Introduction
Homocysteine (Hcy) is a sulfur-containing,
non-protein amino acid that is derived from methionine metabolism
(1). An imbalance in Hcy
metabolism is considered an independent risk factor for many
disorders, such as physical impairment (2), atherosclerosis (3,4),
neurodegenerative diseases (5) and
alcoholic liver disease (ALD) (6,7).
Alcoholic hyperhomocysteinemia (HHcy) is often seen in rodents and
humans with during the development of alcoholic steatohepatitis
(8). In contrast with the adverse
effects of Hcy in the progression of ALD, Hcy may serve a positive
role in cellular antioxidant reactions as an important producer of
cellular total glutathione (GSH) (9,10).
This metabolic link between Hcy and GSH occurs by the
transsulfuration of Hcy to cysteine, a precursor for GSH (11,12).
GSH performs a crucial role in maintaining hepatic
reduction-oxidation (redox) balance in biological systems (13). However, the underlying molecular
mechanism of the positive role that Hcy serves in the progression
of ALD has not been clearly elucidated.
The nuclear factor (erythroid-derived 2)-like 2
(Nrf2) functions as a master regulator of the antioxidant response
by regulating the expression of cytoprotective genes, including
genes involved in the antioxidant GSH pathway (14–16).
Notably, a Hcy-activated Nrf2-antioxidant reaction has been
demonstrated in mouse macrophage and human hepatoma cell lines
(6,9,10).
Therefore, the present study hypothesized that Nrf2 may function to
mediate Hcy-regulated GSH production in hepatocytes, and that the
hepatic Hcy-Nrf2-GSH pathway may serve a protective role in the
progression of ALD. Cellular viability and intracellular GSH
expression levels were assayed to investigate the antioxidant
effects of Hcy and Nrf2 on HepG2 cell injury induced by the hepatic
lipid-peroxidation product 4-hydroxynonenal (4-HNE). Hcy-activated
Nrf2 expression was demonstrated by western blot analysis and
immunofluorescence staining assay. Further research into this area
will provide deeper insights into the association between Hcy and
its potentially protective roles in the progression of ALD.
Materials and methods
Cell culture conditions and MTT
assay
The HepG2 human hepatocarcinoma cell line was
purchased from the American Type Culture Collection (Manassas, VA,
USA) and maintained in Dulbecco's modified Eagle's medium (DMEM)
containing 10% fetal bovine serum (FBS; both from HyClone; GE
Healthcare Life Sciences, Logan, UT, USA) at 37°C in a 5%
CO2 atmosphere.
Immunofluorescence staining
HepG2 cells were grown on 6-well cover slips at a
density of 3×103 cells/well, and treated with Hcy (0,
10, 20, 50 or 100 µM) for 24 h at 37°C. Adherent cells were fixed
to slides with paraformaldehyde for 15 min at 37°C and incubated
with 0.5% Trion-X-100 for 10 min at 37°C. Nonspecific binding was
blocked by 3% BSA for 30 min at 37°C as previously described
(17). Immunostaining was
performed with an anti-Nrf2 antibody overnight at 4°C (1:300;
bs-1074R, Rabbit polyclonal IgG, BIOSS, Beijing, China), followed
by incubation with a cyanine-3 (Cy3)-conjugated secondary antibody
for 2 h at 37°C (1:200. P0183, Goat Anti-Rabbit IgG, Beyotime
Institute of Biotechnology, Shanghai, China). Nuclear staining was
performed by incubating with DAPI (1:100) for 10 min at 37°C.
