Introduction
The oral cavity contains a large number of sites,
which are suitable for the growth and assemblage of various
microorganisms (1). To date, ~700
phylotypes have been identified in the oral cavity and half of
these microbes exist in any individual during their lifetime
(2). These microorganisms, which
are predominantly composed ofbacteria, may prevent oral
colonization from exogenous organisms, indicating that they are
indispensableto health (3). In
addition, many normal oral inhabitants are associated with some
oral diseases including dental caries, gingivitis, periodontitis,
candidiasis, endodontic infections, orthodontic infections and
peri-implantitis (2,4–7).
Therefore, it is crucial to investigate the actual microbe
compositions inhealthy peopleand dental patients for understanding
the relationship between bacteria and various oral diseases.
In general, bacterial infections within the oral
cavity are polymicrobial in nature, and it is quite unusual for any
oral disease caused by a single species (8). However, medical microbiologists have
relied on culture techniques to elucidate the complexity of
infections for decades, and these culture methods can only be used
for identifying the ‘culturable’ bacteria that are growing
relatively quickly and easily in laboratory media (9,10),
and it is technically difficult to separate and identify >3-6
species because of their various requirements of nutrition, pH,
temperature andoxygen, and the interspecies competition
anddifferent concentrations of bacteria on the culture plate also
increase the difficulties of screening (11,12).
To determine the roles of bacteria in oral diseases, it is
necessary to define the full panoply of organisms within the human
mouth, and high-throughput sequencing methods may be a useful
method for alleviating this problem since it can detect almost all
the DNA signatures of microorganisms within a specific environment,
even those present inlow numbers or in a dormant metabolic state
(13–15).
As is already known, dental caries is a destructive
condition of the dental hard tissues that can progress to
inflammation and death of vital pulp tissue, with eventual spread
of infection to the periapical area of the tooth and beyond
(16). Conversely, periodontal
diseases can involve in both the soft and hard tissues and are the
most common inflammatory destructive conditions that affect humans
(8). To eliminate the
overestimation of the importance of species that are easily
cultured and the underestimation of the fastidious organisms that
may be highly prevalent and important in dental caries and
periodontal diseases, high-throughput sequencing analyses were used
in the present study.
Materials and methods
Ethical statement
The study was approved by the Ethical Committee of
the Stomatological Hospital of Nanchang University and all
participants provided written informed consent.
Patient selections and sampling
The patients only with typical dental caries and
periodontal diseases were selected from the Stomatological Hospital
of Nanchang University (Nanchang, China). Patients with dental
caries were recorded if either cumulative caries (dmft/DMFT) or
initial caries score was >0 or the Streptococcus mucans
count was ≥105 CFU/ml (17), and
periodontal disease was defined as two or more teeth with clinical
attachment loss (CAL) or ≥4 mm (18). Then, the patients were divided into
seven groups: Children-Control group (C.C, n=5, 6–18 years old),
Children-Dental caries group (C.DC, n=8, 6–18 years old);
Youth-Control group (Y.C, n=8, 18–35 years old), Youth-Dental
caries group (Y.DC, n=8, 18–35 years old); Adult-Control group
(A.C, n=7, 35–60 years old), Adult-Dental caries group (A.DC, n=6,
35–60 years old), and Adult-Periodontitis group (A.P, n=8, 35–60
years old). All of the subjects were Chinese, and those patients
who were pregnant, lactating or had other systemic conditions were
excluded. In addition, none of the subjects had received systemic
antibiotics or periodontal therapy in the previous 6 months
(19).
Stimulated whole saliva and oropharyngeal samples
were collected over a 6-month period from healthy and dental
patients. Subjects chewed a 1 g piece of paraffin wax for 1 min,
and after swallowing once, they expectorated secreted saliva into a
sterile plastic 50 ml tube several timesand this was kept frozen at
−20°C until processing. The oropharyngeal samples were collected
using dry cotton to gently swab the posterior wall of the
oropharynx in which the samples were directly suspended in a
microtube containing 200 µl lysis buffer [Tris 20 mM, EDTA 2 mM (pH
8), Tween-20 1%, proteinase K (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) 400 µg/ml]. Then, saliva samples with the same
volume of 2X lysis buffer and the oropharyngeal lysates were mixed
in a 1:10 ratio (19,20), and 200 µl mixture of saliva and
oropharyngeal lysates from every patient in same group were mixed
together for DNA extraction (21).
