Introduction
Exposure to high ambient temperatures can result in
heat stroke, a clinical condition characterized by a rapid rise
core temperature above 40°C, which is associated with central
nervous system alterations such as delirium, convulsions or coma
(1). The pathogenesis of tissue
injury and cell death due to heat stroke is not well understood,
which may explain the high morbidity and mortality associated with
heat stroke, as no specific mechanisms can be targeted for
treatment (2,3). Previous studies suggest that systemic
inflammatory response syndrome (SIRS) is directly related to the
severity of the heat insult, which ensues during heat stroke
(4,5). Among the most prominent features of
heat stroke-related SIRS, endothelial injury contributes
significantly to its outcome (6,7).
The activation of coagulation disorders with the
concurrent downregulation of anticoagulant systems and fibrinolysis
are associated with key symptoms, such as hypovolemia and
hypotension. Impaired endothelium promotes platelet adhesion via
the secretion of von Willebrand factor and secretes an
extracellular matrix that supports platelet adhesion following
vascular injury (8). Vascular
injury promotes inflammation, cell adherence to vascular
endothelium and invasion of organs that receive blood supply from
the injured vasculature. Vascular endothelial injury-induced
adhesion, invasion of inflammatory cells and coagulant disorders
contribute to disseminated intravascular coagulation, a syndrome
characterized by widespread intravascular thrombosis that can
compromise adequate blood supply to various organs (9). This serious syndrome has been
implicated in multiple organ dysfunction syndromes (MODSs) and
leads a high mortality of heatstroke (10,11).
Therefore, prevention of endothelial cell injury is a potential
strategy to reduce the high rate of heat shock-induced
morality.
Previous studies have suggested that endothelial
cells are an early target of heat stress injury and that damaged
endothelial cells are prominent features of severe heat stroke and
can be induced to undergo apoptosis during the acute response phase
to heat stress (12,13). NO and equivalent amounts of
citrulline are synthesized from guanidino nitrogen of L-arginine by
nitric oxide synthases (NOS) identified in endothelial cells and
neurons and can be activated in most cell types. NO can also act as
an anti-apoptotic agent (14).
Relatively low concentrations of NO appear to favor cell
proliferation and anti-apoptosis responses, which activate pathways
of cell cycle arrest, mitochondrial respiration and apoptosis
(15). Endothelial NO production
leads to vasodilation and increases tissue perfusion, which leads
to inflammatory cell migration from the vascular to the supported
tissue. This results in an inflammation response in the target
tissue and MODS. Therefore, elevation of NO production in vascular
endothelial cells suppresses inflammation and multi-organ injury
and represents a potential pathway to treat systematic inflammation
response-related diseases including heat stroke.
Sodium tanshinone IIA sulfonate (STS) is a
derivative of tanshinone II A, which is isolated from the root of
Salvia miltiorrhiza. Injection of STS is used widely and
successfully in clinics in China for treating cardiovascular
diseases as a vascular-protecting drug (16). In addition, it is used as a
vascular remodeling inducer and inflammation controller (17). Based on the known functions and
possible mechanisms of STS, the authors tested STS for its ability
to control inflammatory responses due to heat shock. The outcomes
of STS treatment were good, inline with a previous study of the
authors, but the exact mechanism of its therapeutic effect is
poorly understood. Therefore, the present study was conducted to
explore how STS suppresses inflammation, which is associated with
vascular endothelial protection.
Materials and methods
HUVEC culture
HUVECs were obtained from a Chinese Center for Type
Culture Collection (Wuhan, China). Cells were cultured in
Dulbecco's modified Eagle medium (DMEM, Invitrogen, Thermo Fisher
Scientific, Inc., Waltham, MA, USA) with low glucose containing 10%
fetal bovine serum (FBS, Hyclone; GE Healthcare Life Sciences,
Chalfont, UK) and 2 mM L-glutamine (Sigma-Aldrich; Merck KGaA;
G3126) and were grown on tissue culture dishes at 37°C in 5%
CO2 at 100% humidity. Non-adherent cells were removed by
washing with phosphate buffered saline, and the culture liquid was
refreshed every 2 d. Cells were subcultured at 80% confluence.
Primary cultures of HUVECs between passages 5 and 10 were used in
all experiments.
High temperature exposure
HUVECs were grown to ~80% confluence in 100 mm
dishes in DMEM supplemented with 10% FBS (v/v) prior to the start
of the experiments. For the high temperature exposure, the cell
cultures were placed in an incubator chamber flushed with a gas
mixture of 5% CO2 at 42°C for various time periods and
treatments, as needed.
Drug and pharmacological
interventions
The drug STS was used at a final concentration of 2
µg/ml. The following pharmacological inhibitors were used: NOS
inhibitor NGmonomethyl-L-arginine (L-NMMA; 250 mM; EMD Millipore,
Billerica, MA, USA), AKT inhibitor MK-2206 (5 µM; Selleck
Chemicals, Houston, TX, USA), and PI3K inhibitor wortmannin (500
nM; Sigma-Aldrich; Merck KGaA). Each intervention drug was
dissolved in 10% FBS in DMEM and was added to the cell cultures 30
min prior to initiating the high temperature exposure.