Increased Nrf2 expression was observed under an inverted microscope
(Nikon Corporation, Tokyo, Japan). Images were processed using
Image ProPlus 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
Knockdown of Nrf2 by siRNA
Nrf2 expression was silenced by Nrf2-siRNA (Suzhou
GenePharma LLC). A non-targeted control siRNA (NC) was used as a
negative control. The siRNA sense sequences were as follows:
siNrf2, 5′-GCACCUUAUAUCUCGAAGUTT-3′; NC,
5′-UUCUCCGAACGUGUCACGUTT-3′. Briefly, HepG2 cells were grown to
50–70% confluence. A total of 0.27 µg (1 µl) siRNA, 10 µl
INTERFERin Gene siRNA Transfection Reagent (Suzhou GenePharma LLC)
and 400 µl serum-free Opti-MEM-1 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MD, USA) were combined and added to the
cells, which were incubated at room temperature for 10 min,
followed by the subsequent addition of the siRNA/Transfection
Reagent mixture directly onto the cells with 4 ml serum-free
Opti-MEM-1 medium (final siRNA concentration, 1 nM). Following
incubation for 4 h at 37°C, the media was replaced with DMEM
containing 10% FBS. Total RNA was extracted and reverse transcribed
to cDNA using a Golden 1st cDNA Synthesis kit (HaiGene Bio Inc.,
Harbin, China). cDNA was stored at −80°C and transfection
efficiency was later validated by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
MTT assay
HepG2 cells were seeded at a density of
2×103 cells/well in two 96-well plates; one plate was
treated with 70 µM 4-HNE (Cayman Chemical Company, Ann Arbor, MI,
USA), 200 µM buthionine sulfoximine (BSO; Shanghai Aladdin Bio-Chem
Technology Co., Ltd., Shanghai, China), 100 µM Hcy (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany), 4-HNE+Hcy, BSO+Hcy, BSO+4-HNE and
BSO+4-HNE+Hcy for 24 h at 37°C; the second plate was transfected
with 1 nM negative control and Nrf2-targeted small interfering RNA
(siRNA; Suzhou GenePharma LLC, Suzhou, China), followed by addition
of 100 µM Hcy, 70 µM 4-HNE or 4-HNE+Hcy respectively, for 24 h at
37°C. The following day, 20 µl MTT (5 mg/ml), the yellow
mitochondrial dye, was added to the cells, and the plates were
incubated at 37°C for an additional 4 h. The culture medium was
discarded, and any formazan crystals that may have formed were
dissolved in 150 µl DMSO. Absorbance was measured at 570 nm using a
SPECTRA Max 190 (Molecular Devices, LLC, Sunnyvale, CA, USA).
Measurement of reduced GSH and
oxidized GSH levels in HepG2 cells
HepG2 cells (3×105) were treated with
increasing concentrations of Hcy (0–500 µM) and
tert-butylhydroquinone (t-BHQ; 0–100 µM, Sigma-Aldrich; Merck KGaA)
and 200 µM BSO for 24 h, respectively, or transfected with 1 nM
Nrf2- or NC-siRNA for 4 h at 37°C. The media was subsequently
replaced with DMEM containing 10% FBS with or without Hcy for 9 h.
Cells were trypsinized and collected by centrifugation at 170 × g
for 5 min at room temperature, re-suspended in protein removal
reagent M (Beyotime Institute of Biotechnology) and immediately
vortex mixed for 10s. The samples were frozen rapidly with liquid
nitrogen and thawed at 37°C, this was repeated twice, and then the
samples were incubated in an ice-bath for 5 min. The resulting
supernatant was collected by centrifugation at 10,000 × g for 10
min, and analyzed using the sulfhydryl reagent 5,5′-dithiobis
(2-nitrobenzoic acid), which forms the yellow derivative
5′-thio-2-nitrobenzoic acid (TNB), and is measured at 412 nm using
a SPECTRA Max 190 (Molecular Devices, LLC). Levels of reduced and
oxidized GSH were determined using a GSH Assay kit (Beyotime
Institute of Biotechnology), according to the manufacturers
protocol (18).
Western blot analysis
HepG2 cells were treated with Hcy (0–100 µM) for 24
h at 37°C. Cells were homogenized in RIPA lysis buffer (Beyotime
Institute of Biotechnology) containing leupeptin. Protein
concentrations were quantified using a Bicinchoninic Acid Protein
Assay kit (Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. Cellular protein samples (30 µg) were
separated by 10% SDS-PAGE as previously described (19). Proteins were transferred onto
nitrocellulose membranes and the membranes were incubated with an
anti-Nrf2 polyclonal antibody (1:500; bs-1074R, BIOSS, Beijing,
China) or an anti-β-actin monoclonal antibody (1:3,000; TA-09;
Zhongshan Golden Bridge Biotechnology, Beijing, China) overnight at
4°C, followed by horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G secondary antibody (1: 5,000; ZB-2301, Zhongshan
Golden Bridge Biotechnology). Protein bands were visualized by
enhanced chemiluminescence (HaiGene Bio Inc.). Densitometric
analysis of band intensity was calculated using Quantity One
software, version v4.62 (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Relative mRNA expression
quantification by RT-qPCR
Total RNA was extracted from cultured HepG2 cells
(2×105 cells) using TRIzol Reagent (HaiGene Bio Inc.),
according to the manufacturer's protocol (9), and subsequently reverse transcribed
to cDNA using a Golden 1st cDNA Synthesis Kit (HaiGene Bio Inc.).
