STS
significantly attenua ted the increased number of apoptotic aortic
endothelial cells following HS after recovery for 6 h (P<0.05;
Fig. 2A). Furthermore, the number
of apoptotic cells was maintained at an extremely low level in the
rats treated with 40 mg/kg STS, which was significantly lower than
in the other STS-treated groups (P<0.05; Fig. 2B).
STS attenuates DIC in HS
Significant differences were observed in the plasma
PT (Fig. 3A), aPTT (Fig. 3B), platelet counts (Fig. 3C) and D-dimer levels (Fig. 3D) in the NC, HS-6 and STS-6-40
group (40 mg/kg STS-treated heat stroke group with recovery for 6
h). The plasma PT, aPTT, and D-dimer values in the HS-6 group were
significantly increased compared with the NC group. By contrast,
the platelet count was significantly reduced in the HS-6 group
compared with the NC group. In addition, 40 mg/kg STS treatment of
the HS-6 group attenuated the HS-induced increased plasma levels of
PT, aPTT, and D-dimer and the decreased platelet count
(P<0.01).
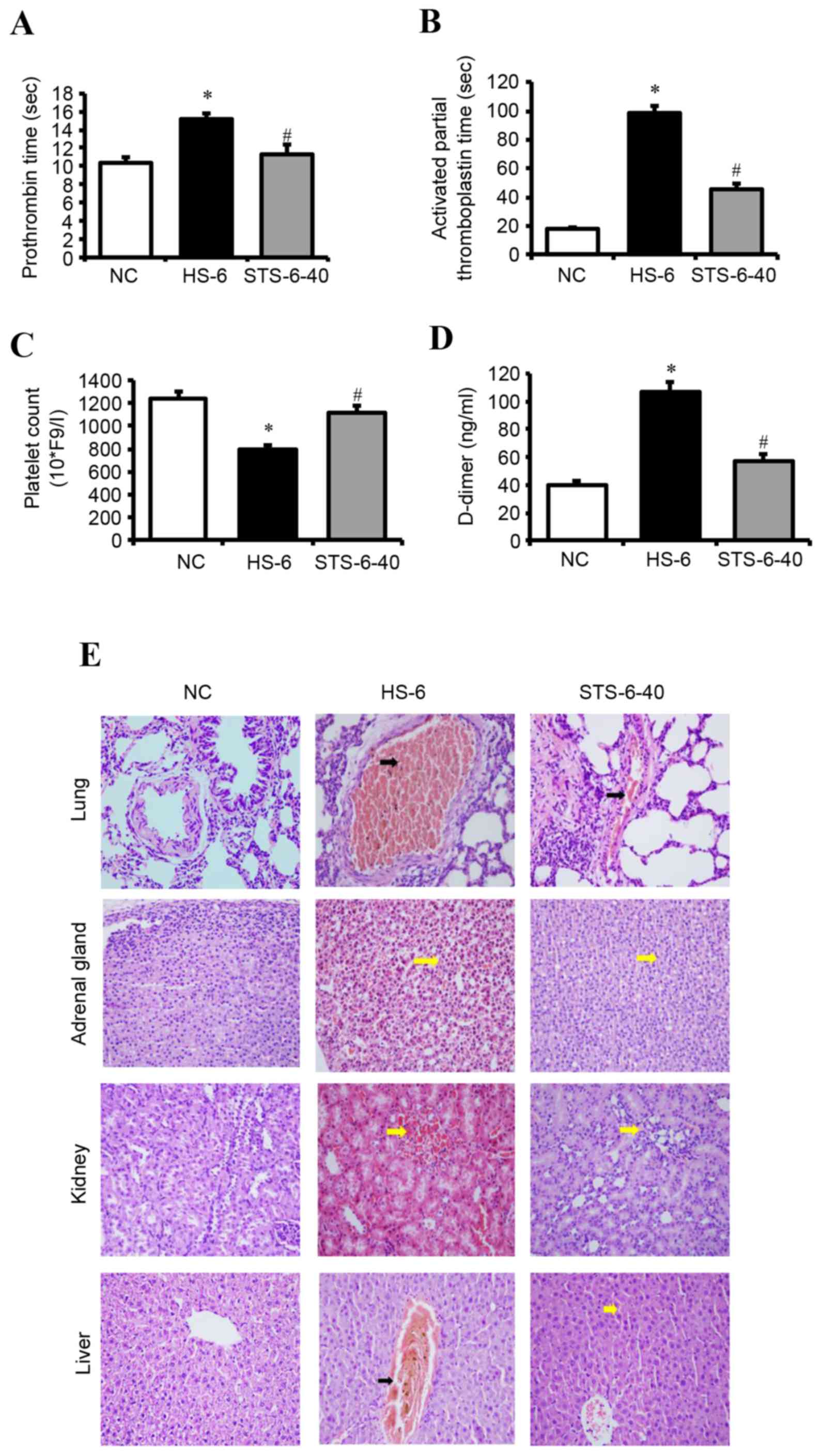 | Figure 3.STS negatively regulates HS-induced
DIC. Rats were exposed to HS with or without 40 mg/kg STS, and
recovered for 6 h. (A) Prothrombin time, (B) activated partial
thromboplastin time, (C) platelet counts and (C) levels of D-dimer
in plasma were determined. Data are presented as the mean ±
standard deviation. *P<0.05 vs. NC; #P<0.05 vs.
