Introduction
The influenza A/California/07/2009 (H1N1) virus
outbreak in Mexico and the United States in 2009 was found to be
genetically correlated with swine influenza viruses (1). The H1N1 virus subsequently spread to
other countries and caused severe outbreaks around the world. Up to
10% of severe influenza infections cause pneumonia with myocarditis
or acute myocardial infarction (2–4).
Individuals with underlying chronic cardiovascular diseases are at
higher risk of influenza A virus (IAV)-induced myocarditis
(5). However, the mechanism
underlying the viral myocarditis remains to be fully
elucidated.
The whole genome of IAV contains eight
negative-sense RNA segments, comprising PB1, PB2, PA, HA,
nucleoprotein (NP), neuraminidase (NA), (M1 and NS1), which encode
>10 proteins. The IAV genome is variable for the low fidelity of
RNA polymerase and recombination between strains (6), allowing viruses to evade host
immunity and outbreak cyclically. Influenza virus infection leads
to the activation of various intracellular signaling pathways,
which not only partially increase the antiviral response, but also
support viral replication simultaneously (7,8).
Several IAV proteins have been implicated in apoptosis in infected
cells. Previous studies have indicated the NS1 protein of IAV
downregulates apoptosis in early infection to support virus
replication (9). NA can activate
transforming growth factor (TGF)-h, a known inducer of apoptosis
(10,11). PB1 and PB2 are also considered to
be involved in the induction of apoptosis in infected cells
(12). NP is phosphorylated and
cleaved by proteases in infected cells, and IAVNP (56 kDa; NP56) is
converted proteolytically into a 53-kDa form (NP53) (13), NP cleavage appears to prevent
packaging of viral ribonucleoprotein into virus particles, with
only uncleaved NP56 found assembled into virions (14). To date, the association between the
level of NP and apoptosis remains to be fully elucidated.
In the present study, the replication of IAV in H9C2
cells was suppressed using short hairpin (sh) RNA against NP, and
the apoptosis induced by virus infection was examined. The results
aimed to determine whether viral replication is associated with the
level of apoptosis induced.
Materials and methods
Cell culture and production of the
shRNA-NP lentivirus
Cardiomyoblast H9C2 cells (American Type Culture
Collection; ATCC; Manassas, VA, USA) and 293T cells were cultured
in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal
calf serum (FCS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin/streptomycin and 2 mM L-glutamine (growth medium), and
were maintained at 37°C in a humidified 5% CO2
atmosphere. For the production of the shRNA-NP lentivirus, cells
were seeded in a T25 tissue culture plate at a density of 2×105
cells/ml, when 50–70% confluent, and were transfected for 6 h with
an shRNA-NP plasmid and a control-shRNA plasmid, using
Lipofectamine 2000 as the transfection reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified 5% CO2
atmosphere. After 48 h, the supernatant was harvested, filtered
using a 0.2 µm filter and stored at −80°C.
IAV virus propagation
The influenza A/H1N1pdm2009 virus (CA07) was
propagated in Madin Darby canine kidney cells (line CV-1; ATCC).
The cells were passaged in DMEM containing 10% bovine FCS. For
infection of the H9C2 cells, 2-day-old confluent cell monolayers
were incubated with the virus at the indicated multiplicity of
infection (MOI) for 1 h at 37°C. Following infection, the cells
(2×104 cells/well) were washed and incubated with DMEM at 37°C for
different durations (0, 8, 12, 24 and 48 h) and were then prepared
for further examination. The titers of infectious virus in the
supernatants were determined using common plaque assays, as
described previously (15,16).
Evaluation of NP mRNA levels using
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analysis
The mRNA level of NP was determined using RT-qPCR
analysis, as described previously (2). Briefly, total RNA was isolated from
cells (1×105 cells/sample) using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The cDNA was then synthesized by RT using a Reverse
Transcription kit (Thermo Fisher Scientific, Inc.) with the
NP-specific primer (5′-cac caa acg atc ata tga ac-3′). qPCR was
performed using the following primers (10 nM): NP, forward 5′-cca
gaa tgt gct ctc taa tg-3′, reverse 5′-tcc ttt cac cgc agc acc
tg-3′, as previously described (2). The quantification cycle (Cq) values
(17) of the target gene were
normalized to β-actin (forward 5′-gta ccc tgg cat tgc cga ca-3′,
reverse 5′-gga ctc gtc ata ctc ctg ctt gct-3′) from the same sample
as relative mRNA levels. Each sample was assessed in
triplicate.
Western blot analysis
The H9C2 cells were collected and lysed with lysis
buffer (Thermo Scientific, Rockford, IL, USA) on ice for 20 min.
The cell lysates were centrifuged at 15,000 × g at 4°C for 30 min.
The supernatant was collected as the total cellular protein
extract. Protein concentration was determined using the BCA Protein
Assay kit (Kangwei Shiji Biotechnology Co., Ltd., Beijing, China).
The samples of total cellular protein (5 µg) were loaded onto a 10%
SDS-PAGE. The separated proteins were electrophoretically
transferred onto polyvinylidene difluoride membranes (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The membranes were blocked
overnight in blocking buffer containing PBS-T and 5% non-fat milk.