cDNA samples were amplified using a DNA thermal cycler (Thermo
Fisher Scientific, Inc.). qPCR reactions were set up by mixing 10X
SYBR Green PCR Master Mix (Roche Diagnostics GmbH, Mannheim,
Germany) with 2 µl 100 fold diluted cDNA template, 0.6 µl each of
forward and reverse primers (0.3 µM) and 6.8 µl PCR-grade water in
a 20 µl reaction volume. The reaction mixtures were amplified using
an ABI 7300 Sequence Detection System (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with an initial denaturation step at 95°C
for 10 min, followed by 40 cycles of: denaturation at 95°C for 15
sec; annealing at 58°C for 20 sec; extension at 72°C for 30 sec;
and melting curve analysis. The primer sequences were as follows:
18S, forward 5′-AACTTTCGATGGTAGTCGCCG−3′, reverse
5′-CCTTGGATGTGGTAGCCGTTT-3′; Nrf2, forward
5′-GCACCUUAUAUCUCGAAGUTT-3′, reverse 5′-ACUUCGAGAUAUAAGGUGCTT-3′;
GCLc, forward 5′-ATGTGGACACCCGATGCAGTATT-3′; reverse
5′-TGTCTTGCTTGTAGTCAGGATGGTTT-3′. Primers were designed using the
GenBank database (https://www.ncbi.nlm.nih.gov/genbank). Nrf2 mRNA and
GCLc mRNA expression were measured by RT-qPCR and normalized to
18s. Data was analyzed using Applied Biosystems SDS V1.4 software
(Thermo Fisher Scientific, Inc.) using the 2−ΔΔCq
method, where ΔΔCq=ΔCq target-ΔCq internal control (20).
Statistical analysis
Statistical analysis was performed using one-way
analysis of variance followed by Newman-keuls test. Analyses were
performed using OriginPro7.5 software (OriginLab, Northampton, MA,
USA) and Graph Pad Prism version 5 (Graph Pad Software Inc., La
Jolla, CA, USA). Data were presented as the mean ± standard error
of the mean. Values of P<0.05 were considered to indicate a
statistically significant difference. All experiments were
performed at least three times and representative results were
presented.
Results
Hcy treatment increases total GSH
expression levels in HepG2 cells
Intracellular levels of the antioxidant thiol GSH
were evaluated using the GSH Assay kit. In response to exogenous
Hcy (10, 20, 100 or 500 µM) treatment, the levels of intracellular
