Introduction
Cardiac conduction disease is the major risk factor
for sudden cardiac death (SCD), and therefore is the primary cause
of mortality from SCD worldwide (1,2). The
principal characteristics of the disease are sudden arrhythmia and
syncope (3). The human
voltage-gated cardiac sodium channel serves a role in cardiac
conduction, and mutations in sodium channel genes have been
demonstrated to be associated with cardiac conduction disease
(4), including sodium
voltage-gated channel-α subunit 5 (SCN5A) (5), sodium voltage-gated channel-β subunit
4 (6) and sodium voltage-gated
channel-β subunit 1 (7).
The gene SCN5A encodes the pore-forming subunit of
the human cardiac sodium channel NaV1.5 (8) and mutations in this gene have been
reported to be associated with a variety of arrhythmogenic
disorders, including type 3 long-QT syndrome (9), atrial fibrillation (10), dilated cardiomyopathy (11), Brugada syndrome (8) and certain complex overlapping
disorders (12).
The present study investigated a
clinically-characterized family with a history of syncope and
arrhythmia. A clear autosomal-dominant inheritance of arrhythmia
had been identified in the family. Using whole-exome sequencing
(WES), in combination with arrhythmia-associated gene filtering, a
novel missense mutation (c.1099C>G/p.R367G) was identified in
SCN5A, which may underlie the pathogenesis of this type of familial
arrhythmia.
Materials and methods
Patients and subjects
The protocol of the present study was approved by
the Review Board of the Second Xiangya Hospital of Central South
University (Changsha, China) and the study participants gave
informed consent. A total of 14 members of the family (5 affected,
7 unaffected and 2 unknown) were enrolled in the present study.
Blood was obtained from the affected probands and family members.
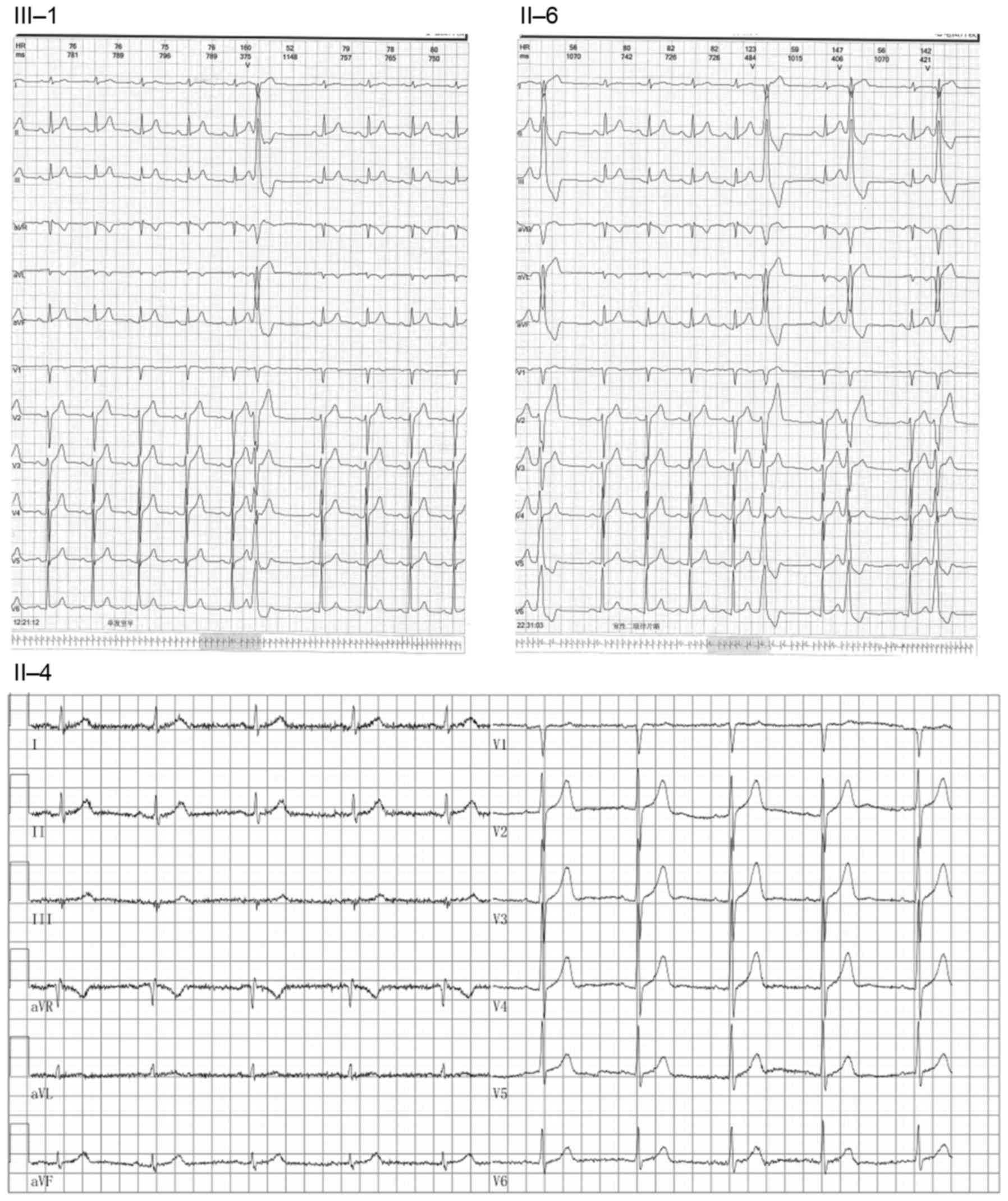
Subjects underwent medical tests, including 12-lead
electrocardiogram (ECG; Fig. 1)
and ultrasonic cardiogram (UCG), and their hospital records were
analyzed. A further 200 healthy people were also enrolled to
exclude the single nucleotide polymorphism (13).