Extraction of genome DNA and
high-throughput sequencing
Genomic DNA of each sample was extracted by a
TIANamp Genomic DNA kit (Tiangen Biotech Co., Ltd., Beijing, China)
combined with bead beating, as previously published (22). The extracted genomicDNA was used as
the template to amplify the V3region of 16S rRNA genesusing primer
of 515F/806R with the barcode. PCR reactions, pyrosequencing of the
PCR amplicons and quality control of raw data were performed as
described previously (23).
Bioinformatics and multivariate
statistics
Paired-end reads from the original DNA fragments
weremerged using FLASH (Fast Length Adjustment of Short Reads to
Improve Genome Assemblies; http://www.cbcb.umd.edu/software/flash) to merge
paired-end reads when at least some of the reads overlap the read
generated from the oppositeend of the same DNA fragment, and
paired-end reads were assigned to each sample according to the
unique barcodes.
Then, sequences analysis was performed by UPARSE
software package version 7.0.1001 (http://drive5.com/uparse/) using the
UPARSE-operational taxonomic unit (OTU) and UPARSE-OTUref
algorithms. In-house Perl scripts were used to analyze alpha
(within samples) and beta (among samples) diversity. Sequences with
≥97% similarity were assigned to the same OTUs. Sequence was picked
as a representative for each OTU, and the Ribosomal Database
Project (RDP; http://rdp.cme.msu.edu/classifier/classifier.jsp)
classifier was used to annotate taxonomic information for each
representative sequence. Cluster analysis was preceded by
unweighted UniFrac distance using the Quantitative Insights into
Microbial Ecology software package version 1.8.0 (QIIME; http://qiime.org/index.html).
Results
Shared genera inhealthy peopleand
dental patients
To compare the microbes in healthypeople and dental
patients, 16S rRNA amplicon sequencing analysis was used to
sequence the V4 hypervariable region, and the sequencing data was
filtered to get the valid data. All the effective tags of all
samples were clustered and those sequences with over 97% similarity
were considered as one OTU. In total, 1,321,995 usable raw
sequences (8,686 unique sequences) and 2,207 OTUs were obtained
from all the samples with an average of 315.3 OTUs per group
(Table I). The Chao1 and Shannon
indexes were almost saturated and the rarefaction curve of every
sample was able to enter the plateau phase (data not shown).
 | Table I.Number of total tags, taxon tags,
unclassified tags, unique tags and OTUs from the oral cavity of
healthy people and patients with dental caries and periodontal
disease by high-throughput sequencing. |
Table I.
Number of total tags, taxon tags,
unclassified tags, unique tags and OTUs from the oral cavity of
healthy people and patients with dental caries and periodontal
disease by high-throughput sequencing.
| Group | Total tags | Taxon tags | Unclassified
tags | Unique tags | OTUs |
|---|
| C.C | 182,851 | 181,759 | 0 | 1,092 | 305 |
| C.DC | 159,863 | 158,641 | 0 | 1,222 | 285 |
| Y.C | 206,886 | 205,408 | 0 | 1,478 | 305 |
| Y.DC | 151,942 | 151,027 | 0 | 915 | 335 |
| A.C | 188,805 | 187,700 | 0 | 1,105 | 288 |
| A.DC | 168,826 | 167,602 | 0 | 1,224 | 331 |
| A.P | 262,822 | 261,172 | 0 | 1,650 | 358 |
| Mean | 188,856 | 187,615.6 | 0 | 1,241 | 315 |
| Total | 1,321,995 | 1,313,309 | 0 | 8,686 | 2,207 |
The Venn figure may reflect the difference between
all groups. As presented in Fig.
1, therewere305 (C.C), 285 (C.DC), 305 (Y.C), 335 (Y.DC), 288
(A.C), 331 (A.DC), and 358 (A.P) OTUs in each group, and there were
237 common OUTs in the C.C, Y.C and A.C groups (healthy group,
Fig. 1D); of which 73, 64, 53, 19
and 18 OUTs belonged to Firmicutes, Bacteroidetes,
Proteobacteria, Actinobacteria and
Fusobacteria respectively (data not shown). The common OUTs
in the C.C, Y.C and A.C groups (disease group, Fig. 1E) was 248, of which 79, 64, 54, 19
and 14 OUTs belonged to Firmicutes, Bacteroidetes,
Proteobacteria, Fusobacteria and
Actinobacteria respectively (data not shown). It should be
noted that there were only threecommon OUTs in the C.DC, A.DC and
Y.DC groups belonging to Spirochaetes (1 OTU) and
Proteobacteria (2 OTUs), and the unique OUTs in the A.P
group was 27, of which Actinobacteria only occupied 1 OUT,
while Proteobacteria and Spirochaetesoccupied 5 and 3
OUTs, respectively.