total GSH (Fig. 1A), oxidized GSH
(Fig. 1B) and reduced GSH
(Fig. 1C) were increased in a
concentration-dependent manner, with a peak at 100 µM Hcy.
Therefore, exogenous Hcy was administered at a concentration of 100
µM for the following tests.
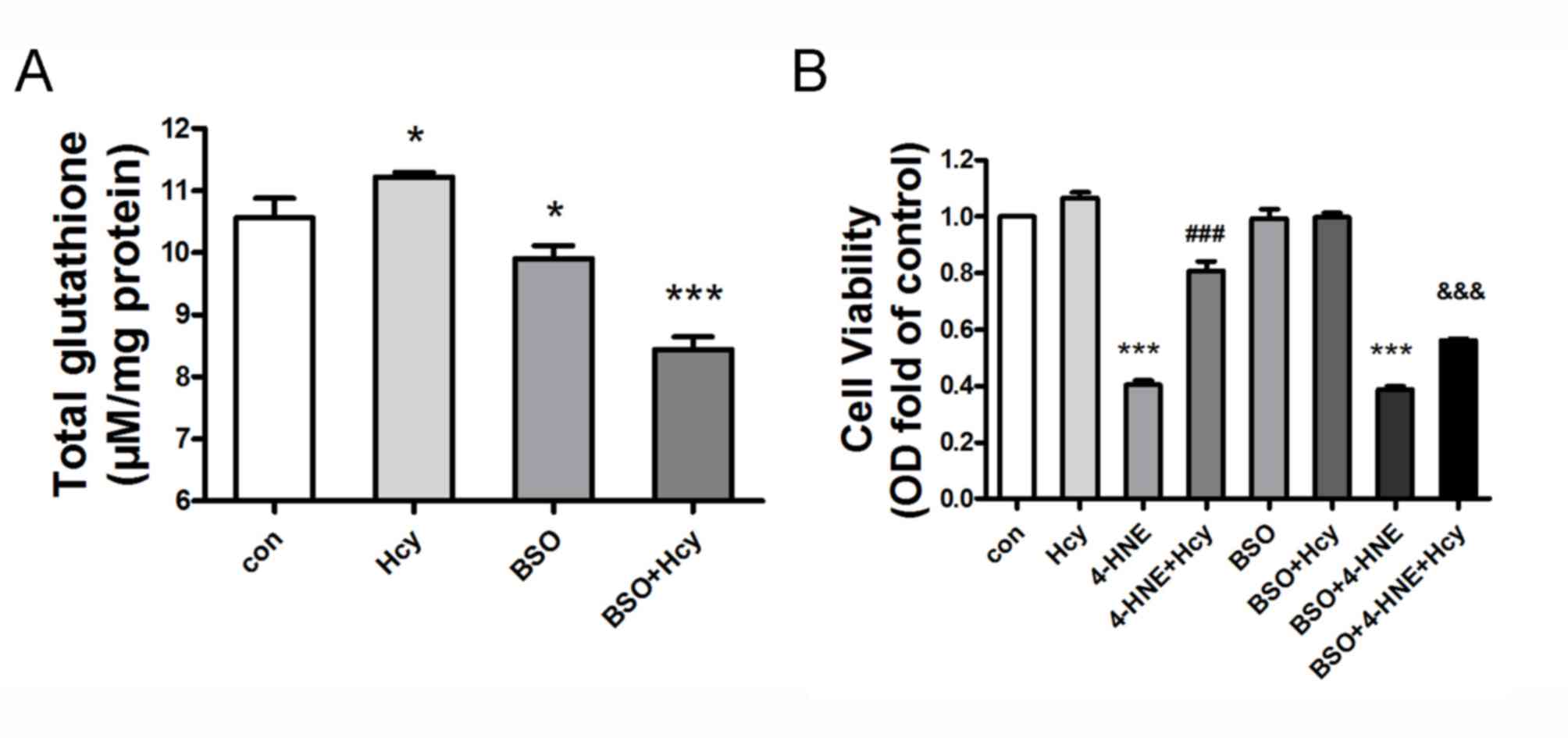
Hcy protects against oxidative
stress-induced cell injury in HepG2 cells
HepG2 cells were pretreated with the specific GSH
production inhibitor BSO (200 µM) for 2 h, and subsequently treated
or co-treated with Hcy (100 µM) for 24 h. Hcy treatment led to
elevated GSH content compared with the untreated control group
(n=6, P<0.05). However, BSO co-treatment significantly abolished
the Hcy-induced GSH expression in HepG2 cells, compared with the
Hcy-treated group (Fig. 2A; n=6;
P<0.05). HepG2 cells were treated with several combinations, as
follows: 4-HNE (70 µM)+Hcy (100 µM); BSO (200 µM)+4-HNE; BSO+Hcy;
BSO+4-HNE+Hcy all together. MTT assay indicated that treatment with
the lipid peroxidation product 4-HNE decreased cell viability, and
no cellular toxicity was observed in HepG2 cells at 10 µM Hcy.
However, Hcy co-treatment attenuated 4-HNE-induced cell injury,
compared with the 4-HNE-treated group (Fig. 2B; n=6; P<0.05). Notably, BSO
pretreatment significantly weakened the protective effect of Hcy on
4-HNE-induced cell injury in HepG2 cells, compared with the
4-HNE+Hcy group (n=6; P<0.05). Conversely, BSO pretreatment had
no effect on viability in Hcy- or 4-HNE-treated cells (Fig. 2B; n=6; P>0.05).
Hcy treatment induces Nrf2 protein
expression and promotes nuclear accumulation in HepG2 cells
HepG2 cells were treated with Hcy (0–100 µM) for 24
h. Western blot analysis revealed that Hcy exposure induced Nrf2
protein expression in a concentration-dependent manner (Fig. 3A). Immunofluorescence analysis with
an anti-Nrf2 antibody revealed that the protein expression of Nrf2
increased with the Hcy concentration (Fig. 3B). In addition, Hcy (0–100 µM)
treatment promoted the expression of glutamate-cysteine ligase
catalytic subunit (GCLc), a downstream target gene of Nrf2
(Fig. 3C).