The membranes were then incubated with primary antibodies against
NP (1:800; cat. no. 11675-V08B-50; Sino Biological, Inc., Beijing,
China), caspase-3 (1:500; cat. no. 3CSP03), cytochrome c (CytC;
1:500; cat. no. sc-7159), B cell lymphoma-2-associated X protein
(Bax; 1:500; cat. no. sc-493) and β-actin (1:500; cat. no.
sc-81178), obtained from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA), for 1 h at 37°C, and then washed with PBS-T four times.
Following incubation with the secondary horseradish
peroxidase-conjugated antibody (1:400; cat. no. Ab131366; Abcam,
Cambridge, UK) for 1 h at room temperature, the membranes were
washed four times, treated with enhanced chemiluminescence reagent
and exposed to X-ray film. Protein bands were semi-quantified using
ImageJ software version 1.43b (National Institutes of Health,
Bethesda, MD, USA).
Determination of apoptosis
The percentages of apoptotic cells were determined
using an Annexin V-FITC Apoptosis Detection kit (EMD Millipore,
Billerica, MA, USA). Briefly, the cells were incubated for 15 min
in the dark with Annexin V-FITC and PI, according to the
manufacturer's protocol. A total of 1.0×106 cells were washed twice
with ice-cold PBS and incubated for 10 min in binding buffer,
containing 5 µl PI and 5 µl Annexin V-FITC. To quantify the
apoptotic rate of cells in each group, at least 100,000 cells from
each treatment group were examined using flow-cytometry, and the
percentage of Annexin V-positive cells or Annexin V-plus-PI
positive cells were calculated. All experiments were performed in
triplicate. The activity of caspase-3 was analyzed using a
caspase-3 activity assay kit (Cell Signaling Technology, Inc.,
Danvers, MA, USA).
MTT cell viability assay
The viability of the H9C2 cells was measured using
the MTT method in 96-well plates. Briefly, following infection with
the A/H1N1pdm2009 virus (multiplicity of infection=1) for 12, 24 or
48 h, H9C2 cells (1×104 cells/well) at 80% confluence, were treated
with 20 µl of MTT solution (at a final concentration of 5 mg/ml)
