Introduction
Trauma is a leading cause of death in people aged
5–44 years (1), and >50% of
early deaths following trauma are due to major hemorrhage (2). The loss of blood during hemorrhagic
shock results in hemodynamic instability, coagulopathy, reduced
tissue perfusion and oxygen delivery, and therefore cellular
hypoxia (3,4). In turn, this results in organ
failure, including the liver (5)
and intestine (6). Management of
hemorrhagic shock includes interventions to control the bleeding,
coagulation support, administration of vasoactive drugs and
resuscitation with fluids to maintain tissue oxygenation (3,4,7).
However, the mortality rate following major hemorrhage remains
high, emphasizing the need for novel treatments. Elucidation of the
cellular alterations induced by hypoxia and major hemorrhage in
animal models may help identify novel therapeutic targets to
minimize the detrimental effects of hemorrhagic shock and therefore
improve survival.
Vascular adhesion protein-1 (VAP-1), a
copper-containing amine oxidase composed of 763 amino acids, is a
member of the semicarbazide-sensitive amine oxidase (SSAO) family
(8). These proteins are primarily
expressed in the vascular endothelial cells, adipose tissue and
smooth muscle cells in humans and a variety of mammals (9,10).
VAP-1 is an ectoenzyme on the vascular endothelial cell surface,
but is additionally expressed in the plasma as soluble VAP-1
(sVAP-1) (11). In the plasma,
sVAP-1 catalyzes the transformation of aromatic and aliphatic
primary amines in the blood into corresponding aldehydes, and
produces H2O2 and NH3 (12–14).
VAP-1 is also a homing-associated molecule, and may mediate
adhesion and effusion between leukocytes and endothelial cells by
antigen-antibody binding (15–20)
or enzyme activity (18,19,21,22).
These dual functions of VAP-1 allow leukocytes to patrol throughout
the body (23). Therefore,
previous studies characterizing VAP-1 have focused mainly on
inflammation.
It has been reported that plasma sVAP-1
concentrations are significantly increased in chronic liver
disease, chronic kidney disease, septic shock and certain other
diseases (24–26). In disease models of peritonitis,
hepatitis, colitis, skin inflammation, stroke, sepsis,
ischemia-reperfusion injury and allograft rejection, anti-VAP-1
monoclonal antibodies (mAbs) and VAP-1 enzyme inhibitors have been
demonstrated to relieve inflammation by preventing leukocyte
migration (17,27–32).
As VAP-1 mediates leukocyte migration and adherence to vascular
endothelium, it has additionally been implicated in cardiovascular
and cerebrovascular diseases, including the dissemination of tumor
cells (33–35) and ischemic stroke (36,37).
Therefore, VAP-1 may prove to be a prognostic biomarker or
potential drug target (23).
However, little is known about VAP-1 regulatory
mechanisms despite increasing evidence suggesting that VAP-1 is
closely associated with the occurrence and development of multiple
diseases. Indeed, VAP-1 expression has been well described in
inflammatory reactions, and inflammation is considered a major
factor that increases sVAP-1 concentration in the plasma. However,
no specific pro-inflammatory factors have been identified that
directly induce VAP-1 expression or stimulate signal transduction
pathways that regulate VAP-1 expression (9,38,39).
Nonetheless, the inflammatory cascade markedly alters the local
microenvironment of endothelial cells, and this may alter their
gene expression to allow them to adapt to the altered environment
(40,41).
In previous animal experiments, it has been
demonstrated that the gene for VAP-1 (AOC3) was
differentially expressed in hepatic and intestinal tissues between
rats surviving severe hemorrhagic shock and those that died within
an hour following induction of shock (42). As hypoxia is one of the major
physiological insults associated with hemorrhagic shock, this
raises the possibility that cellular hypoxia may be an important
factor that regulates VAP-1 expression in the liver and intestine.
Therefore, the present study was designed to assess the importance
of severe shock and cellular hypoxia in the regulation of VAP-1
expression in the liver and intestine. The data suggested that
hypoxia causes upregulation of VAP-1 expression.
Materials and methods
Cell culture
Rat hepatic sinusoidal endothelial cells (RHSECs)
and rat intestinal microvascular endothelial cells (RIMECs; Wuhan
PriCells Biomedical Technology Co., Ltd., Wuhan, China) were
cultured in primary endothelial cell basic culture medium (Wuhan
PriCells Biomedical Technology Co., Ltd.) supplemented with 1%
special additives SUP-0002 (Wuhan PriCells Biomedical Technology
Co., Ltd.), 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and 1% penicillin (Shanghai
Hualan Chemical Technology Co., Ltd., Shanghai, China) and
streptomycin (North China Pharmaceutical Group Corp., Shijiazhuang,
China), at 37°C in a humid environment containing 5%
CO2. As VAP-1/SSAO expression is progressively lost with
passage of cells (43,44), only cells of the first generation
were used in the present study. HEK 293 cells were purchased from
ATCC (Manassas, VA, USA) and cultured in ATCC complete growth
medium, which included ATCC-formulated Eagle's Minimum Essential
Medium (ATCC) and 10% FBS. Cells were maintained in a humidified
environment at 37.0°C with 5% CO2.
Construction of adenovirus vectors and
transduction of adenovirus
The adenovirus vector pAd-IRES-EGFP was constructed
using the Invitrogen BP Recombination and LP Recombination systems
to obtain the adenovirus vector pAd-AOC3-IRES-EGFP, which was used
for overexpression of rat AOC3 (Thermo Fisher Scientific,
Inc.; AOC3 GeneID: NM_031582.2). The vector did not contain
the endogenous rat AOC3 promoter. The pAd-AOC3-IRES-EGFP
adenovirus vector was used to transfect HEK 293 cells for virus
amplification. Cells were transfected using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol.
RHSECs and RIMECs were divided into three
experimental groups: Control, which received no adenoviral
transduction; pAd-IRES-EGFP, transduced with an empty adenovirus
vector (pAd-IRES-EGFP); and pAd-AOC3-IRES-EGFP, transduced with an
adenovirus vector for AOC3 overexpression
(pAd-AOC3-IRES-EGFP). In the two transduction groups, cells in the
logarithmic growth phase were plated on 6-well cell culture plates
at a density of 5×104 cells/well and cultured in
complete medium (primary endothelial cell basic culture medium,
supplemented as above) for 24 h. The medium was replaced with fresh
medium and the cells were transduced (multiplicity of infection
[MOI]=10) with the adenovirus vector for 24 h, and subsequently
cultured under either normoxic (95% air, 5% CO2) or
hypoxic (95% N2, 5% CO2) conditions for 24 h.
AOC3 mRNA expression and VAP-1 protein expression were
subsequently determined using reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting,
respectively.
Animals and grouping
Healthy specific pathogen-free (SPF) male
Sprague-Dawley rats (n=60; weight, 200–220 g; age, 8–9 weeks), were
purchased from Sino-British Sippr/BK Lab Animal Ltd. (Shanghai,
China) and housed for >1 week in a SPF animal room at 20–25°C
and in a 12-h light/dark cycle, to allow for adaptation prior to
experiments. Rats were fed until they reached a desired weight
(300–350 g), after which surgery was performed.
The experiment was repeated 5 times; for each
experiment 12 rats were randomly divided into six groups (n=2 per
group), and each group therefore contained a total of 10 rats
following all 5 repeats. The six groups were as follows: Sham (sham
operation); hemorrhagic shock (HS; hemorrhagic shock with no
resuscitation); HS/R group (hemorrhagic shock/resuscitation
following hemorrhagic shock); sham+2-bromoethylamine (2-BEA;
treatment with the SSAO enzyme inhibitor 2-BEA followed by sham
surgery); HS+2-BEA (treatment with 2-BEA, followed by hemorrhagic
shock with no resuscitation); and HS/R+2-BEA (treatment with 2-BEA,
followed by hemorrhagic shock and resuscitation).
The present study was approved by the Institutional
Animal Care and Use Committee of the Second Military Medical
University (SMMU; Shanghai, China) of the People's Liberation Army
(protocol number 12106). The surgical procedures were conducted at
the Experimental Animal Center, and other work at the Laboratory of
the College of Pharmacy, SMMU (Shanghai, China).
Treatment with 2-BEA prior to
surgery
Starting 4 weeks prior to surgery, rats in the
sham+2-BEA, HS+2-BEA and HS/R+2-BEA groups received intraperitoneal
injections of the reversible competitive SSAO enzyme inhibitor,
2-BEA (20 mg/kg daily; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) (45,46). Rats in the sham, HS and HS/R groups
received daily intraperitoneal injections of the same volume of
deionized water. The body weight of the rats was measured daily,
and the drug dosage was adjusted according to the body weight.
Rat model of hemorrhagic shock and
resuscitation
Surgery was performed 4 weeks following treatment
with 2-BEA or vehicle (water). Rats were fasted for 12 h prior to
surgery, but had free access to water. The rat model of hemorrhagic
shock was established using a modification of Wigger's method
(42). Each rat was anesthetized
by intraperitoneal injection of a 3% solution of sodium
pentobarbital (30 mg/kg; Sigma-Aldrich; Merck KGaA). Rats in the
HS/R group were injected with sodium pentobarbital (20 mg/kg) again
prior to resuscitation, and all rats were sacrificed under
anesthetic, following completion of the experimental procedure.