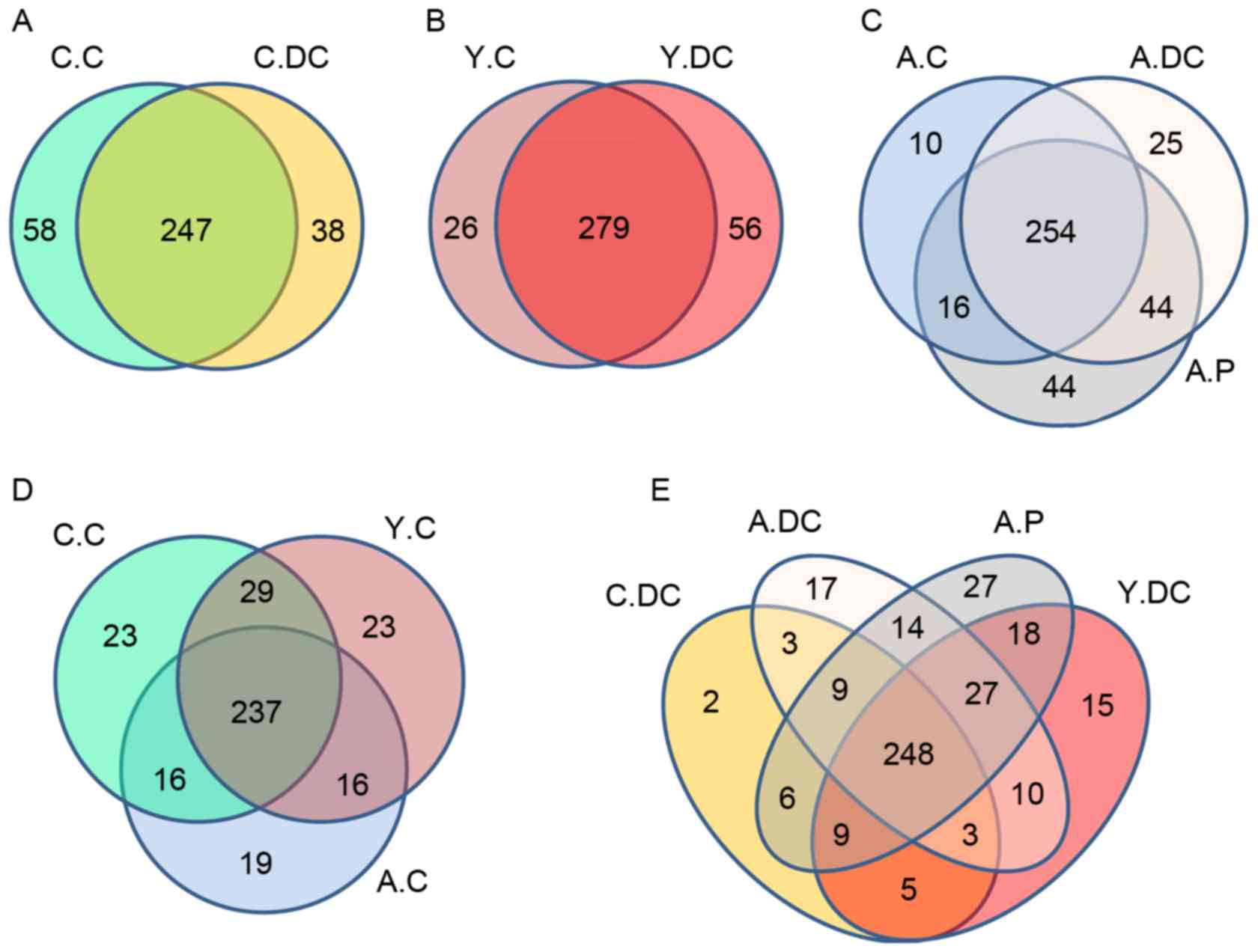 | Figure 1.Scalar-Venn representation of the
oral microbiota among C.C, C.DC, Y.C, Y.DC, A.C, A.DC and A.P
groups. The shared OTUs between (A) C.C and C.DC; (B) Y.C and Y.DC;
(C) A.C, A.DC and A.P; (D) C.C, Y.C and A.C; (E) C.DC, A.DC, A.P
and Y.DC groups. C.C, Children-Control group; C.DC, Children-Dental
caries group; Y.C, Youth-Control group; Y.DC, Youth-Dental caries
group; A.C. Adult-Control group; A.DC, Adult-Dental caries group;
A.P, Adult-Periodontitis group. |
Compositions and relative abundance of
bacterial communities in phylum level
Based on the weighted UniFrac distance in phylum
level, data for the top 10 microorganism populations were analyzed
using unweighted Pair-group Method with Arithmetic Mean (UPGMA) to
check the similarity between different groups (24). As presented in Fig. 2, Firmicutes,
Proteobacteria and Bacteroidetes constituted the
three commonest dominant phyla and accounted for >95% of the
total sequencing number in all groups, of which the percentage of
Firmicutes was ~75% in the Children-Control group (C-C, n=5,
6–18 years old), 48% in Youth-Control group (Y-C, N=8, 18~35 years
old) and 36% in the Adult-Control group (A-C, n=7, 35–60 years
old). The percentage for Proteobacteria was 10% in the C.C
group, 32% in the Y.C group and 50% in the A.C group (Fig. 2), respectively. Notably, the
percentage of Bacteroidetes was relatively stable,
maintaining a ratio of 10% of the total sequencing number.