for 4 h at 37°C. The cell supernatant was carefully removed, and
200 µl of DMSO was added to each well and mixed. The plate was
placed in a 37°C incubator to dissolve air bubbles and the optical
density (OD) 50 value of each well was measured at a 570 nm
wavelength using a microplate reader (Thermo Fisher Scientific,
Inc.). The results were calculated as follows: (A570 control
wells-A570 treated wells)/(A570 control wells-A570 blank wells)
×100%.
Statistical analysis
Data are presented as the mean ± standard deviation
calculated from three independent experiments. For comparison
between two groups, Student's t-test was used. For multiple
comparisons among three or more groups, one-way analysis of
variance was used followed by a post hoc Newman-Keuls test.
Analysis was performed using the SPSS software version 16.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
H1N1pdm2009 infection induces the
apoptosis of cardiomyocytes
The H9C2 cells were infected with the 1 MOI
H1N1pdm2009 strain. The relative mRNA and protein levels of NP to
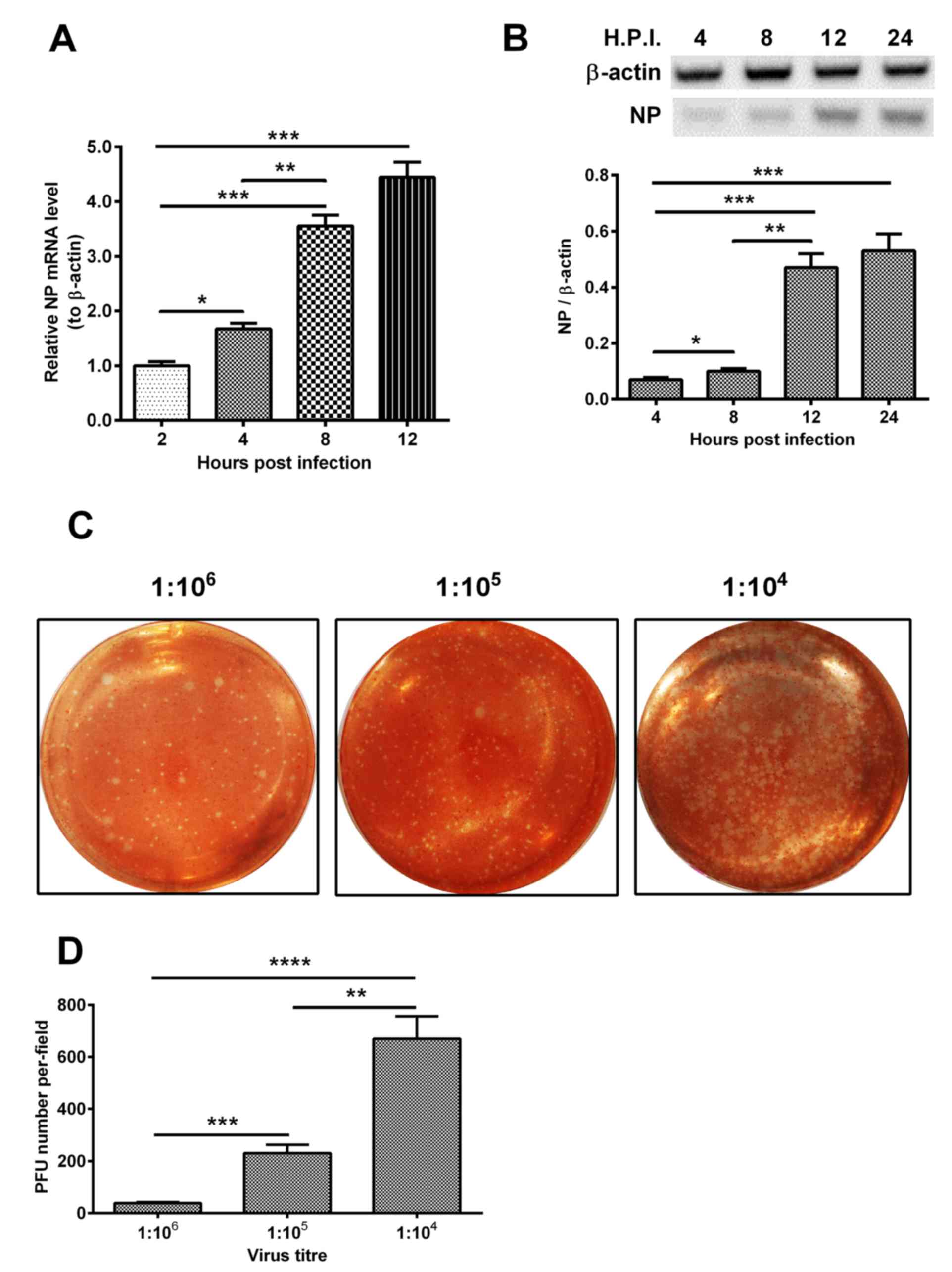
β-actin were measured at 2, 4, 8 and 12 h post-infection. As shown
in Fig. 1A, the expression level
of NP increased 4-fold at 12 h post-infection, compared with the
level at 4 h post-infection (P<0.001). In addition, the level of
NP increased 2-fold at 8 h post-infection, compared with that at 2
h post-infection (P<0.01). Similar results were observed in the
expression level of NP (Fig. 1B).
The plaque forming ability of H1N1pdm2009 in the H9C2 cells was
also assessed at dilutions of 1:104, 1:105 and 1:106, respectively.
As shown in Fig. 1C and D, the PFU
number per-field at the 104 dilution was the highest among the
groups with statistical significance.
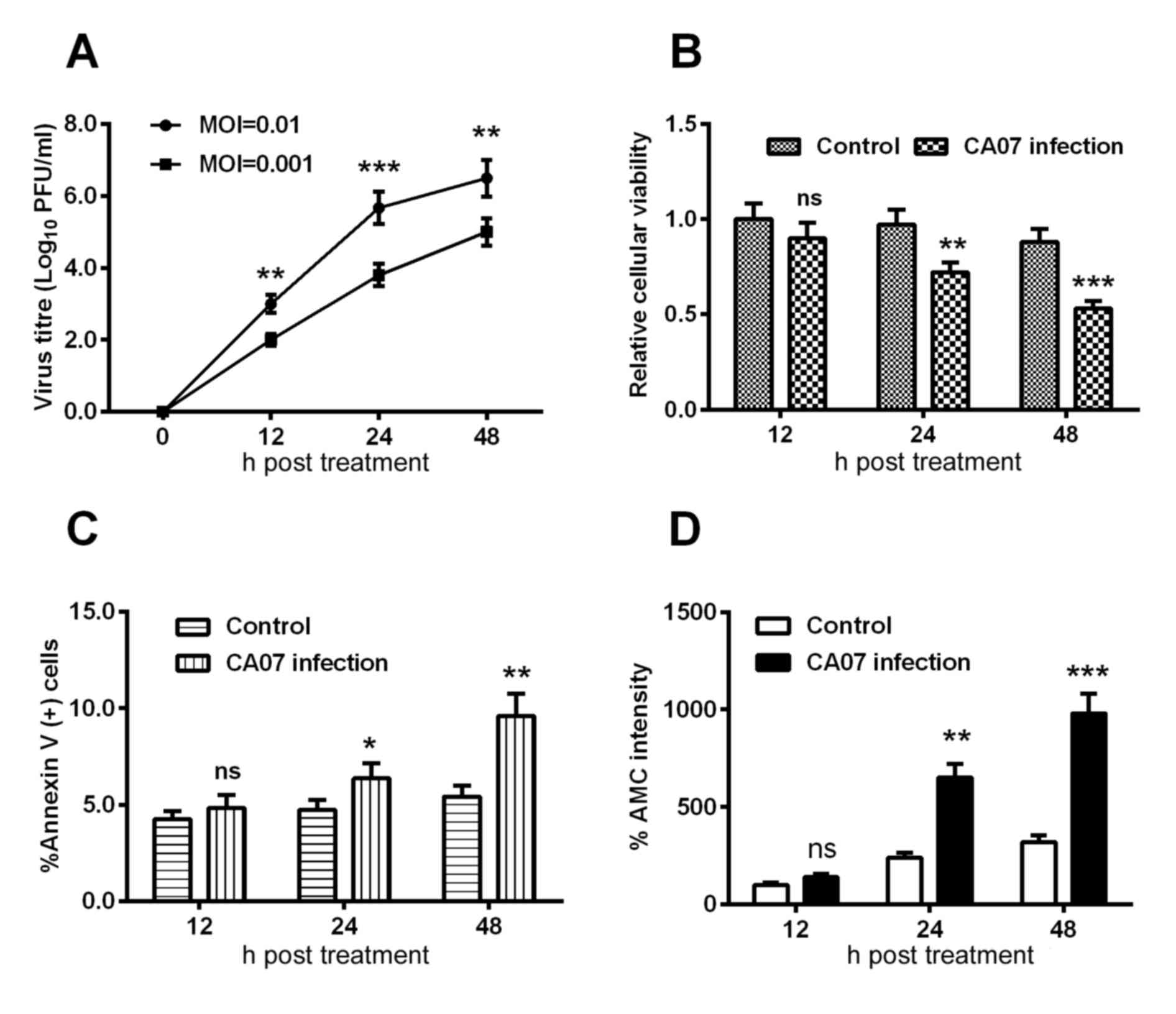
Subsequently, the growth curve of the H1N1pdm2009
virus in H9C2 cells was examined using an MOI of 0.01 and 0.001,
and the viral titer at each time point was determined using a
plaque-forming assay. As shown in Fig.