The left carotid artery was dissected and connected
to a polygraph (MPA-2000; Shanghai Alcott Biotech Co., Ltd.,
Shanghai, China) to allow monitoring of blood pressure. In
addition, the right femoral artery and vein were dissected and
intubated, and the femoral artery was connected to a micro-injector
(ALC-IP900; Shanghai Alcott Biotech Co., Ltd.) for drawing blood.
The tube in the femoral vein was temporarily locked, and was
available for transfusion of blood following shock. Rats in the
sham group received only femoral artery and vein intubation; no
blood was drawn or transfused.
Once a stable state had been reached 10 min
following intubation, blood was drawn from the right femoral artery
of rats in the HS group at 2 ml/kg/min, until the mean arterial
pressure (MAP) reached ≤30 mmHg. Repeat bloodletting was performed
when the MAP reached ≥40 mmHg, and multiple drawings of blood were
performed to maintain the MAP at 30–40 mmHg for 1 h, with no
infusions given. The total volume of blood withdrawn was measured
and recorded. Rats in the HS/R group were treated similarly to
animals in the HS group. However, once a low MAP had been
maintained for 1 h, a transfusion was given consisting of whole
blood (half the original blood loss volume) together with lactated
Ringer's solution (volume, 2X the original blood-loss volume;
Baxter, Deerfield, MA, USA). Following this, rat status and MAP
changes were observed for 1 h.
Following completion of the above procedures, a
laparotomy was performed on sterile drapes. The hepatic left
lateral lobe was cut, and 3 ml of blood was quickly drawn from the
femoral artery for measurements of the partial pressures of
O2 and CO2 (i-STAT 300 blood gas analyzer;
Abbott Laboratories, Chicago, IL, USA). Subsequently, the animals
were sacrificed through further bleeding. Mortality was confirmed
by monitoring the blood pressure and heart rate, via the left
carotid artery. Specimens of the liver were taken and sliced into
small sections (~0.5×0.5 cm). Specimens of the small intestine
(including the mesentery) were obtained, cut along the longitudinal
axis, and then cut into small sections (~1×1 cm). All specimens
were rinsed three times in phosphate-buffered saline (PBS) and
stored in liquid nitrogen. Serum samples were prepared by
centrifugation of the blood (4°C, 3,200 × g, 10 min), and stored in
liquid nitrogen.
The experiment was repeated to determine the 24 h
survival rate with another set of healthy SPF male Sprague-Dawley
rats (n=120; weight, 200–220 g; age, 8–9 weeks), purchased from
Sino-British Sippr/BK Lab Animal Ltd. and housed as aforementioned.
Rats were fed until they reached a desired weight (300–350 g), and
were divided into the following four groups (n=30/group): HS, HS/R,
HS+2-BEA, HS/R+2-BEA. Rats underwent the same surgical procedures
as aforementioned, whereby the rats were aneasthetized and
monitored during the intubation/shock/recovery process. When these
processes were completed, rats that survived the procedure were
extubated, placed into cages, and monitored up to 24 h.
RT-qPCR
Cells (RHSECs or RIMECs) were seeded into 6-well
cell culture plates and were seeded at 5×104 cells/well, and
transduced as aforementioned. Total RNA was isolated from the
cells, using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
Samples of hepatic or intestinal tissue (100 mg; which had been
stored in liquid nitrogen) were transferred into a clean 10 ml
glass tube containing 1 ml TRIzol and homogenized. The above
samples were transferred into 1.5 ml tubes and centrifuged at
12,000 × g at 4°C for 5 min. RNA purity was determined by
spectroscopy [ratio of optical density at 260 nm to that at 280 nm
(OD260/OD280)=1.8–2.0] and agarose gel
electrophoresis (1.0% agarose, 1 ng total RNA/lane). Total RNA (500
ng) was reverse transcribed into cDNA using Reverse Transcriptase
M-MLV. (Takara Bio, Inc., Otsu, Japan). qPCR (TP800 Thermal Cycler
Dice Real Time System; Takara Bio, Inc.) was performed with 2 µl
cDNA, 0.4 µl each of forward and reverse primer, and Takara SYBR
Premix Ex Taq (DRR041A; Takara Bio, Inc.) in a final reaction
volume of 20 µl. The sequence-specific primers for AOC3 and
β-actin (TaKaRa Bio Inc.) were as follows: Forward,
5′-CCTAAGGCCAACCGTGAAAAGATG-3′ and reverse,
5′-GTCCCGGCCAGCCAGGTCCAG-3′ for AOC3; forward,
5′-GCTCCGGCGACACCACTCAG-3′ and reverse, 5′-CGCCAGCACCGAAGAAGAAAG-3′
for β-actin.
The cycling conditions were: 50°C for 2 min, initial
denaturation at 95°C for 10 min followed by 40 cycles of
denaturation at 90°C for 15 sec, and annealing at 60°C for 1 min.
Quantitation cycle (Cq) values of the target gene AOC3 were
normalized to those of β-actin, and relative mRNA expression levels
were calculated using the 2−∆∆Cq method (47). The experiment was repeated three
times.
Western blotting
Cells were lysed with radioimmunoprecipitation assay
buffer (Invitrogen, Thermo Fisher Scientific, Inc.) and centrifuged
at 18,500 × g for 15 min at 4°C. Proteins were extracted from
hepatic and intestinal tissues using Tissue Protein Extraction
Reagent (T-PER; Pierce, Rockford, IL, USA). Frozen tissue (~100 mg)
was added to a mixture of 1 ml T-PER reagent and protease inhibitor
cocktail (Abcam, Cambridge, UK), homogenized, and centrifuged at
4°C for 5 min at 10,000 × g. Total protein content was determined
with a Bicinchoninic Acid protein assay kit (Pierce), and equal
amounts of proteins (100 µg) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The non-specific sites on each blot were blocked with 5% milk
powder diluted in Tris-buffered saline (TBS) with 0.05% Tween-20
(TBST) for 1 h at room temperature. Following washing with TBST,
membranes were incubated overnight at 4°C in the presence of
primary antibodies against glyceraldehyde 3-phosphate dehydrogenase
(rabbit mAb; 1:1,000; #2118; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and VAP-1 (mouse mAb; 1:1,000; ab81718; Abcam).
Blots were subsequently incubated at room temperature for 1 h with
the appropriate horseradish peroxidase (HRP)-conjugated secondary
antibody (anti-rabbit immunoglobulin (Ig)-G (#7074; 1:3,000) or
anti-mouse IgG (#7076; 1:6000); Cell Signaling Technology, Inc.),
and developed using enhanced chemiluminescence western blotting
substrate (Pierce). Band intensity was quantified using TotalLab
Quant v12.0 software (TotalLab Ltd., Newcastle upon Tyne, UK).
Experiments were repeated twice.
Determination of serum SSAO
activity
The Fluorescent SSAO Detection kit (Cell Technology,
Inc., Mountain View, CA, USA) was used to determine SSAO activity
in various specimens of serum, in accordance with the
manufacturer's protocol. Briefly, serum samples were diluted in 1X
reaction buffer, and a monoamine oxidase-B inhibitor was added 30
min prior to the assay, which was performed in the dark in 96-well
plates. Subsequently, 100 µl sample or positive control were loaded
in each well, followed by the addition of 100 µl reaction cocktail.
The mixture was incubated at 37°C for 1–3 h, and the plates were
read using a DU-730 ultraviolet fluorescence spectrophotometer
(Beckman Coulter, Inc., Brea, CA, USA) with excitation at 530 nm
and emission at 590 nm. Serum SSAO activity was calculated
according to a standard curve and expressed in mU/ml. One unit
represents the amount of enzyme required to oxidize 1 µmol of SSAO
substrate (benzylamine) in 1 min at 25°C.
Immunohistochemistry
Immunohistochemistry experiments (labeled
streptavidin-biotin method) were performed using a
Streptavidin-Peroxidase Immunohistochemical Detection kit (SP-9000;
ZSGB-BIO, Beijing, China). In brief, formalin-fixed,
paraffin-embedded sections (thickness: 4 µm) were dewaxed with
fresh xylene and subjected to gradient hydration (100% ethanol, 95%
ethanol, 60% ethanol, purified water). Following elimination of
endogenous peroxidase activity (incubation with 3%
H2O2 for 15 min at room temperature),
microwave antigen retrieval and incubation with normal goat serum
(Abcam) for 15 min at 37°C. The sections were then incubated
overnight at 4°C with a primary antibody against VAP-1 (rabbit
polyclonal; 1:200; ab187202; Abcam). Following 3 washes with PBS,
the specimens were incubated with a biotin-conjugated goat
anti-rabbit (1:1,000; ab6720; Abcam) secondary antibody for 15 min
at 37°C, followed by streptavidin-peroxidase for 15 min at 37°C.