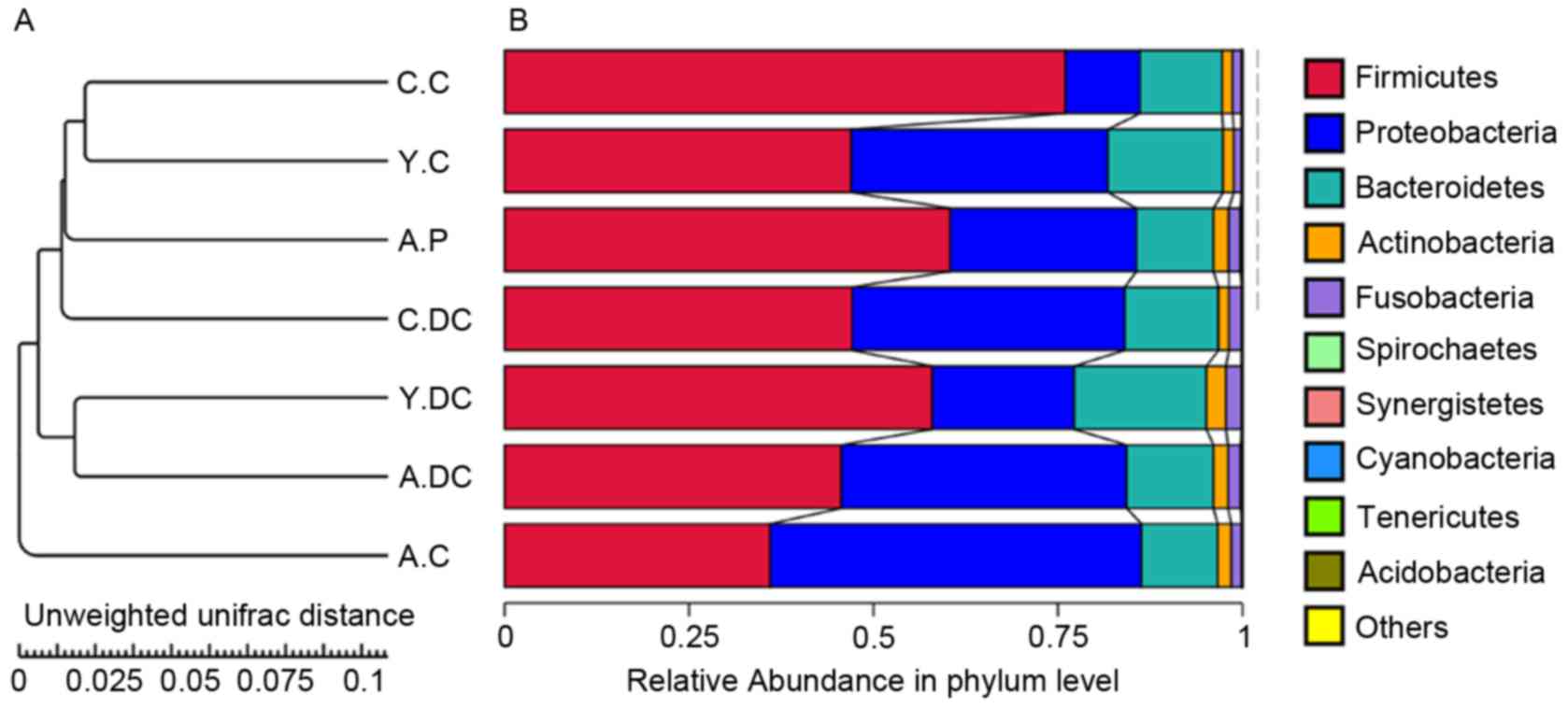 | Figure 2.Composition and relative abundance of
bacterial communities based 16S rDNA sequences in C.C, C.DC, Y.C,
Y.DC, A.C, A.DC and A.P groups. (A) The UPGMA clustering tree Based
on the Weighted UniFrac distance in phylum level. (B) The
composition and relative abundance of bacterial communities in
Kingdom, Phylum, Class, Family, Genus and Species levels. C.C,
Children-Control group; C.DC, Children-Dental caries group; Y.C,
Youth-Control group; Y.DC, Youth-Dental caries group; A.C.