2A, the group with an MOI of 0.01 presented with a higher
titer, compared with that at the MOI of 0.001 at each time point.
In addition, the relative cellular viabilities at each time point
were assessed using an MTT assay; it was found that the cell
viabilities were decreased by 30 and 50% at 24 and 48 h
post-infection, respectively, and these differences were
statistically significant (Fig.
2B).
Regarding the apoptosis induced by H1N1 infection,
the present study quantified the H1N1 infection-induced apoptosis
using an Annexin V kit, and observed a time-dependent increase in
the percentage of Annexin V(+) cells and AMC intensity (Fig. 2C and D), indicating that H1N1
infection induced the apoptosis of H9C2 cells.
shRNA efficiently inhibits the
replication of H1N1pdm2009
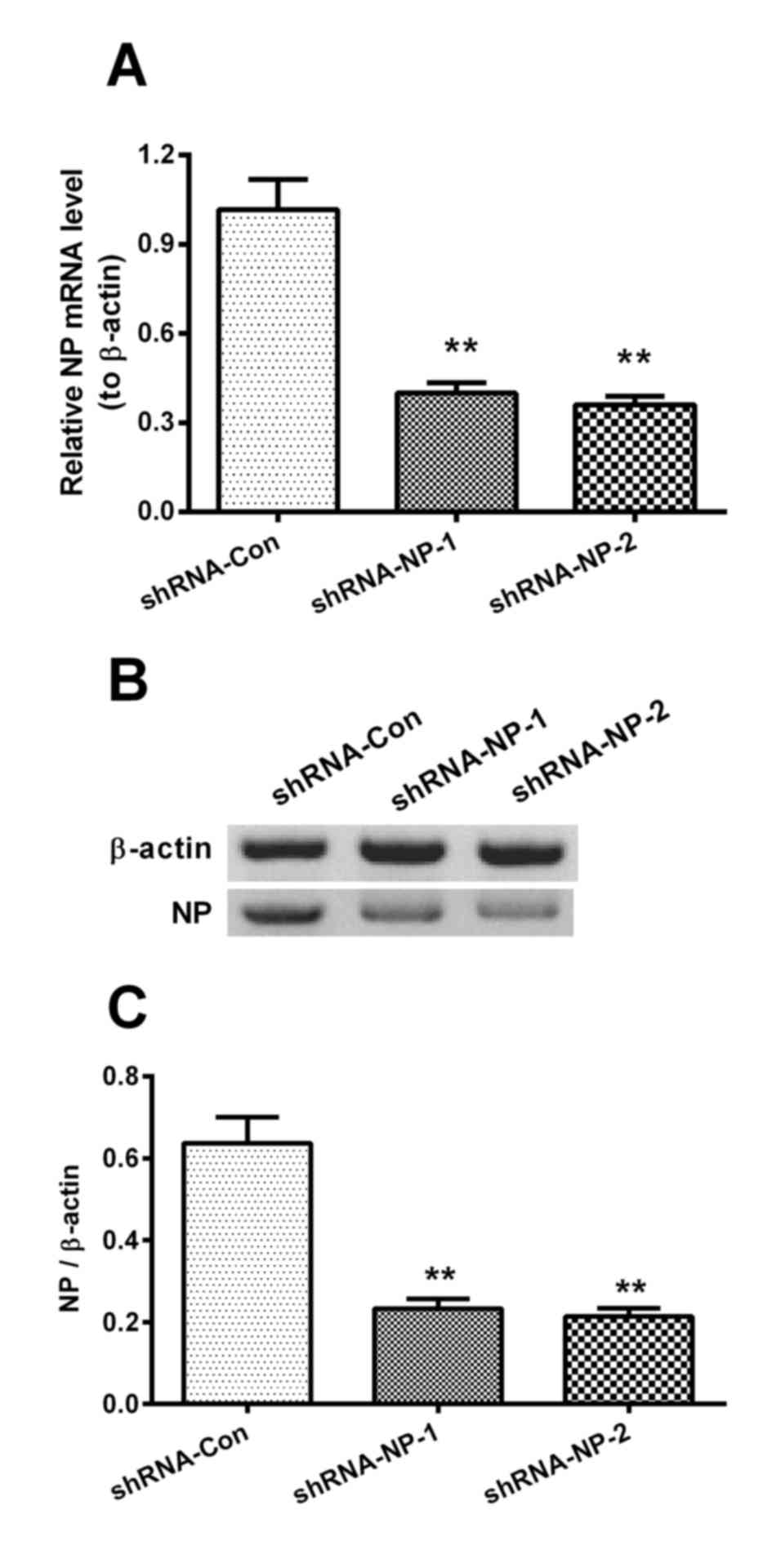
The cells infected with 1 MOI of the H1N1pdm2009
virus were then infected by 1 MOI of Lenti-shRNA-NP-1,
Lenti-shRNA-NP-2 or Lenti-shRNA-Con virus, and the relative mRNA
levels of NP were measured at 12 h post-infection. The relative
mRNA levels of NP in the shRNA-NP-treated groups were decreased by
60 and 65%, compared with that in the shRNA control group,
respectively (Fig. 3A). The
results of the western blot analysis results also confirmed that
shRNA transfection inhibited the expression of NP by 75 and 80%,
respectively (Fig. 3B and C).