HSC. (E) Representative micrographs of hematoxylin and eosin
staining of the lungs, adrenal glands, kidneys and livers.
Interstitial space hemorrhage (yellow arrows) in the adrenal gland,
kidney and liver. Intravascular thrombus (black arrows) in the
central vein of the liver and pulmonary arteriole. Magnification,
×400. HS, heat shock; STS, sodium tanshinone IIA sulfonate; NC,
normothermic control. |
The histopathological findings revealed DIC in the
vital organs of the HS-6 group. Intravascular thrombus formation
was observed in small- and medium-sized blood vessels, and
interstitial space hemorrhage was observed in the lungs, kidneys,
adrenal glands and livers. However, hemorrhage and thrombosis were
significantly alleviated in the STS-6-40 group compared with the
HS-6 group (Fig. 3E).
STS attenuates multiple organ damage
in HS
Significant differences were observed in serum
levels of ALT (Fig. 4A), AST
(Fig. 4B), ALP (Fig. 4C), BUN (Fig. 4D), Cr (Fig. 4E), and LDH (Fig. 4F) in the NC, HS-6 and STS-6-40
groups. The serum levels of ALT, AST, ALP, BUN, Cr and LDH in the
HS-6 group were significantly increased compared with the NC group.
However, 40 mg/kg STS treatment significantly attenuated the
HS-induced increased serum levels of these components
(P<0.01).
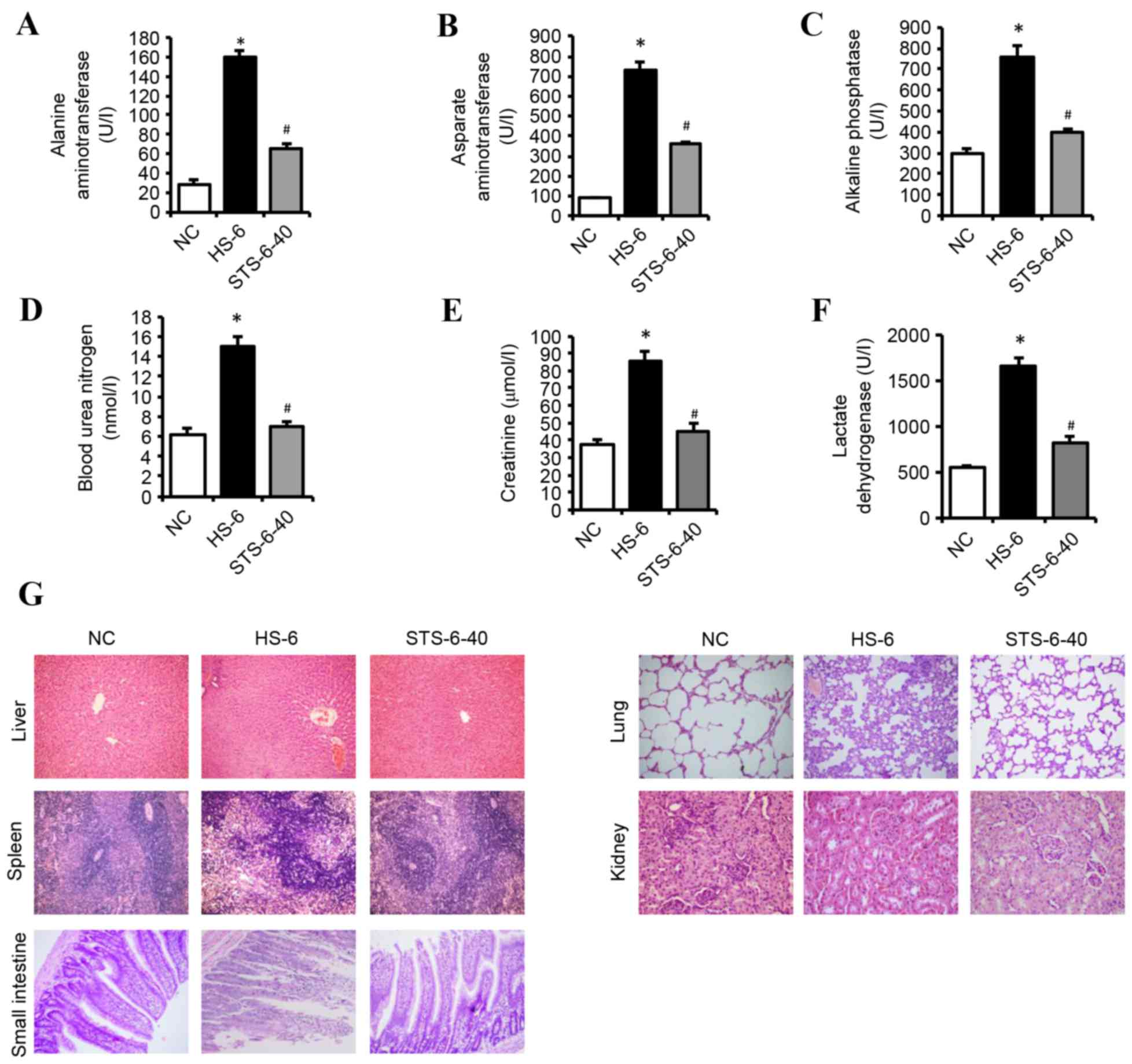 | Figure 4.STS prevents heat stroke-induced
multiple organ damage. Rats were exposed to HS with or without 40
mg/kg STS, and recovered for 6 h. Serum levels of (A) alanine
aminotransferase, (B) aspartate aminotransferase, (C) alkaline
phosphatase, (D) blood urea nitrogen, (E) creatine and (F) lactate
dehydrogenase were determined. Data are presented as the mean ±
standard deviation. *P<0.05 vs. NC; #P<0.05 vs.
HSC. (G) Representative micrographs of hematoxylin and eosin
staining of the livers, spleens, lungs, kidneys, and intestines.
Magnification, ×200 (liver, spleen, lung, small intestine);
magnification, ×400 (kidney). NC, normothermic control; HS, heat
shock; STS, sodium tanshinone IIA sulfonate. |
The histopathological findings revealed unremarkable