The slides were stained with 3,3-diaminobenzidine for 10 min at
room temperature, and re-stained with hematoxylin. The specimens
were subsequently dehydrated in a graded series of ethanol (30, 70
and 100%), sealed with neutral resin, and observed under a light
microscope (Olympus Corporation, Tokyo, Japan). A total of 10
fields were assessed (magnification, ×100) in three independent
experiments.
Statistical analysis
Statistical analysis was performed using SPSS
software version 17.0 (SPSS, Inc., Chicago, IL, USA). All data are
expressed as the mean ± standard deviation derived from at least
three independent experiments. Statistical significance was
evaluated by one-way analysis of variance followed by the least
significant difference post-hoc test, or the independent samples
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of hypoxia on AOC3 mRNA
expression, VAP-1 protein expression, and SSAO activity in RHSECs
and RIMECs
As tissue hypoxia is a major physiologic insult
arising from hemorrhagic shock, in vitro experiments were
performed to investigate the effects of hypoxia on the mRNA
expression of AOC3 and protein expression of VAP-1 in RHSECs
and RIMECs. Fluorescence microscopy revealed the successful
transduction of the AD-AOC3-IRES-EGFP adenovirus vector into RHSECs
(Fig. 1A) and RIMECs (Fig. 1B).
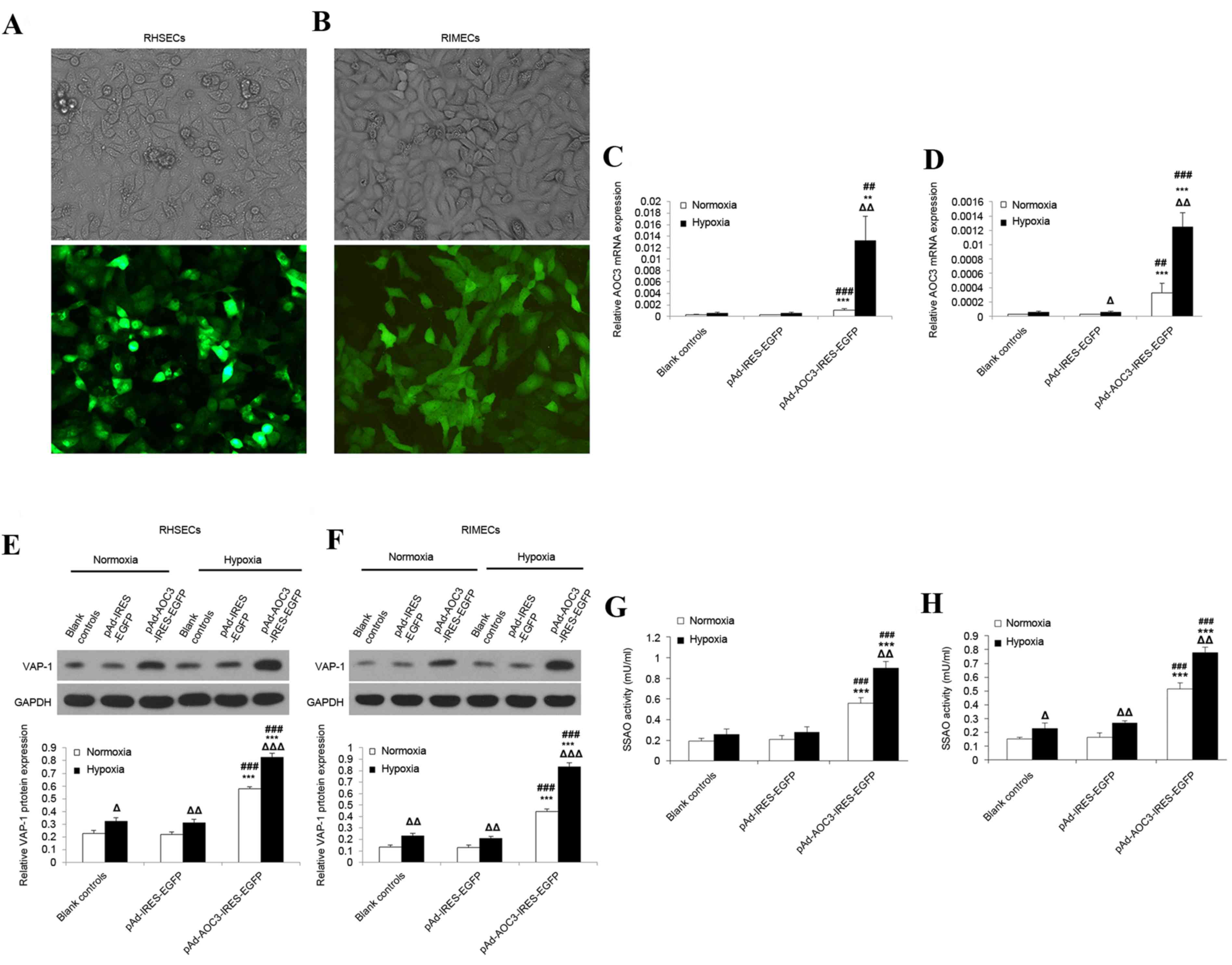 | Figure 1.Effects of hypoxia on VAP-1 protein
expression and AOC3 mRNA expression in RHSECs and RIMECs.
RHSECs and RIMECs were transduced with pAd-AOC3-IRES-EGFP or
pAd-IRES-EGFP adenovirus vectors for 72 h; blank controls were not
exposed to any adenovirus vector. Light and fluorescence microscopy
images confirmed the successful transduction of (A) RHSECs and (B)
RIMECs with pAd-AOC3-IRES-EGFP (magnification, ×200). AOC3
mRNA expression in (C) RHSECs and (D) RIMECs under normoxic and
hypoxic conditions, determined by reverse
transcription-quantitative polymerase chain reaction. β-actin
served as an internal control. VAP-1 protein expression in (E)
RHSECs and (F) RIMECs under normoxic and hypoxic conditions,
determined by western blotting. GAPDH was used as an internal
control. Serum SSAO activity in (G) RHSECs and (H) RIMECs was
determined using a fluorometric assay. Data are presented as the
mean ± standard deviation. *P<0.05, **P<0.01 and
***P<0.001 vs. blank controls; #P<0.05,
##P<0.01 and ###P<0.001 vs.
pAd-IRES-EGFP; ΔP<0.05, ΔΔP<0.01 and
ΔΔΔP<0.001 hypoxia vs. normoxia within the same
group. VAP-1, vascular adhesion protein-1; AOC3, VAP-1 gene;
RHSECs, rat hepatic sinusoidal endothelial cells; RIMECs, rat
intestinal microvascular endothelial cells; SSAO,
semicarbazide-sensitive amine oxidase; pAd-IRES-EGFP, adenovirus
control vector; pAd-AOC3-IRES-EGFP, adenovirus vector for
AOC3 overexpression. |
RT-qPCR revealed that, for RHSECs and RIMECs
cultured under normoxic conditions, AOC3 mRNA expression was
low in blank controls and cells transduced with AD-IRES-EGFP
(RHSECs, Fig. 1C; RIMECs, Fig. 1D). However, RHSECs transduced with
AD-AOC3-IRES-EGFP had significantly increased AOC3 mRNA
expression levels than blank controls (~4.3-fold increase;
P<0.01; n=3) and cells transduced with AD-IRES-EGFP (~4.2-fold
increase; P<0.001; n=3; Fig.
1C). Similarly, RIMECs transduced with AD-AOC3-IRES-EGFP had
significantly increased AOC3 mRNA expression than blank
controls (~12.5-fold increase; P<0.01; n=3) and cells transduced
with AD-IRES-EGFP (~12.5-fold increase; P<0.01; n=3; Fig. 1D). In RHSECs and RIMECs, hypoxia
(culture for 24 h in 0% O2) had little or no effect on
blank controls and cells transduced with AD-IRES-EGFP (Fig. 1C and D). In contrast, hypoxia
caused a significant upregulation of AOC3 mRNA expression in
RHSECs (~12.1-fold increase; P<0.01; n=3) and RIMECs (~3.8-fold
increase; P<0.01; n=3) transduced with AD-AOC3-IRES-EGFP
(Fig. 1C and D, respectively).
Western blotting revealed that under normoxic
conditions, VAP-1 protein expression in RHSECs and RIMECs did not
differ significantly between blank controls and cells transduced
with AD-IRES-EGFP (Fig. 1E and F,
respectively). RHSECs transduced with AD-AOC3-IRES-EGFP had
significantly increased VAP-1 protein expression compared with
blank controls (~3.3-fold increase; P<0.001; n=3) and cells
transduced with AD-IRES-EGFP (~3.4-fold increase; P<0.001; n=3;
Fig. 1E). RIMECs transduced with
AD-AOC3-IRES-EGFP additionally had significantly increased VAP-1
protein expression compared with blank controls (~2.5-fold
increase; P<0.001; n=3) and cells transduced with AD-IRES-EGFP
(~2.6-fold increase; P<0.001; n=3; Fig. 1F). In RHSECs and RIMECs, hypoxia
for 24 h resulted in a moderate (1.4–1.7-fold) but significant
enhancement of VAP-1 protein expression in blank controls
(P<0.05 and P<0.001, respectively; n=3; Fig. 1E and F) and cells transduced with
AD-IRES-EGFP (P<0.001 and P<0.001; n=3; Fig. 1E and F). Hypoxia also caused an
upregulation of VAP-1 protein expression in RHSECs (~1.4-fold
increase; P<0.001; n=3; Fig.