Subsequently, the H9C2 cells were infected with an
MOI of 0.01 or 0.001 viral titer following the infection with 1 MOI
Lenti-shRNA-NP-1, Lenti-shRNA-NP-2 or Lenti-shRNA-Con virus. A
growth curve was mapped at each time point, which was quantified
using the plaque-forming assay. As shown in Fig. 4. A, no statistically significant
differences were found among the groups at 12 h post-infection,
however, the viral titers of the Lenti-shRNA-NP-1 and
Lenti-shRNA-NP-2 groups were downregulated by >40%, compared
with that of the Lenti-shRNA-con group (P<0.01). In addition,
the relative cellular viabilities of the Lenti-shRNA-NP-treated
groups were significantly improved, compared with that in the
control group at 24 or 48 h post-infection (Fig. 4B).
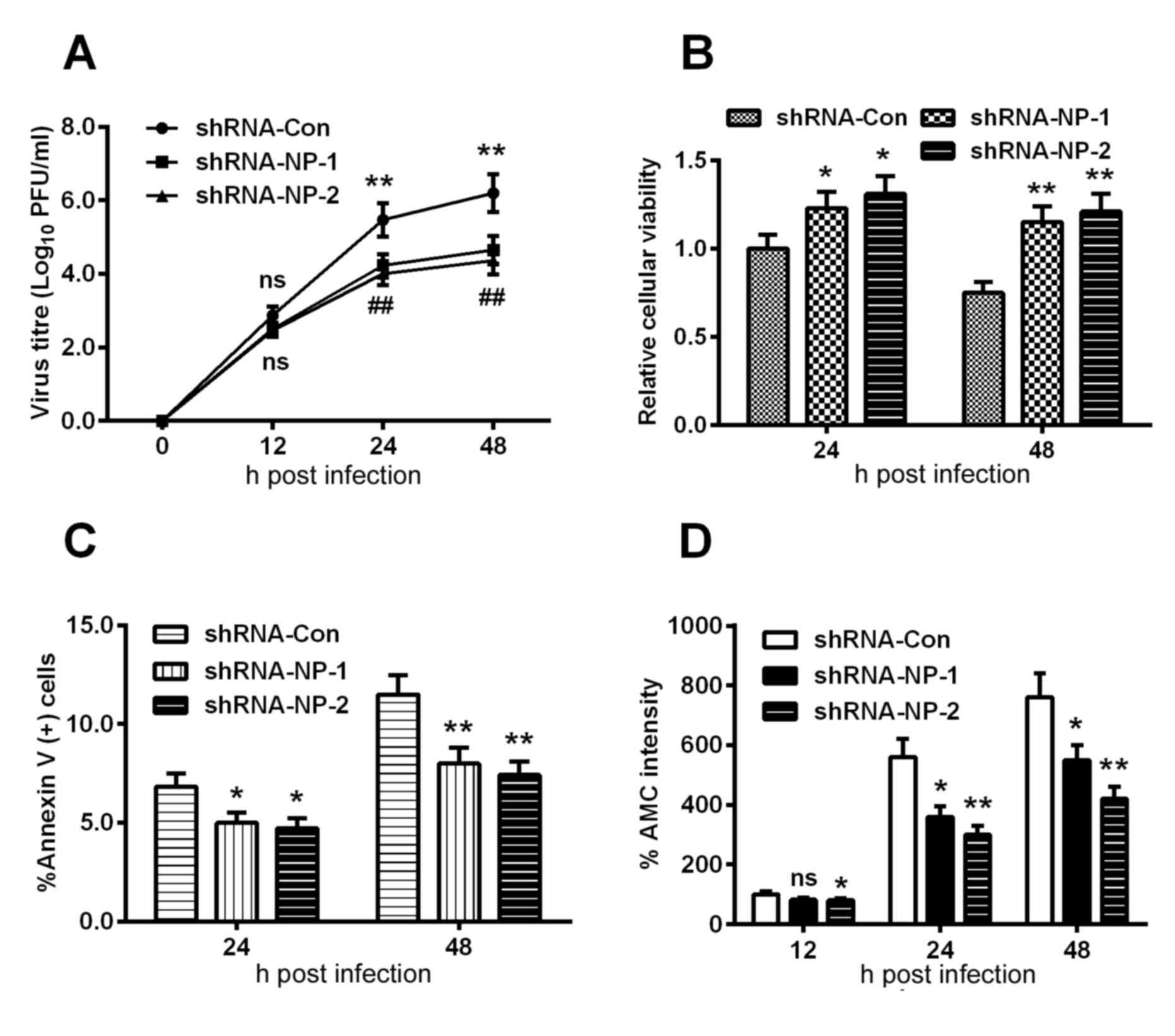 | Figure 4.Apoptosis is induced by proliferation
of the H1N1pdm2009 virus in H9C2 cells following NP knockdown. (A)
Growth curve of H1N1pdm2009 virus in H9C2 cells (MOI of 0.01 and
0.001), the viral titer at each time point was quantified using the
plaque-forming assay following infection with 1 MOI
Lenti-shRNA-NP-1, Lenti-shRNA-NP-2 or Lenti-shRNA-Con virus.