damage to the liver, spleen, lung, kidney, and small intestines in
the NC group. The damage observed in the major organs of the HS-6
group was extensive; however, a marked decrease in this damage was
observed in the STS-6-40 group. The injury to the liver was
multifocal in the HS-6 group, including hepatocellular architecture
disruption, hepatic cell degenerative changes, hepatic sinusoid
congestion and/or hemorrhage, thrombi, and increased inflammatory
cells. The architecture of spleen tissues had become disordered,
and the boundary between the red and white pulp was unclear in the
HS-6 group. Furthermore, the cellularities of the periarterial
lymphatic sheath and the marginal zone were indistinct, although
there was extravagated blood in the marginal zone. The lung tissues
in the HS-6 group exhibited substantial morphological alterations,
including pulmonary edema, alveolar collapse, inflammatory cell
infiltration and extensively thickened pulmonary alveolar septa.
Furthermore, vascular congestion, hemorrhage, and thrombosis were
observed. The kidney injury manifested as tubular epithelial cell
edema, hemorrhage, thrombosis, and inflammatory cell infiltration
in the HS-6 group. In the small intestines, the bowel walls were
swollen and thickened, and the villous architecture exhibited
disorder, degeneration, capillary exposure, congestion, and
inflammatory cell infiltration. The pathologic impairments of the
liver, spleen, lung, kidney and small intestines were significantly
alleviated in the STS-6-40 group (Fig.
4G).
Discussion
The present study demonstrated that HS induced
increased inflammatory mediators, aortic endothelial cell
apoptosis, DIC and multiple organ damage in an experimental rat
model. STS treatment following HS reduced inflammation, aortic
endothelial cell apoptosis, DIC and multiple organ damage.
Previous studies have demonstrated that endothelial
cell injury is one of the most important features in the
pathophysiology of HS (1,22,23).
The direct cytotoxic effects of heat and increased inflammatory
mediators in HS may induce endothelial cell injury.