Whole-exome sequencing
Genomic DNA was extracted using the DNeasy Blood
& Tissue kit (Qiagen, Inc., Valencia, CA, USA). The Novogene
Bioinformatics Institute (Beijing, China) performed the exome
capture, high-throughput sequencing and common filtering. All of
the exomes were captured using SureSelect Human All ExonV5 kits
(Agilent Technologies, Inc., Santa Clara, CA, USA) and were
sequenced using the HiSeq2000 platform (Illumina, Inc., San Diego,
CA, USA). Filtering strategies were as described in a previous
study (13). The effect of
variants were predicted by polyphen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml),
SIFT (http://sift.jcvi.org/) and
MutationTaster (http://www.mutationtaster.org/).
Co-segregation analysis
Segregation analysis was applied in all family
members according to the results of the WES. Primer pairs were
designed using DNASTAR version 6.0 (Madison, WI, USA) and the
sequences of primers are forward: CTA CAC CAG CTT CGA TTC CTT;
reverse: TGA TCC CTT CTC CCT CAG AA.
Results
Clinical features
A Chinese family with a history of arrhythmia and
syncope was identified (Fig. 2A).
The proband, a 21-year-old college student from the Hunan province
of central-southern China, had experienced syncope during strenuous
physical exercise 6 months previously. A family history analysis
demonstrated that the maternal uncle of the proband had experienced
syncope 8 years previously and that the maternal grandmother of the
proband had succumbed to an unknown cause during sleep. ECG and UCG
analysis demonstrated that the maternal uncle of the proband
exhibited sinus bradycardia. The proband and the mother of the
proband exhibited ventricular premature beats (Fig. 1). The ECG of the sister of the
proband was not obtained.
 | Figure 2.SCNA5 sequencing and alignment. (A)
Pedigree of the family affected by arrhythmia. Family members are
identified by generations and numbers. Squares, male family
members; circles, female members; closed symbols, affected members;
open symbols, unaffected members; arrow, proband. (B) Sequencing
results of the SCN5A mutation. The sequence chromatogram indicates
a C to G transition of nucleotide 1099. (C) Alignment of multiple
SCN5A protein sequences across multiple species. The R367 affected
amino acid is located in a highly-conserved amino acid region in
different mammals. Red indicates the R367 site. P. troglodytes,
Pan troglodytes; M. mulatta, Macaca mulatta; F. catus, Felis catus;
M. musculus, Mus musculus; G. gallus, Gallus gallus. |
Genetic analysis
Data filtering excluded shared common variants
present in the 1000 Genomes Project (http://www.internationalgenome.org/), dbSNP132
(https://www.ncbi.nlm.nih.gov/projects/SNP/) and ESP
databases (http://evs.gs.washington.edu/EVS/). A total of 711
unique single-nucleotide polymorphisms were identified in the
proband. Through screening of the variants of arrhythmia-associated
genes, a novel missense mutation (p.R367G) of SCN5A was identified
and Sanger DNA sequencing demonstrated that this mutation was
co-segregated with affected members (Fig. 2B). The newly identified
c.1099C>G mutation in SCN5A was not observed in the 200 control
individuals. The mutation was not present in the dbSNP and Exome
Variant Server databases (evs.gs.washington.edu/EVS). Alignment of SCN5A amino
acid sequences from human, Pan troglodytes, Mus
musculus, Gallus gallus and other genomes (Fig. 2C) demonstrated that the affected
amino acid was evolutionarily conserved. A total of three programs
used for analyzing protein functions, polyphen2, SIFT and
MutationTaster, predicted that the p.R367G variants of SCN5A may be
damaging, deleterious and disease-causing, respectively.