Quantitative data are the average of three independent experiments.
**P<0.01, Lenti-shRNA-NP-1 vs. Lenti-shRNA-Con;
##P<0.01, Lenti-shRNA-NP-2 vs. Lenti-shRNA-Con.
Results of MTT assays for (B) cellular viability, (C) induction of
apoptosis and (D) activity of caspase-3 in H9C2 cells infected with
1 MOI H1N1pdm2009 virus for 12, 24 or 48 h, following infection
with 1 MOI Lenti-shRNA-NP-1, Lenti-shRNA-NP-2 or Lenti-shRNA-Con
virus. Quantitative data are the average of three independent
experiments. *P<0.05, **P<0.01 and ***P<0.001, as
indicated. ns, no significance; NP, nucleoprotein; MOI,
multiplicity of infection; shRNA, short hairpin RNA; con,
control. |
shRNA inhibits H1N1pdm2009-induced
apoptosis
In order to examine the apoptosis inhibited by
shRNA, the apoptotic rate of shRNA-treated cells were determined
using an Annexin V kit, which revealed that apoptosis was
significantly inhibited by >20% s at 24 h post-infection
(Fig. 4C). Similar results were
observed in the fluorescence intensity of AMC (Fig. 4D). The key apoptosis-associated
proteins, including released CytC, cleaved caspase-3 and Bax, were
examined using western blot analysis (Fig. 5A). Treatment with shRNA-NP-1 and
shRNA-NP-2 inhibited the expression levels of released CytC,
cleaved caspase-3 and Bax. Following quantification of each band,
it was found that shRNA-NP-1 and shRNA-NP-2 downregulated the
expression of released CytC by 50% (P<0.05; Fig. 5B). shRNA-NP-1 and shRNA-NP-2
treatment also downregulated the expression of cleaved caspase-3 by
at least 40% (P<0.05; Fig. 5C).
The expression of Bax was significantly decreased by ~50% by
shRNA-NP-1 and shRNA-NP-2 (Fig.
5D).
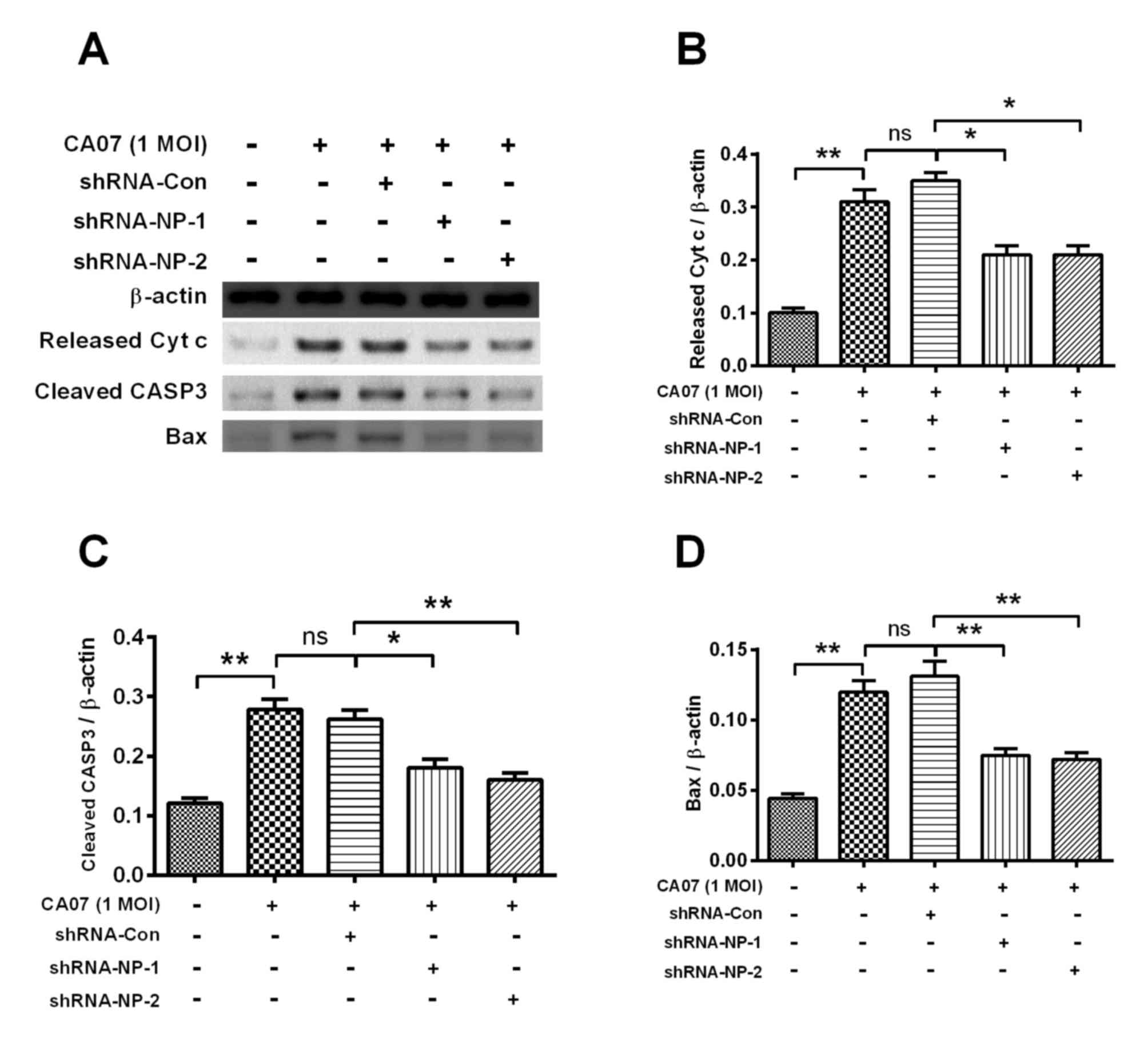 | Figure 5.Western blot analyses of
apoptosis-associated markers in H1N1pdm2009-infected H9C2 cells
with or without NP-knockdown. (A) Representative western blot assay
results of released Cytc from mitochondria, cleaved CASP3 or Bax in
H1N1pdm2009-infected (1 MOI) H9C2 cells with or without
NP-knockdown. Relative levels of (B) released Cytc, (C) cleaved
CASP3 and (D) Bax inH1N1pdm2009-infected H9C2 cells with or without
NP-knockdown. Quantitative data are the averaged of triplicate
independent experiments. *P<0.05 and **P<0.01. ns, no
significance; NP, nucleoprotein; shRNA, short hairpin RNA; Cytc,
cytochrome c, Bax, B cell lymphoma-2-associated X protein;
MOI, multiplicity of infection; con, control. |
Discussion
In the present study, the apoptosis of
cardiomyocytes infected with H1N1pdm2009 was investigated. It was
found that H1N1pdm2009 viral infection promoted the apoptosis of
the H1N1pdm2009-infected H9C2 cells. The NP-specific shRNA
significantly inhibited the viral infection and the virus-induced
apoptosis of the H9C2 cells. The results of the present study
indicated the key pathogenic role of NP in H1N1pdm2009-induced
apoptosis of the cardiomyocytes, and NP-specific shRNA may be an