1E) and RIMECs (~1.9-fold increase; P<0.001; n=3; Fig. 1F) transduced with
AD-AOC3-IRES-EGFP.
Under normoxic conditions, SSAO activity in RHSECs
and RIMECs did not differ significantly between blank controls and
cells transduced with AD-IRES-EGFP (Fig. 1G and H, respectively). RHSECs
transduced with AD-AOC3-IRES-EGFP had significantly higher SSAO
activity than blank controls (~2.9-fold increase; P<0.001; n=3)
and cells transduced with AD-IRES-EGFP (~2.7-fold increase;
P<0.001; n=3; Fig. 1G). RIMECs
transduced with AD-AOC3-IRES-EGFP also had significantly higher
SSAO activity than blank controls (~3.4-fold increase; P<0.001;
n=3) and cells transduced with AD-IRES-EGFP (~3.2-fold increase;
P<0.001; n=3; Fig. 1H). Hypoxia
caused an upregulation of SSAO activity in RHSECs (~1.6-fold
increase; P=0.002; n=3) and RIMECs (~1.5-fold increase; P=0.002;
n=3) transduced with AD-AOC3-IRES-EGFP (Fig. 1G and H, respectively).
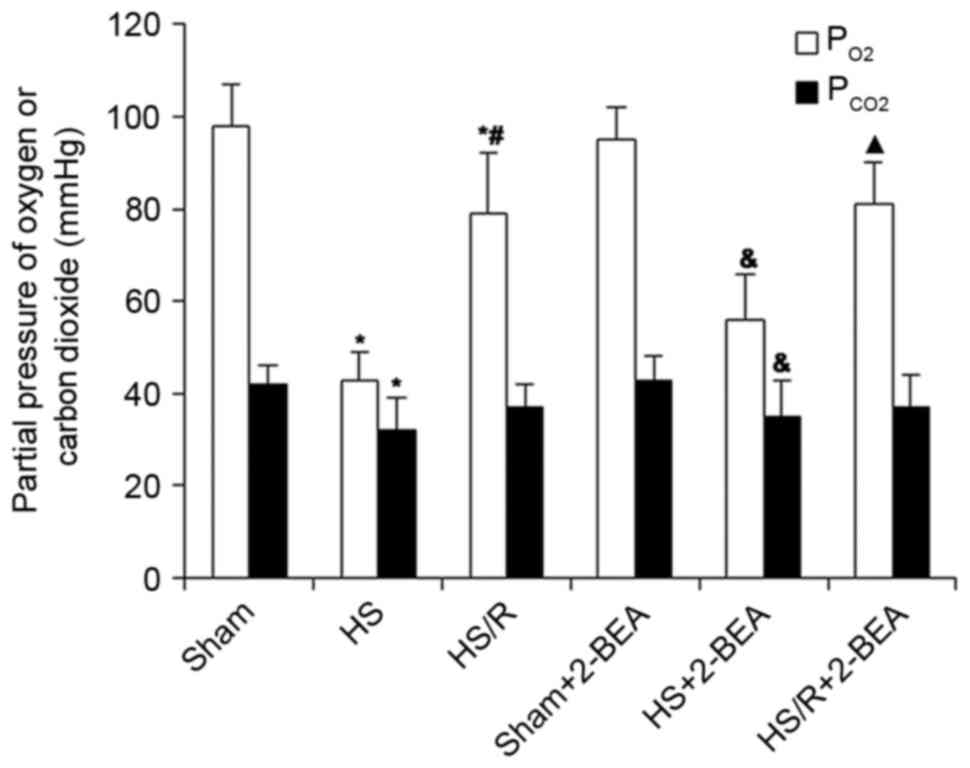
PaO2 and PaCO2
in rats following surgically induced hemorrhagic shock or sham
surgery
In view of the effects of hypoxia in RHSECs and
RIMECs described above, an additional series of experiments was
performed to determine whether changes in AOC3 mRNA
expression and VAP-1 protein expression were observed in
vivo in a rat model of surgically induced hemorrhagic shock.
Normal values for PaO2 and PaCO2 were
measured in the sham group (98±9 and 42±4 mmHg, respectively; n=10;
Fig. 2), whereas those in the HS
group were markedly reduced (43±6 and 32±7 mmHg, respectively;
n=10; Fig 2), consistent with
hemorrhage-induced hypoxia and a compensatory hypocapnia secondary
to hypoxic stimulation of ventilation. The PaO2 and
PaCO2 values in the HS/R group were intermediate between
those of the HS and sham groups (79±13 and 37±5 mmHg, respectively,
n=10; Fig. 2), indicating a
partial reversion of hypoxia by the administration of blood and
fluids. The administration of 2-BEA had no significant effect on
PaO2 and PaCO2 in all three groups
(P>0.05; Fig. 2).
AOC3 mRNA expression and VAP-1 protein
expression in rat hepatic and intestinal tissues following
surgically-induced hemorrhagic shock or sham surgery
The expression of AOC3 mRNA in hepatic tissue
was significantly increased in the HS (~4.0-fold increase;
P<0.001 n=10; Fig. 3A) and HS/R
(~3.2-fold increase; P<0.001; n=10; Fig. 3A) groups compared with the sham
group. Notably, mRNA expression of AOC3 was significantly
reduced in the HS/R group than in the HS group (P<0.05; Fig. 3A). VAP-1 protein expression was
additionally significantly increased in the HS (~2.2-fold increase;
P<0.001; n=10; Fig. 3B) and
HS/R (~1.8-fold increase; P<0.001; n=10; Fig. 3B) groups compared with the sham
group. However, although the level of VAP-1 expression reduced in
the HS/R group compared with the HS group, the difference was not
statistically significant (P=0.073; Fig. 3B).
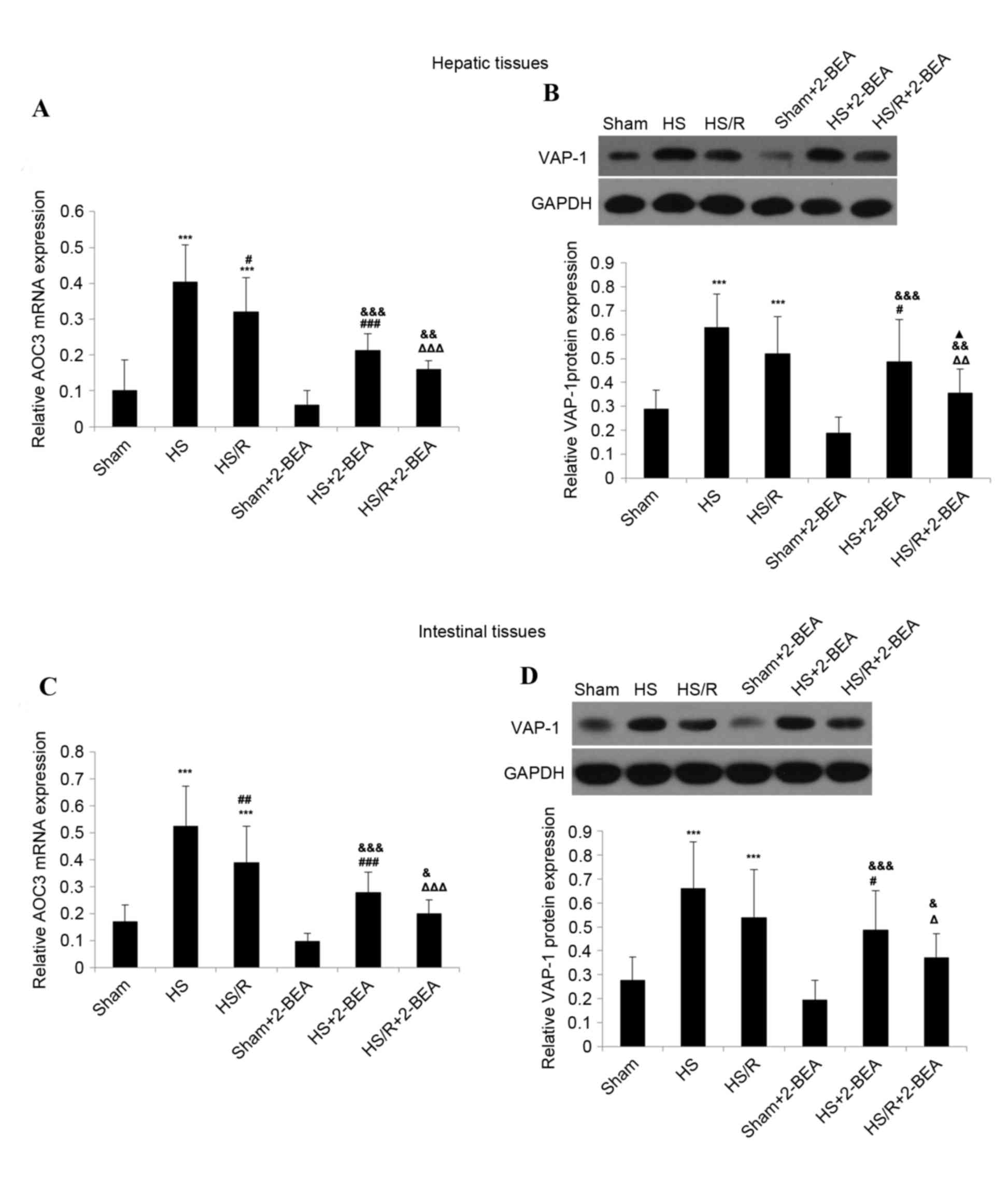 | Figure 3.VAP-1 protein expression and
AOC3 mRNA expression in hepatic and intestinal tissues from
rats of the various experimental groups, and the effects of 2-BEA.