Adult-Control group; A.DC, Adult-Dental caries group; A.P,
Adult-Periodontitis group. |
Compared with the healthy people, the percentages of
Firmicutes and Proteobacteria were reduced from 75 to
45% in dental caries patients, and from 10 to 39% in the children
group (C.C vs. C.DC), whereas the percentage of Firmicutes
was increased from 48 to 58% and Proteobacteria was reduced
from 32 to 19% in the youth group (Y.C vs. Y.DC). In the adult
group, the dental caries made a slight increase in
Firmicutes and a minor reduction in Proteobacteria,
while the percentage of Firmicutes in the periodontitis
group was greatly increased from 36 to 60%, and
Proteobacteria was reduced from 50 to 25%.
As indicatedin Fig.
3, similarities of 0.247 (C.C vs. Y.C), 0.345 (C.C vs. A.C) and
0.136 (Y.C vs. A.C) were obtained in the healthy group, and 0.180
(C.DC vs. Y.DC), 0.088 (C.DC vs. A. DC) and 0.169 (Y.DC vs. A.DC)
were obtained in the dental caries group.
 | Figure 3.The heatmap of beta diversity index
among C.C, C.DC, Y.C, Y.DC, A.C, A.DC and A.P groups. C.C,
Children-Control group; C.DC, Children-Dental caries group; Y.C,
Youth-Control group; Y.DC, Youth-Dental caries group; A.C.
Adult-Control group; A.DC, Adult-Dental caries group; A.P,
Adult-Periodontitis group. |
Compositions and relative abundance of
bacterial communities in genus level
To further investigate the relative abundance in
groups C.C, C.DC, Y.C, Y.DC, A.C, A.DC and A.P, the top 10 genera
were clustered. As demonstrated in Fig. 4, the dominant bacterial genera in
all groups were Streptococcus and Neisseria. When
dental caries occurred, the relative abundance of
Streptococcus reduced from 0.60 to 0.30 and Neisseria
increased from 0.06 to 0.26 in the children group. Notably, the
dental caries posed a slight effect on the relative abundance of
Streptococcus, while Neisseria received a dramatic
reduction in the youth group (from 0.26 to 0.09) and the adult
group (from 0.42 to 0.19).