effective agent to prevent influenza virus-induced myocarditis.
It has been reported that influenza viruses can
induce apoptosis in various cell types (18,19),
and several viral proteins, including M1, NS1 and PB1-F2 have also
been shown to induce or inhibit apoptosis in human cells (20,21).
A previous report showed that the IAV may utilize its NS1 protein
to interact with cellular β-tubulin to induce apoptosis (22). To date, few studies have been
performed to investigate the role of NP in influenza-induced
apoptosis. In the present study, it was found that shRNA targeting
NP efficiently disrupted the production of H1N1pdm2009 virus, and
markedly reduced the apoptotic rates of the infected H9C2 cells. In
addition, key apoptosis-associated molecules, including released
CytC, cleaved caspase-3 and Bax, were downregulated. These results
indicated that NP has a key pathogenic role in H1N1pdm2009-induced
apoptosis of cardiomyocytes.
Caspase-3 has been reported to be essential for
viral replication in MDCK cells (23), and another study reported that the
caspase-mediated cleavage of influenza NP in apoptosis has an
antiviral effect (24). The
results obtained in the present study indicated that shRNA against
NP decreased the viral titer and effectively downregulated the
level of cleaved caspase-3, and consequently suppressed the
apoptotic rates of the cells. These results are in accordance with
the previous conclusions that apoptosis may exert its protective
effects not only by the elimination of infected cells, but also by
the NP cleavage-mediated suppression of viral multiplication
(24).
Numerous studies have indicated that cytokines are
crucial in the induction of apoptosis in the heart (25), and it type I interferons are
considered to cause influenza-induced apoptosis via a
caspase-8-dependent mechanism (26). However, the exact pathogenic roles
of cytokines during H1N1-induced apoptosis remain to be fully
elucidated. The present study did not examine the
apoptosis-associated cytokines in H1N1pdm2009 virus-infected
animals; further investigations are required in animal models.
In conclusion, the present study demonstrated the
vital pathogenic role of NP in the H1N1pdm2009-induced apoptosis of
cardiomyocytes; NP-specific shRNA may be used as a potential
therapeutic strategy to inhibit the influenza virus-induced
apoptosis in cardiomyocytes and prevent influenza virus-associated
acute myocarditis.
References
|
1
|
Fraser C, Donnelly CA, Cauchemez S, Hanage
WP, van Kerkhove MD, Hollingsworth TD, Griffin J, Baggaley RF,
Jenkins HE, Lyons EJ, et al: Pandemic potential of a strain of
influenza A (H1N1): Early findings. Science. 324:1557–1561. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pan HY, Yano M and Kido H: Effects of
inhibitors of Toll-like receptors, protease-activated receptor-2
signalings and trypsin on influenza A virus replication and
upregulation of cellular factors in cardiomyocytes. J Med Invest.