(A) AOC3 mRNA expression and (B) VAP-1 protein expression in
rat hepatic tissue. (C) AOC3 mRNA expression and (D) VAP-1
protein expression in rat intestinal tissue. β-actin and GAPDH were
used as internal controls for reverse transcription-quantitative
polymerase chain reaction and western blotting, respectively. Data
are presented as the mean ± standard deviation (n=10 per group).
***P<0.001 vs. sham group; #P<0.05,
##P<0.01 and ###P<0.001 vs. HS group;
ΔP<0.05, ΔΔP<0.01 and
ΔΔΔP<0.001 vs. HS/R group; &P<0.05,
&&P<0.01 and
&&&P<0.001 vs. sham-2-BEA group;
▲P<0.05 vs. HS-2-BEA group. VAP-1, vascular adhesion protein-1;
AOC3, VAP-1 gene; 2-BEA, 2-bromoethylamine; sham, sham
operation; HS, surgically-induced hemorrhagic shock with no
resuscitation; HS/R, surgically-induced hemorrhagic shock with
resuscitation. |
Similar results were obtained in intestinal tissue.
AOC3 mRNA expression was significantly increased in the HS
(~3.1-fold increase; P<0.001; n=10; Fig. 3C) and HS/R (~2.3-fold increase;
P<0.001; n=10; Fig. 3C) groups
compared with the sham group, and significantly lower in the HS/R
group than in the HS group (P<0.01; Fig. 3C). VAP-1 protein expression was
additionally significantly increased in the HS (~2.4-fold increase;
P<0.001; n=10; Fig. 3D) and
HS/R (~2.0-fold increase; P<0.001; n=10; Fig. 3D) groups compared with the sham
group, but did not differ significantly between the HS/R and HS
groups (P=0.089; Fig. 3D).
Effect of the SSAO enzyme inhibitor,
2-BEA, on AOC3 mRNA expression and VAP-1 protein expression in rat
hepatic and intestinal tissues
For hepatic and intestinal tissues, there were no
significant differences in AOC3 mRNA expression or VAP-1
protein expression between the sham and sham-2-BEA groups (Fig. 3). However, in hepatic tissue,
AOC3 mRNA expression was reduced ~1.9-fold in the HS-2BEA
group compared with the HS group (P<0.001; n=10; Fig. 3A), and ~2.0-fold in the HS/R-2-BEA
group compared with the HS/R group (P<0.001; n=10; Fig. 3A). Consistent with the changes in
mRNA expression, VAP-1 protein expression in hepatic tissue was
decreased ~1.3-fold in the HS-2BEA group compared with the HS group
(P<0.05; n=10; Fig. 3B), and
~1.5-fold in the HS/R-2BEA group compared with the HS/R group
(P<0.01; n=10; Fig. 3B).
Similar findings were obtained in intestinal tissue. AOC3
mRNA expression in intestinal tissue was decreased ~1.9-fold in the
HS-2BEA group compared with the HS group (P<0.001; n=10;
Fig. 3C), and ~1.9-fold in the
HS/R-2BEA group compared with the HS/R group (P<0.001; n=10;
Fig. 3C). VAP-1 protein expression
in intestinal tissue was reduced ~1.4-fold in the HS-2BEA group
compared with the HS group (P<0.05; n=10; Fig. 3D), and ~1.5-fold in the HS/R-2BEA
group compared with the HS/R group (P<0.05; n=10;
Fig. 3D).
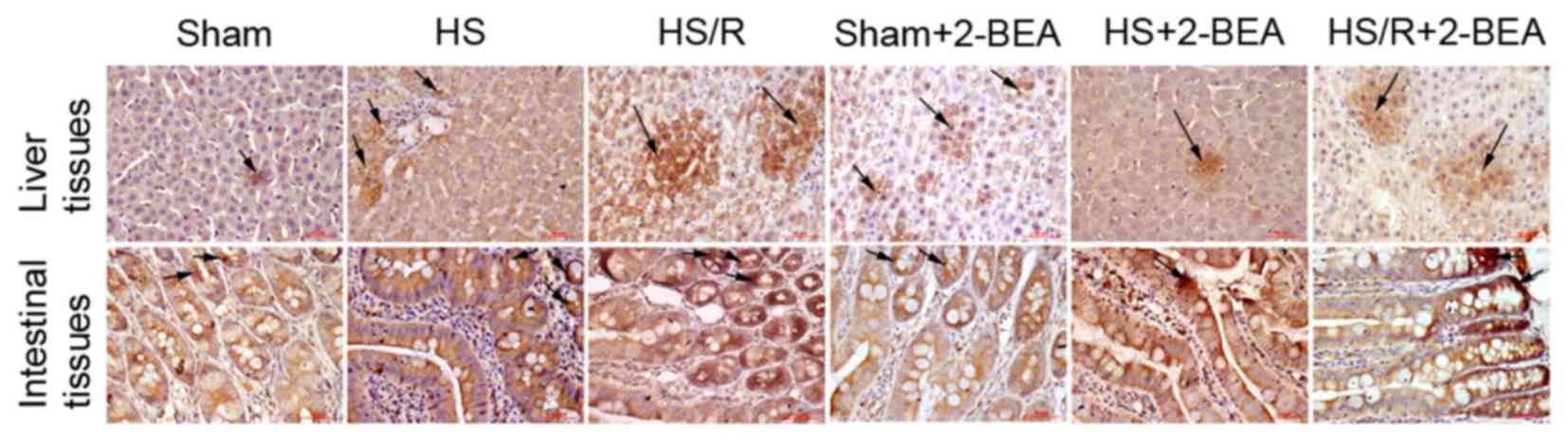
Histological observations of VAP-1
protein expression
Representative immunohistologically stained sections
comparing VAP-1 protein expression between the groups are presented
in Fig. 4. VAP-1 was mainly
distributed in the vascular endothelial cells, primarily in tissues
rich in small blood vessels including the liver blood sinus and
intestinal mucosal vascular endothelium. The results were similar
to those obtained via western blotting.
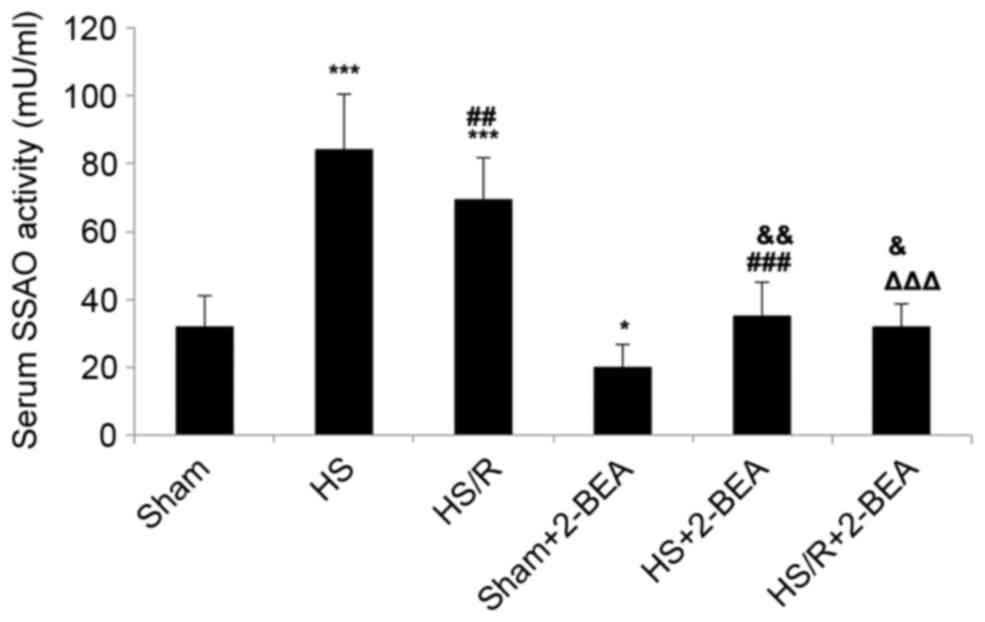
Serum SSAO activity in rats following
surgically-induced hemorrhagic shock or sham surgery, and the
effects of 2-BEA
Further experiments were undertaken to establish
whether alterations in AOC3 mRNA expression and VAP-1
protein expression described above were reflected by alterations in
serum SSAO activity in rats. Serum SSAO activity was significantly
increased in the HS (~2.7-fold increase; P<0.001; n=10; Fig. 5) and HS/R (~2.2-fold increase;
P<0.001; n=10; Fig. 5) groups
than in the sham group, and significantly reduced in the HS/R group
than in the HS group (P<0.01; Fig.