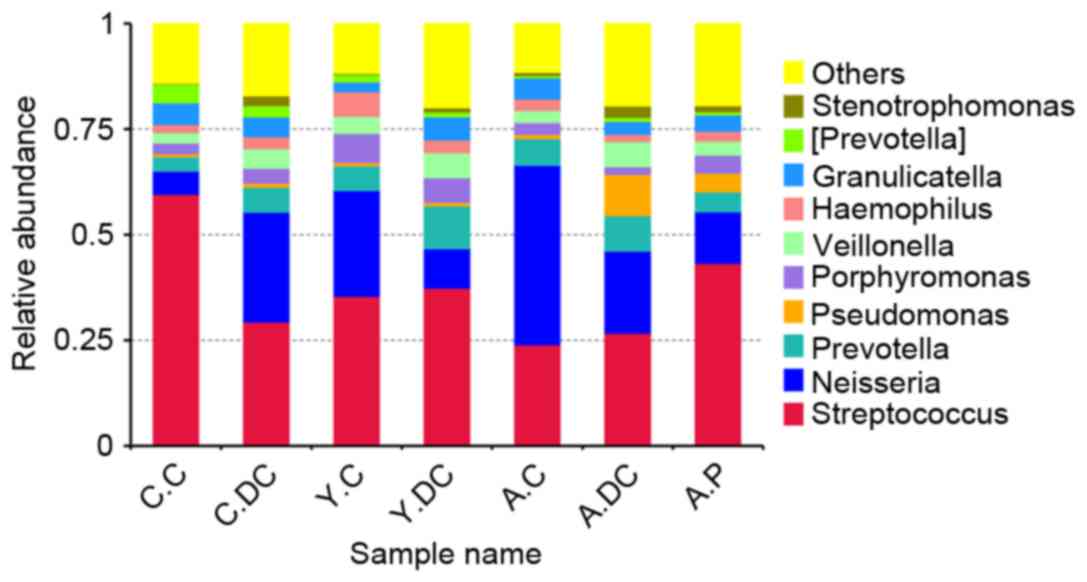 | Figure 4.The relative abundance of the
bacteria among C.C, C.DC, Y.C, Y.DC, A.C, A.DC and A.P groups at
the genus level. C.C, Children-Control group; C.DC, Children-Dental
caries group; Y.C, Youth-Control group; Y.DC, Youth-Dental caries
group; A.C. Adult-Control group; A.DC, Adult-Dental caries group;
A.P, Adult-Periodontitis group. |
Discussion
Oral diseases appear to be the outcome of multiple
microorganisms, and the complexity of the microbial community in
the oral cavity has hampered the identification of a single
etiological agent for dental caries (25). Although it has been demonstrated
that Streptococcus sobrinus and Streptococcus mutans
are acidogenic and serve an important role in caries initiation,
the molecular techniques revealed that a high proportion of samples
from cavities do not contain Mutans streptococci, whereas
other acid-producing bacteria, e.g. Lactobacillus,
Actinomyces or Bifidobacterium, existed (26,27).
Therefore, it is important to investigate the actual microbial
diversity in oral disease as a cooperation of microorganisms,
rather than as individual organisms or species in the human mouth
(28).
In previous studies, high-throughput sequencing had
been used to investigate the microbiota in human and canine oral
cavities (5,19), which may ignore the importance of
the age bracket on the tooth development and oral diseases.
Moreover, the authors suggested that the low sample size in their
studies may overlook inter-animal variations, as well as temporal
changes that possibly occur, and that larger studies were required
(5). To avoid the biodiversity
loss during the DNA extraction process (the small size of each
sample will make a huge loss of DNA) and to low the temporal
changes of the oral microbiota, all the samples in the same age
bracket were mixed and the mean number of 188,856.4 usable
sequences (6–9 folds greater than the single sample in previous
studies) and the saturated Chao1 index and Shannon index (Table I) ensured their reliability for the
future analysis.
In Fig. 1, the Venn
figure demonstrated that a mean of 315 OTUs were obtained in each
group, and 73, 64, 53, 19 and 18 common OUTs belonging to
Firmicutes, Bacteroidetes, Proteobacteria,
Actinobacteria and Fusobacteria, respectively, were
identified in healthy people. As is already known,
Firmicutes can survive in extreme conditions and made up the
largest portion of the gut flora involved in energy resorption and
obesity (29), Bacteroides
are related to the metabolism of fat by absorbing nutrition and
producing short-chain fatty acids (22), and Proteobacteria area major
group of gram-negative bacteria (which includes a wide variety of
pathogens) responsible for nitrogen fixation (30). Most of bacteria in these three
phyla are regarded as pathogenic microorganisms, therefore the
traditional treatment strategy to eradicate all the microorganisms
in oral cavities using antibiotic drugs may not be suitable, as the
‘pathogens’ composed primarily of oral microorganisms and may serve
a key role in oral health.
Based on the weighted UniFrac distance in phylum
level, Firmicutes, Proteobacteria and
Bacteroidetes accounted for >95% of the total sequencing
number in each group, and their total occupancy received little
change between healthy and disease conditions (Fig. 2). However, the reduction of
Firmicutesfrom 75 to 45% and the increase of
Proteobacteria from 10 to 39% indicated the dental caries in
the children group characterized by the raising of
Firmicutes. Conversely, the dental caries made an increase
of Firmicutes and a reduction of Proteobacteria in
youth and adult groups, indicating that the age bracket was crucial
for tooth development and microbial development. Considering the
immature tooth development in the children group (6–18 years old)
and the physiological depression in the adult group (35–60 years
old), the microbial community in the youth group may be the most
reasonable microbiota. Moreover, the high dissimilarity of 0.345
(C.C vs. A.C) in healthy patients and the high similarity of 0.088
(C.DC vs. A.DC) in diseased patients also confirmed the instability
of the children and adult groups, when compared with the youth
group (Fig. 3).