Discussion
In the present study, a novel heterozygous mutation,
c.1099C>G, was identified in the SCN5A gene, causing arrhythmia
and syncope. Cosegregation analysis demonstrated the involvement of
the mutation in the pathogenesis of the arrhythmic phenotype
exhibited by the family in the present study. The diagnosis of
arrhythmia in the proband was based on the ECG and UCG, which
demonstrated a ventricular premature beat, as was additionally
observed in the mother of the proband. The maternal uncle of the
proband exhibited sinus bradycardia with a history of syncope and
arrhythmia. In order to identify the disease-causing gene in the
proband, WES analysis was used, in combination with
arrhythmia-associated gene filtering, to explore the possible
causative genes. A novel missense mutation (c.1099C>G) in exon 9
of the SCN5A gene was identified, which resulted in an amino acid
change from arginine to glycine at position 367 in the DI-S6
subunit of the NaV1.5 channel (14,15).
Bioinformatics analysis predicted that this mutation was
disease-causing, and the site (R367) of SCN5A is evolutionarily
conserved. The mutation was further investigated using
co-segregation analysis, which demonstrated that all affected
individuals in the present study carried the mutation, while the
wild-type allele individuals did not. The results of the present
study suggested that the SCN5A mutation led to the dysfunction of
the NaV1.5 channel, which has been demonstrated to cause syncope
during strenuous physical exercise (16).
The NaV1.5 channel α subunit consists of four
homologous domains, and each domain contains six α-helical
transmembrane repeats (14). In
the present study, the substituted amino acid p.R367G was located
at the six α-helical transmembrane segment of domain I-S6. The
substitution of basic amino acid Arg by neutral amino acid Gly in
position 367 of the SCN5A may affect transmembrane sodium transport
(17). Numerous mutations in this
site and region have been reported. For example, p.R367L and
p.R367C were demonstrated to be associated with Brugada syndrome
(18,19). The mutation p.R367H was previously
demonstrated to be involved in sudden unexplained nocturnal death
syndrome (20). In addition,
p.M369L, p.T370M and p.W374G were observed in Brugada syndrome and
other arrhythmogenic disorders (18,19,21).
These previous results demonstrated that the site (R367) exerts an
important function in the I-S6 domain of the NaV1.5 channel α
subunit, and that this domain serves a role in the regulation of
the gating properties of the channel.
Multiple lines of evidence support the notion that
the mutation identified in the present study is associated with
arrhythmia and syncope: i) This base was not identified in 200
control subjects, suggesting that it is not a common polymorphism;
ii) no other meaningful mutations were identified in SCN5A or other
genes; iii) this base change was not identified in other siblings
who were not diagnosed with arrhythmia and syncope; iv) the
mutation is evolutionally conserved in multiple animal species,
suggesting that any substitution at this codon is not tolerated;
and v) the same site mutations (p.R367H, p.R367L and p.R367C) have
been previously identified to cause arrhythmia and SCD (18–20).
In conclusion, using whole-exome sequencing in
combination with an arrhythmia-associated gene filtering method, a
novel missense mutation (c.1099G>C/p.R367G) in SCN5A was
identified to be a possible cause of a case of familial arrhythmia.
The present study increases the understanding of SCN5A mutations,
and contributes to potential genetic diagnosis and counseling of
families with arrhythmia.
Acknowledgements
The authors of the present study would like to
acknowledge the State Key Laboratory of Medical Genetics of China
(Changsha, China), for technical assistance. The present study was
supported by the Fundamental Research Funds for Central
Universities of Central South University (grant no.
2016zzts163).
References
|
1
|
Smits JP, Veldkamp MW and Wilde AA:
Mechanisms of inherited cardiac conduction disease. Europace.
7:122–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kawaguchi T, Hayashi H, Miyamoto A,
Yoshino T, Taniguchi A, Naiki N, Sugimoto Y, Ito M, Xue JQ,
Murakami Y and Horie M: Prognostic implications of progressive
cardiac conduction disease. Circ J. 77:60–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
den Hoed M, Eijgelsheim M, Esko T, Brundel
BJ, Peal DS, Evans DM, Nolte IM, Segrè AV, Holm H, Handsaker RE, et
al: Identification of heart rate-associated loci and their effects
on cardiac conduction and rhythm disorders. Nat Genet. 45:621–631.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beltran-Alvarez P, Tarradas A, Chiva C,
Pérez-Serra A, Batlle M, Pérez-Villa F, Schulte U, Sabidó E,
Brugada R, Pagans S, et al: Identification of N-terminal protein
acetylation and arginine methylation of the voltage-gated sodium
channel in end-stage heart failure human heart. J Mol Cell Cardiol.