5). Compared with the corresponding groups not administered
with an SSAO inhibitor, 2-BEA was associated with significant
reductions in serum SSAO activity in the sham-2-BEA (~1.6-fold
reduction; P<0.05; n=10), HS-2-BEA (~2.6-fold reduction;
P<0.001; n=10) and HS/R-2-BEA (~2.2-fold reduction; P<0.001;
n=10) groups (Fig. 5).
Effect of the SSAO inhibitor, 2-BEA,
on the 24 h survival rate of rats following surgically-induced
hemorrhagic shock or sham surgery
The 24 h survival rate was 100% (30/30 animals) in
the sham and sham-2-BEA groups. In contrast, the 24 h survival rate
was only 40% (12/30) in the HS group, but was 83.3% (25/30) in the
HS/R group. Furthermore, the 24 h survival rate was 56.7% (17/30)
in the HS-2-BEA group and 86.6% (26/30) in the HS/R-2-BEA group
(Table I).
 | Table I.Effect of semicarbazide-sensitive
amine oxidase enzyme inhibitor 2-BEA on 24 h survival rate in a rat
model of hemorrhagic shock and resuscitation. |
Table I.
Effect of semicarbazide-sensitive
amine oxidase enzyme inhibitor 2-BEA on 24 h survival rate in a rat
model of hemorrhagic shock and resuscitation.
| Group
(n=30/group) | 24 h survival rate,
n (%) |
|---|
| HS | 12 (40.0) |
| HS/R | 25 (83.3) |
| HS+2-BEA | 17 (56.7) |
| HS/R+2-BEA | 26 (86.6) |
Discussion
The main findings of the present study were that
AOC3 mRNA expression and VAP-1 protein expression were
upregulated by hypoxia in RHSECs and RIMECs cultured in
vitro, and in hepatic and intestinal tissue obtained from an
in vivo rat model of hemorrhagic shock. Furthermore, hypoxia
additionally enhanced serum SSAO activity in rats subjected to
experimental hemorrhagic shock. In addition, the SSAO inhibitor,
2-BEA, reduced AOC3 mRNA expression and VAP-1 protein
expression in hepatic and intestinal tissues from rats subjected to
experimental hemorrhagic shock, and may improve 24 h survival in
rats that did not receive resuscitation following hemorrhagic
shock. Taken together, these observations suggest that
hypoxia-induced upregulation of VAP-1 may be one of the mechanisms
that contributes to the detrimental physiological effects of
hemorrhagic shock.
VAP-1 exists in two distinct forms, as a
transmembrane protein in endothelial cells and as a serum protein
(9–11). For this reason, VAP-1 protein
expression was studied in solid organs containing blood vessels
(liver and intestine) and in the serum. The liver and small
intestine (including mesentery) were chosen as parenchymal organs
that are known to be susceptible to damage following hemorrhagic
shock (5,6). The liver, which collects venous blood
from abdominal organs, contains abundant vascular endothelial
tissue in the hepatic sinusoids (11,24),
while high endothelial venules in the mesentery of the small
intestine are regarded as having high levels of VAP-1 (9,45,46).
In addition, hepatic and intestinal tissues in the abdominal cavity
are easily collected, adding to their suitability for the in
vivo study of VAP-1 expression.
The in vitro experiments were performed using
RHSECs and RIMECS transduced with the AOC3 gene, since the
expression of AOC3/VAP-1 by these cells in culture is
normally very low (18,43). The use of a recombinant adenovirus
expressing VAP-1 permitted the study of the effects of hypoxia on
VAP-1 expression. These in vitro studies revealed that
culture of RHSECs or RIMECS under hypoxic conditions resulted in a
significant upregulation of AOC3 mRNA expression and VAP-1
protein expression. This suggested that hypoxia directly induces an
increase in VAP-1 levels via enhanced transcription of the
AOC3 gene. These data were supported by those obtained from
the in vivo rat model of hemorrhagic shock: Maintenance of a
low MAP (30–40 mmHg) for 1 h resulted in an upregulation of
AOC3 mRNA expression and VAP-1 protein expression in hepatic
and intestinal tissue, and this enhanced expression was inhibited
by prompt resuscitation with blood and fluids. These findings
indicated that regulation of VAP-1 expression occurred rapidly over
a short timescale, and were consistent with a previous study, which
reported that hepatic and intestinal AOC3 expression
differed between rats surviving hemorrhagic shock and those that
died within 1 h (42). Notably, a
previous investigation in endothelial cells expressing VAP-1
demonstrated that VAP-1 increased the susceptibility of these cells
to deprivation of oxygen and glucose, and enhanced tissue damage
(48). As VAP-1 also mediates
leukocyte recruitment via its SSAO activity, the enhanced VAP-1
expression following hemorrhagic shock/hypoxia observed in the
present study may increase the susceptibility of the liver and
intestines, and potentially other organs, to damage and
necrosis.
The mechanism by which hypoxia upregulates
AOC3 mRNA and VAP-1 protein expression remains unclear.
Acute hypoxia rapidly activates endothelial cells to release
inflammatory mediators and growth factors, and one hypothesis is
that the regulation of VAP-1 expression by hypoxia may be mediated
by hypoxia-inducible factors (HIFs). HIFs are key transcription
factors that alter gene expression in hypoxic environments, and
activate multiple cytokines and chemokines via regulation of signal
transduction pathways (49,50).
However, the pathophysiologic processes underlying severe
hemorrhagic shock are complicated and its consequences are
reflected in multiple aspects, including alterations in the immune
response, coagulation disorders, initiation of a variety of signal
transduction pathways and release of numerous pro-inflammatory and
anti-inflammatory cytokines. Therefore, cellular hypoxia
constitutes just one step in this complex process, and the
upregulation of VAP-1 expression may be due to factors other than
hypoxia. Nonetheless, the in vitro experiments support the
idea that hypoxia is the primary factor that causes increased
expression of VAP-1.
A notable finding of the in vivo experiments
was that hemorrhagic shock was associated with an increase in the
activity of serum SSAO, and that this enhancement was attenuated by
prompt resuscitation. This suggested that hemorrhagic shock results
in the detachment of VAP-1 from the surface of endothelial cells,
raising the possibility that circulating VAP-1 may be involved in
severe shock. In support of these observations, deprivation of
oxygen and glucose has been demonstrated to stimulate the release
of sVAP-1, via a mechanism partially mediated by matrix
metalloproteinase-2 (48). In
addition, an increase in plasma sVAP-1/SSAO activity following
intracranial hemorrhage has been reported in humans, and serum
sVAP-1/SSAO activity <2.7 pmol/min/mg is an independent
predictor of neurologic improvement following hemorrhagic stroke
(51).
For competitive inhibitors, increasing the dose of
the drug results in the enzyme being almost completely inhibited.
2-BEA is a reversible competitive enzyme inhibitor, and enzyme
function is associated with the concentration of 2-BEA. The action
time of 2-BEA is short and it needs regular, continuous
administration in order to maintain stable blood drug concentration
and effect (52,53). In the pre-experiment, three doses
were tested: 10, 20 and 30 mg/kg. There were no significant
differences in SSAO activity between the 10 mg/kg and control
groups. The mortality rate of rats injected with 30 mg/kg was high.
Therefore, the 20 mg/kg dose was selected. As expected, the
administration of the SSAO inhibitor, 2-BEA, resulted in a decrease
in serum SSAO activity in rats, irrespective of whether they were
subjected to hemorrhagic shock (with or without resuscitation).
However, a notable finding in animals that had received
surgically-induced hemorrhagic shock was that 2-BEA additionally
reduced the expression of AOC3 mRNA and VAP-1 protein in
intestinal and liver tissues, while no such effect of 2-BEA was
observed in the sham group. This implied that, at least under the
conditions of hemorrhagic shock, a degree of positive feedback
exists whereby VAP-1 is able to upregulate the expression of its
gene. The potential mechanisms underlying this effect remain
unknown. However, previous studies have demonstrated that VAP-1 not
only serves a pro-inflammatory function through its SSAO activity,
but rapidly regulates the expression of inflammatory factors
including E-selectin, intracellular adhesion molecule 1 (ICAM-1),
vascular cell adhesion protein-1 and C-X-C motif chemokine ligand
8, tvia activation of signaling pathways including nuclear
factor-κB, phosphoinositide 3-kinase and mitogen-activated protein
kinases (54,55). Nevertheless, there is a possibility
that chronic treatment with 2-BEA may alter other molecular targets
or molecular pathways responsible for the decreased AO3C and VAP-1
expression. Further studies are warranted to elucidate the
mechanisms by which the SSAO activity of VAP-1 leads to
upregulation of AOC3 mRNA and VAP-1 protein expression
during hemorrhagic shock.