Activated/injured endothelial cells may result in the release of
cytokines, thereby further amplifying the inflammatory response
(3,6,24,25).
Previous studies demonstrated that the levels of inflammatory
factors vary with the recovery time following heat stress. The
levels of inflammatory factors are additionally associated with the
severity of HS (1,26,27).
In these preliminary results, the levels of TNF-α, IL-1β and IL-6
varied with the recovery time following heat exposure, and were
highest in the HS-6 group. Previous studies have suggested that
intense heat stress may induce endothelial cell apoptosis, and that
endothelial cells exhibit significant apoptosis during the
acute-phase response to heat stress (28,29).
A previous study by Gu et al (29) demonstrated that in vitro,
the rate of apoptosis in human umbilical vein endothelial cells is
associated with time after heat treatment, and apoptosis increased
most markedly when human umbilical vein endothelial cells were
treated at 43°C for 2 h, followed by replacement with fresh media
and further incubation for 6 h. In the present study, the numbers
of apoptotic aortic endothelial cells were highest in the HS-6
group. These results suggested that this time period is likely to
represent an important phase in the pathophysiology of HS in which
inflammatory mediators, endothelial cell apoptosis and multiple
organ damage reach peak levels. Furthermore, in this study, the
highest levels of inflammatory mediators and numbers of apoptotic
aortic endothelial cells were present at consistent time points;
although this may be a coincidence, it demonstrated that
inflammatory responses are associated with endothelial cell injury
in the pathophysiology of HS (1).
Therefore, the HS-6 group was selected as a heat stroke control
compared with STS treatment in this study. STS has been reported to
have endothelial cell protective abilities and reduce the release
of inflammatory factors (30–32).
In the present study, STS treatment was demonstrated to
dose-dependently decrease the levels of inflammatory mediators and
endothelial cell apoptosis.
There is a close association between coagulant
activity and the status of endothelial cells (33). Apoptotic endothelial cells exhibit
disordered coagulation because of the loss of anticoagulant
membrane components, which subsequently leads to procoagulant
activation. In addition, apoptotic endothelial cells exhibit
decreased expression of coagulation inhibitors, such as
thrombomodulin, heparin sulfates and tissue factor pathway
inhibitor (25,34). Systemic inflammation may
additionally contribute to activation of the coagulation system and
inhibition of anticoagulant mechanisms and fibrinolysis (35). Imbalance between the procoagulant
and anticoagulant systems causes DIC. Previous studies have
demonstrated that DIC during HS primarily manifests as prolongation
of aPTT and PT, increased D-dimer levels, low platelet counts, and
widespread hemorrhage and thrombosis in histological examinations
(22,36–38).
In the present study, aPTT and PT were prolonged, D-dimer was
raised, and platelet counts were decreased in the HS-6 group. In
addition, histological examinations revealed intravascular thrombus
formation and interstitial space hemorrhage in the vital organs in
the HS-6 group. As previously mentioned, STS may reduce the
inflammatory response and inhibit endothelial cell apoptosis in HS
rats, factors that may lead to DIC. Therefore, STS may attenuate
DIC in HS rats. According to these results, STS is considered
beneficial in DIC in HS rats.
A complex interplay among the direct cytotoxic
effects of heat and the inflammatory and coagulation responses
results in multi-organ dysfunction syndrome in HS (1,3,6). Lin
et al (39) demonstrated
that serum organ injury indicators including Cr, BUN, ALT, AST, ALP
and LDH are increased in HS rats. Roberts et al (22) demonstrated that pathological damage
to tissues led to vascular congestion, hemorrhage, thrombosis,
increased inflammatory cells and disruption of architecture in the
vital organs. In the present study, the results of organ damage in
the HS-6 group were consistent with previous studies. Multiple
organ damage was greatly diminished in the STS-6-40 group. To date,
STS has been applied to investigate the effects of HS on DIC and
multiple organ damage in rats. STS may attenuate multiple organ
damage in CHS by reducing the inflammatory response, aortic
endothelial cell apoptosis and DIC. However, there are other
potential underlying mechanisms of action in STS treatment of
multiple organ damage in CHS, and further studies are required.
In conclusion, the present study demonstrated that
STS treatment improves the development of inflammatory processes,
aortic endothelial cell apoptosis, DIC and multiple organ damage
following CHS. These results suggested that STS treatment may be
beneficial for patients with HS.
References
|
1
|
Bouchama A and Knochel JP: Heat stroke. N
Engl J Med. 346:1978–1988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sucholeiki R: Heatstroke. Semin Neurol.