76:126–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiong Q, Cao L, Hu J, Marian AJ and Hong
K: A rare loss-of-function SCN5A variant is associated with
lidocaine-induced ventricular fibrillation. Pharmacogenomics J.
14:372–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu
X, Fang WY and Yang YQ: Mutations of the SCN4B-encoded sodium
channel β4 subunit in familial atrial fibrillation. Int J Mol Med.
32:144–150. 2013.PubMed/NCBI
|
|
7
|
Riuró H, Campuzano O, Arbelo E, Iglesias
A, Batlle M, Pérez-Villa F, Brugada J, Pérez GJ, Scornik FS and
Brugada R: A missense mutation in the sodium channel β1b subunit
reveals SCN1B as a susceptibility gene underlying long QT syndrome.
Heart Rhythm. 11:1202–1209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bezzina CR, Barc J, Mizusawa Y, Remme CA,
Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi
F, et al: Common variants at SCN5A-SCN10A and HEY2 are associated
with Brugada syndrome, a rare disease with high risk of sudden
cardiac death. Nat Genet. 45:1044–1049. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Blich M, Efrati E, Marai I, Suleiman M,
Gepstein L and Boulous M: Novel Clinical Manifestation of the Known
SCN5A D1790G Mutation. Cardiology. 132:228–232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ilkhanoff L, Arking DE, Lemaitre RN,
Alonso A, Chen LY, Durda P, Hesselson SE, Kerr KF, Magnani JW,
Marcus GM, et al: A common SCN5A variant is associated with PR
interval and atrial fibrillation among African Americans. J
Cardiovasc Electrophysiol. 25:1150–1157. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mann SA, Castro ML, Ohanian M, Guo G,
Zodgekar P, Sheu A, Stockhammer K, Thompson T, Playford D, Subbiah
R, et al: R222Q SCN5A mutation is associated with reversible
ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol.
60:1566–1573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hothi SS, Ara F and Timperley J: p.Y1449C
SCN5A mutation associated with overlap disorder comprising
conduction disease, Brugada syndrome, and atrial flutter. J
Cardiovasc Electrophysiol. 26:93–97. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu R, Liu L, Chen C and Shen JM: Exome
Sequencing Identifies a Novel DES Mutation (R227C) in a Chinese
Dilated Cardiomyopathy Family. Cardiology. 137:78–82. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adsit GS, Vaidyanathan R, Galler CM, Kyle
JW and Makielski JC: Channelopathies from mutations in the cardiac
sodium channel protein complex. J Mol Cell Cardiol. 61:34–43. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Amin AS, Asghari-Roodsari A and Tan HL:
Cardiac sodium channelopathies. Pflugers Arch. 460:223–237. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shy D, Gillet L and Abriel H: Cardiac
sodium channel NaV1.5 distribution in myocytes via interacting
proteins: The multiple pool model. Biochim Biophys Acta.
1833:886–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Hoorn F, Campian ME, Spijkerboer A,
Blom MT, Planken RN, van Rossum AC, de Bakker JM, Wilde AA,
Groenink M and Tan HL: SCN5A mutations in Brugada syndrome are
associated with increased cardiac dimensions and reduced
contractility. PLoS One. 7:e420372012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smits JP, Eckardt L, Probst V, Bezzina CR,
Schott JJ, Remme CA, Haverkamp W, Breithardt G, Escande D,
Schulze-Bahr E, et al: Genotype-phenotype relationship in Brugada
syndrome: Electrocardiographic features differentiate SCN5A-related
patients from non-SCN5A-related patients. J Am Coll Cardiol.
40:350–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kapplinger JD, Tester DJ, Alders M, Benito
B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A,
Harris-Kerr C, et al: An international compendium of mutations in
the SCN5A-encoded cardiac sodium channel in patients referred for
Brugada syndrome genetic testing. Heart Rhythm. 7:33–46. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vatta M, Dumaine R, Varghese G, Richard
TA, Shimizu W, Aihara N, Nademanee K, Brugada R, Brugada J,
Veerakul G, et al: Genetic and biophysical basis of sudden
unexplained nocturnal death syndrome (SUNDS), a disease allelic to
Brugada syndrome. Hum Mol Genet. 11:337–345. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hofman-Bang J, Behr ER, Hedley P,
Tfelt-Hansen J, Kanters JK, Haunsøe S, McKenna WJ and Christiansen
M: High-efficiency multiplex capillary electrophoresis single
strand conformation polymorphism (multi-CE-SSCP) mutation screening
of SCN5A: A rapid genetic approach to cardiac arrhythmia. Clin
Genet. 69:504–511. 2006. View Article : Google Scholar : PubMed/NCBI
|