Another notable finding was that 2-BEA improved 24 h
survival in rats subjected to hemorrhagic shock without
resuscitation. This finding is consistent with numerous studies
demonstrating a benefit of SSAO inhibition in intracranial
hemorrhage/ischemia. In a rat model of subarachnoid hemorrhage,
SSAO inhibition has been reported to decrease leukocyte trafficking
and improve neurologic outcome (56), and prevent dysfunction of
arteriolar dilation, potentially via suppression of neutrophil
recruitment (57). Similarly, SSAO
inhibition has been demonstrated to reduce neutrophil extravasation
and infarct volume in a rat middle cerebral artery occlusion model
(58). In addition, inhibitors of
SSAO have been demonstrated to reduce neurobehavioral deficits and
brain edema following intracerebral hemorrhage, potentially via a
mechanism involving reduced neutrophil infiltration,
microglia/macrophage activation, and expression of ICAM-1, monocyte
chemoattractant protein-1 and tumor necrosis factor-α (59). Previous clinical data have revealed
increased VAP-1 expression in the inflammatory response of local
vasculature (9,11,18).
Furthermore, anti-VAP-1 mAbs and SSAO inhibitors have been
demonstrated to partially restrain endovascular extravasation of
lymphocytes, neutrophils, and macrophages into the peritoneum,
liver, pancreas and other tissues during an inflammatory response
(28,29,60),
and to suppress the inflammatory response in conditions including
colitis, skin infection, ischemia-reperfusion injury, sepsis,
pulmonary infection, arthritis and experimental allergic
cerebrospinal meningitis (19,22,30,31).
Thus, VAP-1 is an important factor in the inflammatory cascade
(17,61), and its upregulation during
hemorrhagic shock may contribute to the detrimental processes that
result in tissue damage.
In conclusion, the present study provided evidence
that VAP-1 expression is upregulated by hypoxia during acute
hemorrhagic shock, and that inhibition of VAP-1 may be beneficial
in the setting of hemorrhagic shock. Further studies are required
to confirm these findings and to establish whether VAP-1 may be a
valid target for the development of novel therapies for hemorrhagic
shock.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81101419).
References
|
1
|
Kauvar DS, Lefering R and Wade CE: Impact
of hemorrhage on trauma outcome: An overview of epidemiology,
clinical presentations, and therapeutic considerations. J Trauma.
60 6 Suppl:S3–S11. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rossaint R, Cerny V, Coats TJ, Duranteau
J, Fernández-Mondéjar E, Gordini G, Stahel PF, Hunt BJ, Neugebauer
E and Spahn DR: Key issues in advanced bleeding care in trauma.
Shock. 26:322–331. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Angele MK, Schneider CP and Chaudry IH:
Bench-to-bedside review: Latest results in hemorrhagic shock. Crit
Care. 12:2182008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gann DS and Drucker WR: Hemorrhagic shock.
J Trauma Acute Care Surg. 75:888–895. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karmaniolou II, Theodoraki KA, Orfanos NF,
Kostopanagiotou GG, Smyrniotis VE, Mylonas AI and Arkadopoulos NF:
Resuscitation after hemorrhagic shock: The effect on the liver-a
review of experimental data. J Anesth. 27:447–460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Novosad VL, Richards JL, Phillips NA, King
MA and Clanton TL: Regional susceptibility to stress-induced
intestinal injury in the mouse. Am J Physiol Gastrointest Liver
Physiol. 305:G418–G426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Spahn DR, Cerny V, Coats TJ, Duranteau J,
Fernández-Mondéjar E, Gordini G, Stahel PF, Hunt BJ, Komadina R,
Neugebauer E, et al: Management of bleeding following major trauma:
A European guideline. Crit Care. 11:R172007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith DJ, Salmi M, Bono P, Hellman J, Leu
T and Jalkanen S: Cloning of vascular adhesion protein 1 reveals a
novel multifunctional adhesion molecule. J Exp Med. 188:17–27.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salmi M, Kalimo K and Jalkanen S:
Induction and function of vascular adhesion protein-1 at sites of
inflammation. J Exp Med. 178:2255–2260. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morris NJ, Ducret A, Aebersold R, Ross SA,
Keller SR and Lienhard GE: Membrane amine oxidase cloning and
identification as a major protein in the adipocyte plasma membrane.
J Biol Chem. 272:9388–9392. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kurkijarvi R, Adams DH, Leino R, Möttönen
T, Jalkanen S and Salmi M: Circulating form of human vascular
adhesion protein-1 (VAP-1): Increased serum levels in inflammatory
liver diseases. J Immunol. 161:1549–1557. 1998.PubMed/NCBI
|
|
12
|
Klinman JP and Mu D: Quinoenzymes in
biology. Annu Rev Biochem. 63:299–344. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lyles GA: Mammalian plasma and
tissue-bound semicarbazide-sensitive amine oxidases: Biochemical,
pharmacological and toxicological aspects. Int J Biochem Cell Biol.
28:259–274. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilmot CM, Hajdu J, McPherson MJ, Knowles
PF and Phillips SE: Visualization of dioxygen bound to copper
during enzyme catalysis. Science. 286:1724–1728. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McNab G, Reeves JL, Salmi M, Hubscher S,
Jalkanen S and Adams DH: Vascular adhesion protein 1 mediates
binding of T cells to human hepatic endothelium. Gastroenterology.
110:522–528. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jaakkola K, Jalkanen S, Kaunismäki K,
Vänttinen E, Saukko P, Alanen K, Kallajoki M, Voipio-Pulkki LM and
Salmi M: Vascular adhesion protein-1, intercellular adhesion
molecule-1 and P-selectin mediate leukocyte binding to ischemic
heart in humans. J Am Coll Cardiol. 36:122–129. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tohka S, Laukkanen M, Jalkanen S and Salmi
M: Vascular adhesion protein 1 (VAP-1) functions as a molecular
brake during granulocyte rolling and mediates recruitment in vivo.
FASEB J. 15:373–382. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lalor PF, Edwards S, McNab G, Salmi M,
Jalkanen S and Adams DH: Vascular adhesion protein-1 mediates
adhesion and transmigration of lymphocytes on human hepatic
endothelial cells. J Immunol. 169:983–992. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koskinen K, Vainio PJ, Smith DJ,
Pihlavisto M, Ylä-Herttuala S, Jalkanen S and Salmi M: Granulocyte
transmigration through the endothelium is regulated by the oxidase
activity of vascular adhesion protein-1 (VAP-1). Blood.
103:3388–3395. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aspinall AI, Curbishley SM, Lalor PF,
Weston CJ, Blahova M, Liaskou E, Adams RM, Holt AP and Adams DH:
CX(3)CR1 and vascular adhesion protein-1-dependent recruitment of
CD16(+) monocytes across human liver sinusoidal endothelium.
Hepatology. 51:2030–2039. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stolen CM, Marttila-Ichihara F, Koskinen
K, Yegutkin GG, Turja R, Bono P, Skurnik M, Hänninen A, Jalkanen S
and Salmi M: Absence of the endothelial oxidase AOC3 leads to
abnormal leukocyte traffic in vivo. Immunity. 22:105–115. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Marttila-Ichihara F, Smith DJ, Stolen C,
Yegutkin GG, Elima K, Mercier N, Kiviranta R, Pihlavisto M,
Alaranta S, Pentikäinen U, et al: Vascular amine oxidases are
needed for leukocyte extravasation into inflamed joints in vivo.
Arthritis Rheum. 54:2852–2862. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salmi M and Jalkanen S: Homing-associated
molecules CD73 and VAP-1 as targets to prevent harmful
inflammations and cancer spread. FEBS Lett. 585:1543–1550. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weston CJ and Adams DH: Hepatic
consequences of vascular adhesion protein-1 expression. J Neural
Transm (Vienna). 118:1055–1064. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin MS, Li HY, Wei JN, Lin CH, Smith DJ,
Vainio J, Shih SR, Chen YH, Lin LC, Kao HL, et al: Serum vascular
adhesion protein-1 is higher in subjects with early stages of
chronic kidney disease. Clin Biochem. 41:1362–1367. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sallisalmi M, Tenhunen J, Yang R, Oksala N
and Pettilä V: Vascular adhesion protein-1 and syndecan-1 in septic
shock. Acta Anaesthesiol Scand. 56:316–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martelius T, Salaspuro V, Salmi M,
Krogerus L, Höckerstedt K, Jalkanen S and Lautenschlager I:
Blockade of vascular adhesion protein-1 inhibits lymphocyte
infiltration in rat liver allograft rejection. Am J Pathol.