25:307–314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leon LR and Helwig BG: Heat stroke: Role
of the systemic inflammatory response. J Appl Physiol (1985).
109:1980–1988. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
al-Mashhadani SA, Gader AG, al Harthi SS,
Kangav D, Shaheen FA and Bogus F: The coagulopathy of heat stroke:
Alterations in coagulation and fibrinolysis in heat stroke patients
during the pilgrimage (Haj) to Makkah. Blood Coagul Fibrinolysis.
5:731–736. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bouchama A, Bridey F, Hcimmami MM, Lacombe
C, al-Shail E, al-Ohali Y, Combe F, al-Sedairy S and de Prost D:
Activation of coagulation and fibrinolysis in heatstroke. Thromb
Haemost. 76:909–915. 1996.PubMed/NCBI
|
|
6
|
Leon LR: Heat stroke and cytokines. Prog
Brain Res. 162:481–524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levine M, LoVecchio F, Ruha AM, Chu G and
Roque P: Influence of drug use on morbidity and mortality in
heatstroke. J Med Toxicol. 8:252–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bouchama A: Heatstroke: A new look at an
ancient disease. Intensive Care Med. 21:623–625. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang CK, Chang CP, Chiu WT and Lin MT:
Prevention and repair of circulatory shock and cerebral
ischemia/injury by various agents in experimental heatstroke. Curr
Med Chem. 13:3145–3154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen YW, Chen SH, Chou W, Lo YM, Hung CH
and Lin MT: Exercise pretraining protects against cerebral
ischaemia induced by heat stroke in rats. Br J Sports Med.
41:597–602. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu CC, Shih MF, Wen YS, Lai YH and Yang
TH: Dexamethasone improves heat stroke-induced multiorgan
dysfunction and damage in rats. Int J Mol Sci. 15:21299–21313.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Niu KC, Lin MT and Chang CP: Hyperbaric
oxygen improves survival in heatstroke rats by reducing multiorgan
dysfunction and brain oxidative stress. Eur J Pharmacol.
569:94–102. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shang Q, Xu H and Huang L: Tanshinone IIA:
A promising natural cardioprotective agent. Evid Based Complement
Alternat Med. 2012:7164592012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li W, Li J, Ashok M, Wu R, Chen D, Yang L,
Yang H, Tracey KJ, Wang P, Sama AE and Wang H: A cardiovascular
drug rescues mice from lethal sepsis by selectively attenuating a
late-acting proinflammatory mediator, high mobility group box 1. J
Immunol. 178:3856–3864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou GY, Zhao BL, Hou JW, Ma GE and Xin
WJ: Protective effects of sodium tanshinone IIA sulphonate against
adriamycin-induced lipid peroxidation in mice hearts in vivo and in
vitro. Pharmacol Res. 40:487–491. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang MQ, Zheng YL, Chen H, Tu JF, Shen Y,
Guo JP, Yang XH, Yuan SR, Chen LZ, Chai JJ, et al: Sodium
tanshinone IIA sulfonate protects rat myocardium against
ischemia-reperfusion injury via activation of PI3K/Akt/FOXO3A/Bim
pathway. Acta Pharmacol Sin. 34:1386–1396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang R, Liu A, Ma X, Li L, Su D and Liu J:
Sodium tanshinone IIA sulfonate protects cardiomyocytes against
oxidative stress-mediated apoptosis through inhibiting JNK
activation. J Cardiovasc Pharmacol. 51:396–401. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang L, Zou X, Liang Q, Chen H, Feng J,
Yan L, Wang Z, Zhou D, Li S, Yao S and Zheng Z: Sodium tanshinone
IIA sulfonate depresses angiotensin II-induced cardiomyocyte
hypertrophy through MEK/ERK pathway. Exp Mol Med. 39:65–73. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu TW, Zeng H, Fung KP, Wu J, Pang H, Grey
AA, Weisel RD and Wang JY: Effect of sodium tanshinone IIA
sulfonate in the rabbit myocardium and on human cardiomyocytes and
vascular endothelial cells. Biochem Pharmacol. 46:2327–2332. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bombeli T, Karsan A, Tait JF and Harlan
JM: Apoptotic vascular endothelial cells become procoagulant.