Nrf2 mediates Hcy-induced GSH
expression and cell protection
To explore whether Nrf2 participates in the
Hcy-induced increase in GSH expression in HepG2 cells, cells were
treated with the Nrf2 activator t-BHQ (0, 10, 20, 50 or 100 µM) for
24 h. Cells exposed to t-BHQ exhibited an increase in GSH
expression levels in a concentration-dependent manner (Fig. 4A). Additionally, Nrf2 gene
expression was knocked down in HepG2 cells by siRNA, which resulted
in the decreased expression of Nrf2 mRNA levels by ~50% (Fig. 4B; n=3; P<0.001 vs. NC treated
cells). Hcy+NC treatment for 24 h slightly elevated the
intracellular GSH content in comparison with the NC group. This
small increase may be attributed to invasion of the exogenous siRNA
into the serum-starved cells, which may have resulted in decreasing
the cell sensitivity to the following Hcy treatment. Nrf2-siRNA
exposure resulted in decreased GSH expression levels, compared with
the NC-treated HepG2 cells. Furthermore, Hcy-induced GSH expression
in HepG2 cells was also suppressed by Nrf2 siRNA, compared with the
NC+Hcy group (Fig. 4C; n=3;
P<0.05). Analysis of cell viability with MTT indicated that
Nrf2-siRNA pretreatment reduced the protective role of Hcy in
limiting 4-HNE-induced cell injury in HepG2 cells (Fig. 4D; n=3; P<0.05). In brief, Nrf2
was observed to mediate Hcy-induced GSH expression and the
protective effect of Hcy on cell viability.
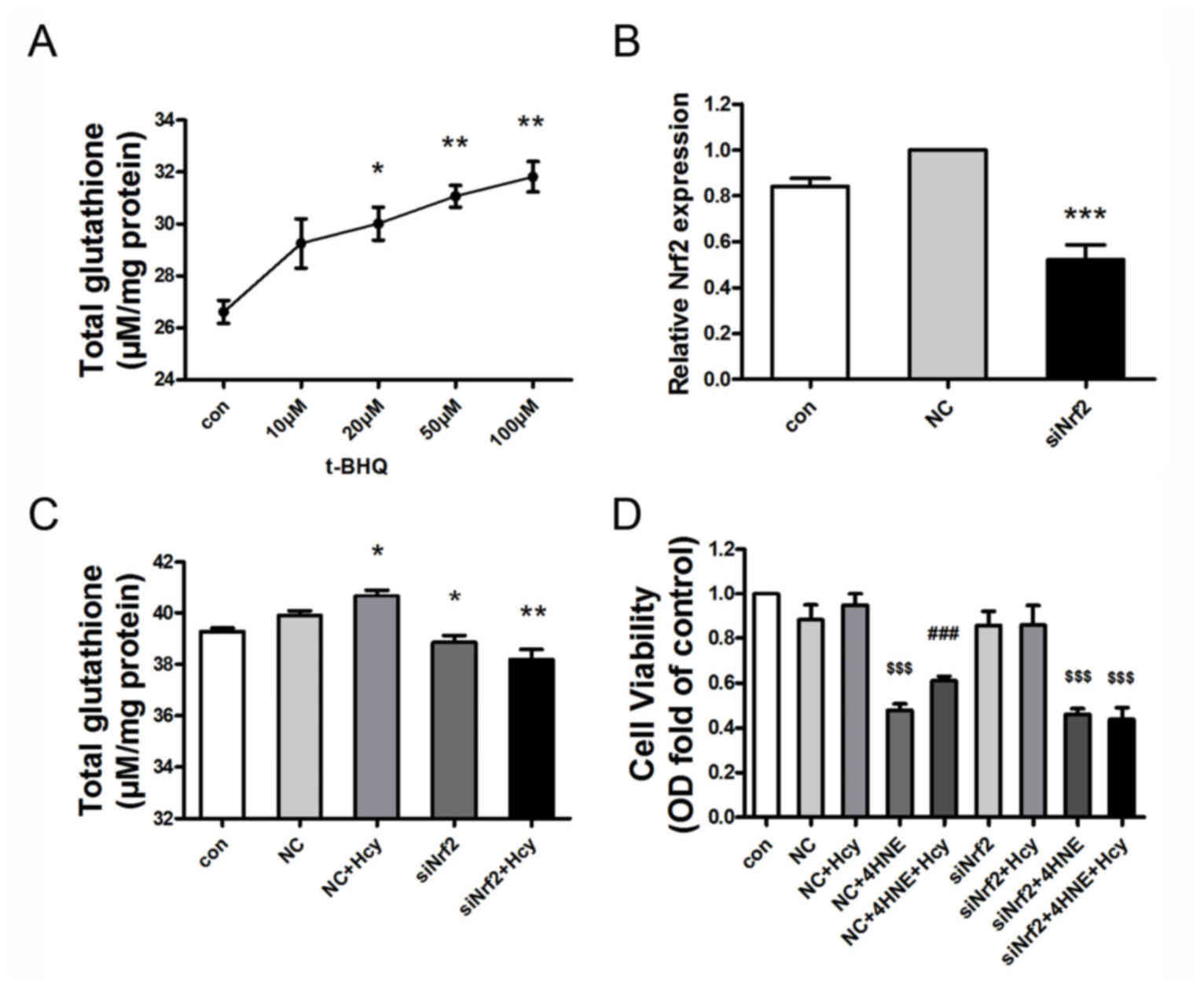 | Figure 4.Nrf2 mediates Hcy-induced GSH
expression and cellular protection in 4-HNE-stressed HepG2 cells.