To further investigate the relative abundance among
groups, the top 10 genera were clustered in Fig. 4, and Streptococcus and
Neisseria were identified as the dominant bacteria genus in
all groups. As with Fig. 2, the
children group was characterized with the increase of
Neisseria, while the youth group (from 0.26 to 0.09) and the
adult group (from 0.42 to 0.19) were characterized by the reduction
in Neisseria. The well-distributed bacteria were beneficial
for the host and defended against invading pathogens (31), thus the more even microbial
distribution in the Y.C group also confirmed their beneficial
potential for pathogenic invasion.
In the present study, the microbial community of the
human oral cavity had been explored using high-throughput
sequencing, and these results indicated that the oral microbial
communities were mainly composed of traditional ‘pathogenic
bacteria’, and their ratio had been largely influenced by human age
and presence of oral diseases. Therefore, it is necessary to
further investigate the actual microbial diversity in healthy
people and oral disease patients on a larger scale and identify the
bacteria at species level; the unique bacteria should be screened
and separated to study their role in maintaining the eco-balance of
the oral cavity.
Acknowledgements
This work was supported by the National Natural
Science Foundation of China (grant nos. 91639106, 81270202 and
91339113 to H-B Xin, 31560264 and 81503364 to Tingtao Chen and
21461015 to Xiaolei Wang); the National Basic Research Program of
China (grant no. 2013CB531103 to Hongbo Xin); the Open Foundation
Of Hubei Key Laboratory Of Lipid Chemistry And Nutrition (grant no.
201602 to Tingtao Chen); the Science Foundation of Jiangxi
Provincial Department of Education (grant nos. KJLD14010 and
20153BCB23035 to Xiaolei Wang and 20151BBG70239 to Yan Shi); and,
the major program of Natural Science Foundation of Jiangxi Province
(grant no. 20161ACB21002 to Xiaolei Wang).
References
|
1
|
Xie G, Chain PS, Lo CC, Liu KL, Gans J,
Merritt J and Qi F: Community and gene composition of a human
dental plaque microbiota obtained by metagenomic sequencing. Mol
Oral Microbiol. 25:391–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Palmer RJ Jr: Composition and development
of oral bacterial communities. Periodontol 2000. 64:20–39. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marsh PD: Microbial ecology of dental
plaque and its significance in health and disease. Adv Dent Res.
8:263–271. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seymour GJ, Ford PJ, Cullinan MP, Leishman
S and Yamazaki K: Relationship between periodontal infections and
systemic disease. Clin Microbiol Infec. 13:(Suppl 4). S3–S10. 2007.
View Article : Google Scholar
|
|
5
|
Sturgeon A, Stull JW, Costa MC and Weese
JS: Metagenomic analysis of the canine oral cavity as revealed by
high-throughput pyrosequencing of the 16S rRNA gene. Vet Microbiol.
162:891–898. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sibley CD, Peirano G and Church DL:
Molecular methods for pathogen and microbial community detection
and characterization: Current and potential application in
diagnostic microbiology. Infect Genet Evol. 12:505–521. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Botelho MP, Maciel SM, Cerci Neto A, Dezan
CC, Fernandes KB and de Andrade FB: Cariogenic microorganisms and
oral conditions in asthmatic children. Caries Res. 45:386–392.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Allaker RP and Ian Douglas CW:
Non-conventional therapeutics for oral infections. Virulence.
6:196–207. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McGuckin M, Goldman R, Bolton L and
Salcido R: The clinical relevance of microbiology in acute and
chronic wounds. Adv Skin Wound Care. 16:12–23; quiz 24–25. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomson PD: Immunology, microbiology, and
the recalcitrant wound. Ostomy Wound Manage. 46:(1A Suppl).
77S–82S; quiz 83S-84S. 2000.PubMed/NCBI
|
|
11
|
Whitley R: The new age of molecular
diagnostics for microbial agents. N Engl J Med. 358:988–989. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davies CE, Hill KE, Wilson MJ, Stephens P,
Hill CM, Harding KG and Thomas DW: Use of 16S ribosomal DNA PCR and
denaturing gradient gel electrophoresis for analysis of the
microfloras of healing and nonhealing chronic venous leg ulcers. J
Clin Microbiol. 42:3549–3557. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kobayashi N, Bauer TW, Tuohy MJ, Lieberman
IH, Krebs V, Togawa D, Fujishiro T and Procop GW: The comparison of
pyrosequencing molecular Gram stain, culture, and conventional Gram
stain for diagnosing orthopaedic infections. J Orthop Res.