Blood. 89:2429–2442. 1997.PubMed/NCBI
|
|
21
|
Clark JD, Baldwin RL, Bayne KA, Manyland
R, Brown MJ, Gebhart GF, Gonder JC, Gwathmey JK, Keeling ME, Kohn
DF, et al: Guide for the Care and Use of Laboratory Animals. The
national academies press; 1996
|
|
22
|
Roberts GT, Ghebeh H, Chishti MA,
Al-Mohanna F, El-Sayed R, Al-Mohanna F and Bouchama A:
Microvascular injury, thrombosis, inflammation, and apoptosis in
the pathogenesis of heatstroke: A study in baboon model.
Arterioscler Thromb Vasc Biol. 28:1130–1136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bouchama A, Hammami MM, Haq A, Jackson J
and al-Sedairy S: Evidence for endothelial cell activation/injury
in heatstroke. Crit Care Med. 24:1173–1178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tong H, Wan P, Zhang X, Duan P, Tang Y,
Chen Y, Tang L and Su L: Vascular endothelial cell injury partly
induced by mesenteric lymph in heat stroke. Inflammation. 37:27–34.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lovren F and Verma S: Evolving role of
microparticles in the pathophysiology of endothelial dysfunction.
Clin Chem. 59:1166–1174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamakawa K, Matsumoto N, Imamura Y, Muroya
T, Yamada T, Nakagawa J, Shimazaki J, Ogura H, Kuwagata Y and
Shimazu T: Electrical vagus nerve stimulation attenuates systemic
inflammation and improves survival in a rat heatstroke model. PLoS
One. 8:e567282013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leon LR, Blaha MD and DuBose DA: Time
course of cytokine, corticosterone, and tissue injury responses in
mice during heat strain recovery. J Appl Physiol (1985).
100:1400–1409. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brinton MR, Tagge CA, Stewart RJ, Cheung
AK, Shiu YT and Christensen DA: Thermal sensitivity of endothelial
cells on synthetic vascular graft material. Int J Hyperthermia.
28:163–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu ZT, Wang H, Li L, Liu YS, Deng XB, Huo
SF, Yuan FF, Liu ZF, Tong HS and Su L: Heat stress induces
apoptosis through transcription-independent p53-mediated
mitochondrial pathways in human umbilical vein endothelial cell.
Sci Rep. 4:44692014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou ZW, Xie XL, Zhou SF and Li CG:
Mechanism of reversal of high glucose-induced endothelial nitric
oxide synthase uncoupling by tanshinone IIA in human endothelial
cell line EA.hy926. Eur J Pharmacol. 697:97–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jang SI, Jeong SI, Kim KJ, Kim HJ, Yu HH,
Park R, Kim HM and You YO: Tanshinone IIA from salvia miltiorrhiza
inhibits inducible nitric oxide synthase expression and production
of TNF-alpha, IL-1beta and IL-6 in activated RAW 264.7 cells.
Planta Med. 69:1057–1059. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jang SI, Kim HJ, Kim YJ, Jeong SI and You
YO: Tanshinone IIA inhibits LPS-induced NF-kappaB activation in RAW
264.7 cells: Possible involvement of the NIK-IKK, ERK1/2, p38 and
JNK pathways. Eur J Pharmacol. 542:1–7. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vallet B and Wiel E: Endothelial cell
dysfunction and coagulation. Crit Care Med. 29:(Suppl 7). S36–S41.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bouvy C, Gheldof D, Chatelain C, Mullier F
and Dogné JM: Contributing role of extracellular vesicles on
vascular endothelium haemostatic balance in cancer. J Extracell
Vesicles. 3:Jul 11–2014.doi: 10.3402/jev.v3.24400. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schouten M, Wiersinga WJ, Levi M and van
der Poll T: Inflammation, endothelium, and coagulation in sepsis. J
Leukoc Biol. 83:536–545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen CM, Hou CC, Cheng KC, Tian RL, Chang
CP and Lin MT: Activated protein C therapy in a rat heat stroke
model. Crit Care Med. 34:1960–1966. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ockelford PA and Carter CJ: Disseminated
intravascular coagulation: The application and utility of
diagnostic tests. Semin Thromb Hemost. 8:198–216. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sohal RS, Sun SC, Colcolough HL and Burch
GE: Heat stroke. An electron microscopic study of endothelial cell
damage and disseminated intravascular coagulation. Arch Intern Med.
122:43–47. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin XJ, Mei GP, Liu J, Li YL, Zuo D, Liu
SJ, Zhao TB and Lin MT: Therapeutic effects of melatonin on
heatstroke-induced multiple organ dysfunction syndrome in rats. J
Pineal Res. 50:436–444. 2011. View Article : Google Scholar : PubMed/NCBI
|