(A) t-BHQ treatment increased total GSH expression levels in a
concentration-dependent manner in HepG2 cells. HepG2 cells were
treated with t-BHQ (0–100 µM) for 24 h (n=3). (B) Nrf2 mRNA
expression was downregulated by ~50% following transfection with
Nrf2-siRNA. Nrf2 siRNA transfection was performed for 4 h; the
transfection media was subsequently removed and replaced with
medium containing 10% fetal bovine serum for 9 h (n=3). (C)
Nrf2-siRNA transfection reduced Hcy-induced total GSH elevation.
HepG2 cells were transfected with NC-siRNA or Nrf2-siRNA for 4 h,
then co-treated with Hcy for 9 h (n=3). (D) Nrf2 siRNA attenuated
the protective role of Hcy in 4-HNE-induced cell injury in HepG2
cells. HepG2 cells were pretreated with 1 nM NC siRNA or 1 nM Nrf2
siRNA for 4 h, followed by Hcy (100 µM) and/or 4-HNE (70 µM) for 24
h. Cellular viability was assessed by MTT Assay (n=6). Values are
presented as the mean ± standard error of the mean. *P<0.05,
**P<0.01 and ***P<0.001 vs. control group;
$$$P<0.001 vs. NC group; ###P<0.001 vs.
NC+4-HNE-treated cells. 4-HNE, 4-hydroxynonenal; Con, control; GSH,
glutathione; Hcy, homocysteine; NC, non-targeted control siRNA;
Nrf2, nuclear factor erythroid-derived 2-like 2; siRNA, small
interfering RNA; t-BHQ, tert-butylhydroquinone |
Discussion
The present study is the first, to the best of our
knowledge, to demonstrate the protective role of exogenous Hcy in
4-HNE-induced cell injury in HepG2 cells. Furthermore, the present
study investigated the underlying molecular mechanism of this
process, which involves an increase in GSH expression mediated by
the antioxidant transcriptional factor Nrf2.
Previous studies indicated that circulating and
hepatic Hcy levels were elevated in alcohol-fed mice, and aberrant
Hcy metabolism elicited oxidative stress and hepatic GSH depletion,
which has been demonstrated in both experimental animals and in
humans (21,22). One study indicated that Hcy
treatment was able to trigger acute GSH depletion in HepG2 cells
within 45 min (6). By contrast,
increased GSH expression that was induced by Hcy has also been
demonstrated in HepG2 cells (9)
and macrophages (10); this may be
due to increased activity via the transsulfuration pathway or de
novo synthesis of GSH. Consistent with these reports, the
present study demonstrated that 24 h Hcy treatment increased the
levels of intracellular GSH expression in a concentration-dependent
manner. GSH is a crucial mediator in various antioxidant responses;
therefore, the data suggested that the GSH metabolism-related amino
acid Hcy may be involved in the antioxidant process, and may
protect against oxidative stress-induced cell injury in HepG2
cells.
HHcy is a risk factor for cardiovascular disease and
ALD, and patients with HHcy may develop hepatic steatosis and
fibrosis (1,3,23,24).
However, the balance of Hcy metabolism and its role in the
progression of these diseases is not clearly understood. In
contrast to the adverse effects of elevated Hcy in these
pathological conditions, the present study indicated that exogenous
Hcy treatment may protect HepG2 cells against cell injury induced
by the lipid peroxidation product 4-HNE. Notably, a previous in
vitro investigation indicated that exogenous Hcy exposure had
no effect on HepG2 cell viability (6). Consistent with this observation, the
present study demonstrated that Hcy exposure did not affect HepG2
cell viability in non-stress conditions. Also of note, the GSH
biosynthesis inhibitor BSO significantly lowered the Hcy-induced
increase in GSH expression levels and the Hcy-mediated cellular
protection against 4-HNE. Collectively, these data indicated that
Hcy protects against 4-HNE-indued cell injury in HepG2 cells, via
increased expression of intracellular GSH levels.