24:1641–1649. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang C, Zhang M, Pang X, Zhao Y, Wang L
and Zhao L: Structural resilience of the gut microbiota in adult
mice under high-fat dietary perturbations. ISME J. 6:1848–1857.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang
X, Jia W, Cai S and Zhao L: Structural segregation of gut
microbiota between colorectal cancer patients and healthy
volunteers. ISME J. 6:320–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Selwitz RH, Ismail AI and Pitts NB: Dental
caries. Lancet. 369:51–59. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Näse L, Hatakka K, Savilahti E, Saxelin M,
Pönkä A, Poussa T, Korpela R and Meurman JH: Effect of long-term
consumption of a probiotic bacterium, Lactobacillus rhamnosus GG,
in milk on dental caries and caries risk in children. Caries Res.
35:412–420. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goepfert AR, Jeffcoat MK, Andrews WW,
Faye-Petersen O, Cliver SP, Goldenberg RL and Hauth JC: Periodontal
disease and upper genital tract inflammation in early spontaneous
preterm birth. Obstet Gynecol. 104:777–783. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu B, Faller LL, Klitgord N, Mazumdar V,
Ghodsi M, Sommer DD, Gibbons TR, Treangen TJ, Chang YC, Li S, et
al: Deep sequencing of the oral microbiome reveals signatures of
periodontal disease. PLoS One. 7:e379192012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Faveri M, Mayer MP, Feres M, de Figueiredo
LC, Dewhirst FE and Paster BJ: Microbiological diversity of
generalized aggressive periodontitis by 16S rRNA clonal analysis.
Oral Microbiol Immunol. 23:112–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hirotomi T, Yoshihara A, Ogawa H, Ito K,
Igarashi A and Miyazaki H: A preliminary study on the relationship
between stimulated saliva and periodontal conditions in
community-dwelling elderly people. J Dent. 34:692–698. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu X, Wu X, Qiu L, Wang D, Gan M, Chen X,
Wei H and Xu F: Analysis of the intestinal microbial community
structure of healthy and long-living elderly residents in Gaotian
Village of Liuyang City. Appl Microbiol Biotechnol. 99:9085–9095.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu J, Lian F, Zhao L, Zhao Y, Chen X,
Zhang X, Guo Y, Zhang C, Zhou Q, Xue Z, et al: Structural
modulation of gut microbiota during alleviation of type 2 diabetes
with a Chinese herbal formula. ISME J. 9:552–562. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng M, Qian L, Shen G, Bian G, Xu T, Xu
W, Shen G and Hu S: Microbiota modulate tumoral immune surveillance
in lung through a γδT17 immune cell-dependent mechanism. Cancer
Res. 74:4030–4041. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alcaraz LD, Belda-Ferre P, Cabrera-Rubio
R, Romero H, Simón-Soro A, Pignatelli M and Mira A: Identifying a
healthy oral microbiome through metagenomics. Clin Microbiol
Infect. 18:(Suppl 4). S54–S57. 2012. View Article : Google Scholar
|
|
26
|
Loesche WJ: Role of Streptococcus-mutans
in human dental decay. Microbiol Rev. 50:353–380. 1986.PubMed/NCBI
|
|
27
|
Corby PM, Lyons-Weiler J, Bretz WA, Hart
TC, Aas JA, Boumenna T, Goss J, Corby AL, Junior HM, Weyant RJ and
Paster BJ: Microbial risk indicators of early childhood caries. J
Clin Microbiol. 43:5753–5759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Costerton JW, Stewart PS and Greenberg EP:
Bacterial biofilms: A common cause of persistent infections.
Science. 284:1318–1322. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ley RE, Turnbaugh PJ, Klein S and Gordon
JI: Microbial ecology: Human gut microbes associated with obesity.
Nature. 444:1022–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stackebrandt E, Murray R and Trüper H:
Proteobacteria classis nov., a name for the phylogenetic taxon that
includes the ‘purple bacteria and their relatives’. Int J Syst
Bacteriol. 38:321–325. 1988. View Article : Google Scholar
|
|
31
|
Frank DN and Pace NR:
Molecular-phylogenetic analyses of human gastrointestinal
microbiota. Curr Opin Gastroenterol. 17:52–57. 2001. View Article : Google Scholar : PubMed/NCBI
|