165:1993–2001. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Merinen M, Irjala H, Salmi M, Jaakkola I,
Hänninen A and Jalkanen S: Vascular adhesion protein-1 is involved
in both acute and chronic inflammation in the mouse. Am J Pathol.
166:793–800. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bonder CS, Norman MU, Swain MG, Zbytnuik
LD, Yamanouchi J, Santamaria P, Ajuebor M, Salmi M, Jalkanen S and
Kubes P: Rules of recruitment for Th1 and Th2 lymphocytes in
inflamed liver: A role for alpha-4 integrin and vascular adhesion
protein-1. Immunity. 23:153–163. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salter-Cid LM, Wang E, O'Rourke AM, Miller
A, Gao H, Huang L, Garcia A and Linnik MD: Anti-inflammatory
effects of inhibiting the amine oxidase activity of
semicarbazide-sensitive amine oxidase. J Pharmacol Exp Ther.
315:553–562. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu HL, Salter-Cid L, Linnik MD, Wang EY,
Paisansathan C and Pelligrino DA: Vascular adhesion protein-1 plays
an important role in postischemic inflammation and neuropathology
in diabetic, estrogen-treated ovariectomized female rats subjected
to transient forebrain ischemia. J Pharmacol Exp Ther. 317:19–29.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
O'Rourke AM, Wang EY, Miller A, Podar EM,
Scheyhing K, Huang L, Kessler C, Gao H, Ton-Nu HT, Macdonald MT, et
al: Anti-inflammatory effects of LJP 1586
[Z-3-fluoro-2-(4-methoxybenzyl)allylamine hydrochloride], an
amine-based inhibitor of semicarbazide-sensitive amine oxidase
activity. J Pharmacol Exp Ther. 324:867–875. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Irjala H, Salmi M, Alanen K, Grénman R and
Jalkanen S: Vascular adhesion protein 1 mediates binding of
immunotherapeutic effector cells to tumor endothelium. J Immunol.
166:6937–6943. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marttila-Ichihara F, Auvinen K, Elima K,
Jalkanen S and Salmi M: Vascular adhesion protein-1 enhances tumor
growth by supporting recruitment of Gr-1+CD11b+ myeloid cells into
tumors. Cancer Res. 69:7875–7883. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yasuda H, Toiyama Y, Ohi M, Mohri Y, Miki
C and Kusunoki M: Serum soluble vascular adhesion protein-1 is a
valuable prognostic marker in gastric cancer. J Surg Oncol.
103:695–699. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Airas L, Lindsberg PJ,
Karjalainen-Lindsberg ML, Mononen I, Kotisaari K, Smith DJ and
Jalkanen S: Vascular adhesion protein-1 in human ischaemic stroke.
Neuropathol Appl Neurobiol. 34:394–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hernandez-Guillamon M, Garcia-Bonilla L,
Solé M, Sosti V, Parés M, Campos M, Ortega-Aznar A, Domínguez C,
Rubiera M, Ribó M, et al: Plasma VAP-1/SSAO activity predicts
intracranial hemorrhages and adverse neurological outcome after
tissue plasminogen activator treatment in stroke. Stroke.
41:1528–1535. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arvilommi AM, Salmi M and Jalkanen S:
Organ-selective regulation of vascular adhesion protein-1
expression in man. Eur J Immunol. 27:1794–1800. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Martelius T, Salmi M, Wu H, Bruggeman C,
Höckerstedt K, Jalkanen S and Lautenschlager I: Induction of
vascular adhesion protein-1 during liver allograft rejection and
concomitant cytomegalovirus infection in rats. Am J Pathol.
157:1229–1237. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Galley HF and Webster NR: The
immuno-inflammatory cascade. Br J Anaesth. 77:11–16. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lamb RE and Goldstein BJ: Modulating an
oxidative-inflammatory cascade: Potential new treatment strategy
for improving glucose metabolism, insulin resistance, and vascular
function. Int J Clin Pract. 62:1087–1095. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiaojun Y, Cheng Q, Yuxing Z and Zhiqian
H: Microarray analysis of differentially expressed background genes
in rats following hemorrhagic shock. Mol Biol Rep. 39:2045–2053.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Solé M and Unzeta M: Vascular cell lines
expressing SSAO/VAP-1: A new experimental tool to study its
involvement in vascular diseases. Biol Cell. 103:543–557. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun P, Esteban G, Inokuchi T,
Marco-Contelles J, Weksler BB, Romero IA, Couraud PO, Unzeta M and
Solé M: Protective effect of the multitarget compound DPH-4 on
human SSAO/VAP-1-expressing hCMEC/D3 cells under oxygen-glucose
deprivation conditions: An in vitro experimental model of cerebral
ischaemia. Br J Pharmacol. 172:5390–5402. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Salmi M and Jalkanen S: Different forms of
human vascular adhesion protein-1 (VAP-1) in blood vessels in vivo
and in cultured endothelial cells: Implications for
lymphocyte-endothelial cell adhesion models. Eur J Immunol.
25:2803–2812. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Johansson EL, Rudin A, Wassén L and
Holmgren J: Distribution of lymphocytes and adhesion molecules in
human cervix and vagina. Immunology. 96:272–277. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sun P, Solé M and Unzeta M: Involvement of
SSAO/VAP-1 in oxygen-glucose deprivation-mediated damage using the
endothelial hSSAO/VAP-1-expressing cells as experimental model of
cerebral ischemia. Cerebrovasc Dis. 37:171–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Imtiyaz HZ and Simon MC: Hypoxia-inducible
factors as essential regulators of inflammation. Curr Top Microbiol
Immunol. 345:105–120. 2010.PubMed/NCBI
|
|
50
|
Greer SN, Metcalf JL, Wang Y and Ohh M:
The updated biology of hypoxia-inducible factor. EMBO J.
31:2448–2460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hernandez-Guillamon M, Solé M, Delgado P,
García-Bonilla L, Giralt D, Boada C, Penalba A, García S, Flores A,
Ribó M, et al: VAP-1/SSAO plasma activity and brain expression in
human hemorrhagic stroke. Cerebrovasc Dis. 33:55–63. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kinemuchi H, Kobayashi N, Takahashi K,
Takayanagi K, Arai Y, Tadano T, Kisara K and Oreland L: Inhibition
of tissue-bound semicarbazide-sensitive amine oxidase by two
haloamines, 2-bromoethylamine and 3-bromopropylamine. Arch Biochem
Biophys. 385:154–161. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kobayashi N, Takahashi K, Takayanagi K,
Takahashi K, Masuko S, Tadano T, Kisara K and Kinemuchi H:
Molecular characteristics of tissue-bound semicarbazide-sensitive
amine oxidase (SSAO) in guinea pig tissues. Life Sci. 70:1519–1531.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lalor PF, Sun PJ, Weston CJ, Martin-Santos
A, Wakelam MJ and Adams DH: Activation of vascular adhesion
protein-1 on liver endothelium results in an NF-kappaB-dependent
increase in lymphocyte adhesion. Hepatology. 45:465–474. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lalor PF, Tuncer C, Weston C,
Martin-Santos A, Smith DJ and Adams DH: Vascular adhesion protein-1
as a potential therapeutic target in liver disease. Ann N Y Acad
Sci. 1110:485–496. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xu HL, Garcia M, Testai F, Vetri F,
Barabanova A, Pelligrino DA and Paisansathan C: Pharmacologic
blockade of vascular adhesion protein-1 lessens neurologic
dysfunction in rats subjected to subarachnoid hemorrhage. Brain
Res. 1586:83–89. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Xu H, Testai FD, Valyi-Nagy T, N Pavuluri
M, Zhai F, Nanegrungsunk D, Paisansathan C and Pelligrino DA: VAP-1
blockade prevents subarachnoid hemorrhage-associated
cerebrovascular dilating dysfunction via repression of a neutrophil
recruitment-related mechanism. Brain Res. 1603:141–149. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Watcharotayangul J, Mao L, Xu H, Vetri F,
Baughman VL, Paisansathan C and Pelligrino DA: Post-ischemic
vascular adhesion protein-1 inhibition provides neuroprotection in
a rat temporary middle cerebral artery occlusion model. J
Neurochem. 123 Suppl 2:S116–S124. 2012. View Article : Google Scholar
|
|
59
|
Ma Q, Manaenko A, Khatibi NH, Chen W,
Zhang JH and Tang J: Vascular adhesion protein-1 inhibition
provides antiinflammatory protection after an intracerebral
hemorrhagic stroke in mice. J Cereb Blood Flow Metab. 31:881–893.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
O'Rourke AM, Wang EY, Salter-Cid L, Huang
L, Miller A, Podar E, Gao HF, Jones DS and Linnik MD: Benefit of
inhibiting SSAO in relapsing experimental autoimmune
encephalomyelitis. J Neural Transm (Vienna). 114:845–849. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jalkanen S and Salmi M: VAP-1 and CD73,
endothelial cell surface enzymes in leukocyte extravasation.
Arterioscler Thromb Vasc Biol. 28:18–26. 2008. View Article : Google Scholar : PubMed/NCBI
|