Nrf2 is critical for maintaining the GSH redox state
and protecting cells against oxidative stress (19). High levels of Hcy treatment
suppressed Nrf2-dependent antioxidant protection, but lower levels
of Hcy (100 µM) upregulated the expression of Nrf2 in U373
astroglial cells (25). In
addition, HepG2 cells exposed to 50 µM Hcy exhibited an increase in
Nrf2 protein expression in a time-dependent manner (9). In the present study, HepG2 cells
exposed to Hcy (0–100 µM) for 24 h exhibited increased Nrf2 protein
expression in a concentration-dependent manner. These results
indicated that nontoxic concentrations of Hcy may activate the
antioxidant transcriptional factor Nrf2 in HepG2 cells.
Previous studies have reported that Nrf2 affects GSH
homeostasis by regulating de novo synthesis and salvage
pathways, to protect cells against oxidative stress (15,16,25,26).
Consistent with these reports, the present study demonstrated that
the Nrf2 activator t-BHQ increased total intracellular GSH levels
in a concentration-dependent manner. By contrast, Nrf2-siRNA
treatment inhibited the Hcy-induced GSH elevation. Furthermore,
Nrf2-siRNA treatment reduced the ability of Hcy to protect against
4-HNE-induced cell injury. Collectively, these data suggested that
Nrf2 mediates Hcy-induced GSH expression and cellular protection in
4-HNE-treated HepG2 cells.
In conclusion, the present study demonstrated that
Hcy treatment may protect HepG2 cells against cell injury induced
by the lipid peroxidation product 4-HNE, by inducing the expression
of GSH, and this was mediated by the antioxidant transcriptional
factor Nrf2. Hcy has been regarded as an independent risk factor
for the progression of arteriosclerosis, hypertension or ALD
(3,24,27,28).
The concentration of Hcy (<100 µM) applied in the present study
represented mild - moderate HHcy, which is usually a characteristic
symptom in the early stages of these diseases; an initial elevation
of Hcy may serve a compensatory protective effect on vulnerable
cells. Therefore, the results indicated that hepatic Hcy elevation
may be involved in an antioxidant mechanism, particularly in the
early stage of these pathological conditions.
Acknowledgements
This work was supported by The National Natural
Science Foundation (grant no. 81370523), The Postdoctoral Science
Foundation Special Project (grant no. 201104420), The China
Postdoctoral Science Foundation General Project (grant no.
20100471022) and The Heilongjiang Young Key Academic Staff support
program (grant no. 1251G039).
References
|
1
|
Ganguly P and Alam SF: Role of
homocysteine in the development of cardiovascular disease. Nutr J.
14:62015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Veeranki S and Tyagi SC: Defective
homocysteine metabolism: Potential implications for skeletal muscle
malfunction. Int J Mol Sci. 14:15074–15091. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang D, Fang P, Jiang X, Nelson J, Moore
JK, Kruger WD, Berretta RM, Houser SR, Yang X and Wang H: Severe
hyperhomocysteinemia promotes bone marrow-derived and resident
inflammatory monocyte differentiation and atherosclerosis in
LDLr/CBS-deficient mice. Circ Res. 111:37–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamat PK, Vacek JC, Kalani A and Tyagi N:
Homocysteine induced cerebrovascular dysfunction: A link to
alzheimer's disease etiology. Open Neurol J. 9:9–14. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma M, Tiwari M and Tiwari RK:
Hyperhomocysteinemia: Impact on neurodegenerative diseases. Basic
Clin Pharmacol Toxicol. 117:287–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mani M, Golmohammadi T, Khaghani S, Zamani
Z, Azadmanesh K, Meshkani R and Pasalar P: Homocysteine induces
heme oxygenase-1 expression via transcription factor Nrf2
activation in HepG2 cell. Iran Biomed J. 17:93–100. 2013.PubMed/NCBI
|
|
7
|
Ji C: Mechanisms of alcohol-induced
endoplasmic reticulum stress and organ injuries. Biochem Res Int.
2012:2164502012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esfandiari F, Medici V, Wong DH, Jose S,
Dolatshahi M, Quinlivan E, Dayal S, Lentz SR, Tsukamoto H, Zhang
YH, et al: Epigenetic regulation of hepatic endoplasmic reticulum
stress pathways in the ethanol-fed cystathionine beta
synthase-deficient mouse. Hepatology. 51:932–941. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mani M, Khaghani S, Mohammadi Gol T,
Zamani Z, Azadmanesh K, Meshkani R, Pasalar P and Mostafavi E:
Activation of Nrf2-antioxidant response element mediated glutamate
cysteine ligase expression in hepatoma cell line by homocysteine.