58:19–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mamas MA, Fraser D and Neyses L:
Cardiovascular manifestations associated with influenza virus
infection. Int J Cardiol. 130:304–309. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warren-Gash C, Smeeth L and Hayward AC:
Influenza as a trigger for acute myocardial infarction or death
from cardiovascular disease: A systematic review. Lancet Infect
Dis. 9:601–610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Davis MM, Taubert K, Benin AL, Brown DW,
Mensah GA, Baddour LM, Dunbar S and Krumholz HM; American Heart
Association, : American College of Cardiology: Influenza
vaccination as secondary prevention for cardiovascular disease: A
science advisory from the American Heart Association/American
College of Cardiology. Circulation. 114:1549–1553. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Steinhauer DA, Domingo E and Holland JJ:
Lack of evidence for proofreading mechanisms associated with an RNA
virus polymerase. Gene. 122:281–288. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ludwig S, Planz O, Pleschka S and Wolff T:
Influenza-virus-induced signaling cascades: Targets for antiviral
therapy? Trends Mol Med. 9:46–52. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ludwig S, Pleschka S, Planz O and Wolff T:
Ringing the alarm bells: Signalling and apoptosis in influenza
virus infected cells. Cell Microbiol. 8:375–386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhirnov OP, Konakova TE, Wolff T and Klenk
HD: NS1 protein of influenza A virus down-regulates apoptosis. J
Virol. 76:1617–1625. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mohsin MA, Morris SJ, Smith H and Sweet C:
Correlation between levels of apoptosis, levels of infection and
haemagglutinin receptor binding interaction of various subtypes of
influenza virus: Does the viral neuraminidase have a role in these
associations. Virus Res. 85:123–131. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schultz-Cherry S and Hinshaw VS: Influenza
virus neuraminidase activates latent transforming growth factor
beta. J Virol. 70:8624–8629. 1996.PubMed/NCBI
|
|
12
|
Morris SJ, Nightingale K, Smith H and
Sweet C: Influenza A virus-induced apoptosis is a multifactorial
process: Exploiting reverse genetics to elucidate the role of
influenza A virus proteins in virus-induced apoptosis. Virology.
335:198–211. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhirnov O and Bukrinskaya AG:
Nucleoproteins of animal influenza viruses, in contrast to those of
human strains, are not cleaved in infected cells. J Gen Virol.
65:1127–1134. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhirnov OP and Bukrinskaya AG: Two forms
of influenza virus nucleoprotein in infected cells and virions.
Virology. 109:174–179. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ludwig S, Pleschka S and Wolff T: A fatal
relationship-influenza virus interactions with the host cell. Viral
Immunol. 12:175–196. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pleschka S, Wolff T, Ehrhardt C, Hobom G,
Planz O, Rapp UR and Ludwig S: Influenza virus propagation is
impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat
Cell Biol. 3:301–305. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mori I, Komatsu T, Takeuchi K, Nakakuki K,
Sudo M and Kimura Y: In vivo induction of apoptosis by influenza
virus. J Gen Virol. 76:2869–2873. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roulston A, Marcellus RC and Branton PE:
Viruses and apoptosis. Annu Rev Microbiol. 53:577–628. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chanturiya AN, Basañez G, Schubert U,
Henklein P, Yewdell JW and Zimmerberg J: PB1-F2, an influenza A
virus-encoded proapoptotic mitochondrial protein, creates variably
sized pores in planar lipid membranes. J Virol. 78:6304–6312. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stasakova J, Ferko B, Kittel C, Sereinig
S, Romanova J, Katinger H and Egorov A: Influenza A mutant viruses
with altered NS1 protein function provoke caspase-1 activation in
primary human macrophages, resulting in fast apoptosis and release
of high levels of interleukins 1beta and 18. J Gen Virol.
86:185–195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han X, Li Z, Chen H, Wang H, Mei L, Wu S,
Zhang T, Liu B and Lin X: Influenza virus A/Beijing/501/2009(H1N1)
NS1 interacts with β-tubulin and induces disruption of the
microtubule network and apoptosis on A549 cells. PloS One.
7:e483402012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wurzer WJ, Planz O, Ehrhardt C, Giner M,
Silberzahn T, Pleschka S and Ludwig S: Caspase 3 activation is
essential for efficient influenza virus propagation. EMBO J.
22:2717–2728. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhirnov OP, Konakova TE, Garten W and
Klenk H: Caspase-dependent N-terminal cleavage of influenza virus
nucleocapsid protein in infected cells. J Virol. 73:10158–10163.
1999.PubMed/NCBI
|
|
25
|
Yajima T and Knowlton KU: Viral
myocarditis: From the perspective of the virus. Circulation.
119:2615–2624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Balachandran S, Roberts PC, Kipperman T,
Bhalla KN, Compans RW, Archer DR and Barber GN: Alpha/beta
interferons potentiate virus-induced apoptosis through activation
of the FADD/Caspase-8 death signaling pathway. J Virol.
74:1513–1523. 2000. View Article : Google Scholar : PubMed/NCBI
|