Hepat Mon. 13:e83942013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bea F, Hudson FN, Neff-Laford H, White CC,
Kavanagh TJ, Kreuzer J, Preusch MR, Blessing E, Katus HA and
Rosenfeld ME: Homocysteine stimulates antioxidant response
element-mediated expression of glutamate-cysteine ligase in mouse
macrophages. Atherosclerosis. 203:105–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu SC: Regulation of glutathione
synthesis. Mol Aspects Med. 30:42–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu SC: Glutathione synthesis. Biochim
Biophys Acta. 1830:3143–3153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Niu LY, Guan YS, Chen YZ, Wu LZ, Tung CH
and Yang QZ: A turn-on fluorescent sensor for the discrimination of
cystein from homocystein and glutathione. Chem Commun (Camb).
49:1294–1296. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Soeur J, Eilstein J, Léreaux G, Jones C
and Marrot L: Skin resistance to oxidative stress induced by
resveratrol: From Nrf2 activation to GSH biosynthesis. Free Radic
Biol Med. 78:213–223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bell KF, Fowler JH, Al-Mubarak B,
Horsburgh K and Hardingham GE: Activation of Nrf2-regulated
glutathione pathway genes by ischemic preconditioning. Oxid Med
Cell Longev. 2011:6895242011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Harvey CJ, Thimmulappa RK, Singh A, Blake
DJ, Ling G, Wakabayashi N, Fujii J, Myers A and Biswal S:
Nrf2-regulated glutathione recycling independent of biosynthesis is
critical for cell survival during oxidative stress. Free Radic Biol
Med. 46:443–453. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ho HK, White CC, Fernandez C, Fausto N,
Kavanagh TJ, Nelson SD and Bruschi SA: Nrf2 activation involves an
oxidative-stress independent pathway in
tetrafluoroethylcysteine-induced cytotoxicity. Toxicol Sci.
86:354–364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jayakumar S, Kunwar A, Sandur SK, Pandey
BN and Chaubey RC: Differential response of DU145 and PC3 prostate
cancer cells to ionizing radiation: Role of reactive oxygen
species, GSH and Nrf2 in radiosensitivity. Biochim Biophys Acta.
1840:485–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen CC, Chen HL, Hsieh CW, Yang YL and
Wung BS: Upregulation of NF-E2-related factor-2-dependent
glutathione by carnosol provokes a cytoprotective response and
enhances cell survival. Acta Pharmacol Sin. 32:62–69. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gong Z, Yan S, Zhang P, Huang Y and Wang
L: Effects of S-adenosylmethionine on liver methionine metabolism
and steatosis with ethanol-induced liver injury in rats. Hepatol
Int. 2:346–352. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Paul R and Borah A: The potential
physiological crosstalk and interrelationship between two sovereign
endogenous amines, melatonin and homocysteine. Life Sci.
139:97–107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schalinske KL and Smazal AL: Homocysteine
imbalance: A pathological metabolic marker. Adv Nutr. 3:755–762.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Medici V, Peerson JM, Stabler SP, French
SW, Gregory JF III, Virata MC, Albanese A, Bowlus CL, Devaraj S,
Panacek EA, et al: Impaired homocysteine transsulfuration is an
indicator of alcoholic liver disease. J Hepatol. 53:551–557. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Steele ML, Fuller S, Patel M, Kersaitis C,
Ooi L and Münch G: Effect of Nrf2 activators on release of
glutathione, cysteinylglycine and homocysteine by human U373
astroglial cells. Redox Biol. 1:441–445. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gong P and Cederbaum AI: Transcription
factor Nrf2 protects HepG2 cells against CYP2E1 plus arachidonic
acid-dependent toxicity. J Biol Chem. 281:14573–14579. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pacana T, Cazanave S, Verdianelli A, Patel
V, Min HK, Mirshahi F, Quinlivan E and Sanyal AJ: Dysregulated
hepatic methionine metabolism drives homocysteine elevation in
diet-induced nonalcoholic fatty liver disease. PLoS One.
10:e01368222015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou Y, Zhao L, Zhang Z and Lu X:
Protective effect of enalapril against methionine-enriched
diet-induced hypertension: Role of endoplasmic reticulum and
oxidative stress. Biomed Res Int. 2015:7248762015. View Article : Google Scholar : PubMed/NCBI
|