Western blotting
HUVECs were lysed in 300 µl lysis buffer (50 mM
Tris, pH 7.4, 1% Triton X-100, 0.25% Na deoxycholate, 150 mM NaCl,
1 mM EGTA, 1 mM PMSF, 1 mM NaF, 10 mg/ml aprotinin, 1 mg/ml
leupeptin and 1 mM orthovanadate). The total protein concentration
was determined using a BCA protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). A measure of 10 mg/well proteins was subjected
to SDS-PAGE on 10% polyacrylamide gels and transferred to
nitrocellulose membranes. The membranes were blocked with 5% bovine
serum albumin at 4°C overnight prior to incubation with antibodies.
Immunoblotting was performed with rabbit anti-eNOS (1:1,000;
catalog no. 32027S; Cell Signaling Technology, Inc., Danvers, MA,
USA), rabbit anti-phospho-eNOS (Ser1177, 1:1,000, catalog no.
9570S; Cell Signaling Technology, Inc.), rabbit anti-Akt (1:1,000,
catalog no. 9272; Cell Signaling Technology, Inc.) or rabbit
anti-phospho-AKT antibody (Ser473, 1:1,000 dilution, catalog no.
sc-7985-R; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
overnight at 4°C. The membrane was then washed and incubated with
horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2,500;
catalog no. 554021; BD Biosciences, Franklin Lakes, NJ, USA) at
room temperature for 1.5 h. The membranes were developed using the
Western Blot Chemiluminescence Detection reagent (PerkinElmer,
Inc., Waltham, MA, USA).
Cell counting kit-8 (CCK-8) assay
The viability of stimulated HUVEC cells was
determined using a CCK-8 assay (Beyotime Institute of
Biotechnology, Haimen, China) following the manufacturer's
protocol. Optical density (OD) at a wavelength of 450 nm (formation
of formazan) was measured using a Thermo Scientific microplate
reader and SkanIt™ software version 3.1 (Thermo Fisher Scientific,
USA).
Flow cytometry
For detection of apoptosis, stimulated HUVEC cells
(1×106) were stained with Annexin V-conjugated to fluorescein
isothiocyanate (FITC) and propidium iodide (PI) by using a FITC
Annexin V/Dead Cell Apoptosis kit (BioVision, Inc., Milpitas, CA,
USA) following the manufacturer's protocol. Briefly, cells were
suspended in 500 µl Annexin V binding buffer and incubated with 5
µl Annexin V and 5 µl PI for 5 min at room temperature in the dark.
Percentages of cells undergoing apoptosis were determined by
dual-color analysis. Immediately after staining, the cells were
analyzed on a flow cytometer using 488 nm excitation and a 525 nm
band pass filter for FITC and a 620 nm filter for PI detection. At
least 20,000 cells were acquired using a Beckman Coulter flow
cytometer and Beckman Summit version 6.1.0 software (Beckman
Coulter, Inc., Brea, CA, USA).
Immunoprecipitation of eNOS and
blotting with phospho-eNOS
HUVECs were washed and incubated in lysis buffer
containing 20 mM Tris-HCl (pH=7.4), 150 mM NaCl, 1 mM EGTA, 1 mM
EDTA, 1% Triton X-100, 1 mM PMSF, 1 mM NaF, 10 mg/ml aprotinin, 1
mg/ml leupeptin and 1 mM orthovanadate for 30 min on ice and
briefly sonicated. Immunoprecipitation was carried out by
incubating the lysates with eNOS monoclonal antibody (1:1,000;
catalog no. 32027S; Cell Signaling Technology, Inc.) (2 mg/mg of
total cell protein) for 16 h at 4°C, followed by a 2 h incubation
period with 1:1 protein A:protein G-sepharose slurry. Following
centrifugation at 4°C and 13,400 × g for 15 min, the
immunoprecipitates were washed with lysis buffer, re-suspended in
loading buffer, boiled for 5 min, and subjected to SDS-PAGE on 10%
polyacrylamide gels and transferred to a nitrocellulose membrane.
The primary antibody used for immunoblotting was anti-phospho-eNOS
(Ser1177, 1:1,000 Cell Signaling Technology, Inc.). The membrane
was then washed and incubated with secondary antibodies conjugated
to horseradish peroxidase (1:2,500; catalog no. 554021; BD
Biosciences) and processed as above.
Measurement of no
HUVECs were grown to ~80% confluence in six-well
plates (1×106 cells per well) and exposed to hypoxia for 30 min.
Next, NO release was measured using nitrate/nitrite colorimetric
assay kits (Cayman Chemical Co, Ann Arbor, MI, USA). Briefly, cell
culture supernatants were incubated with nitrate reductase for 3 h
to convert nitrates into nitrites. The total amount of NO was then
determined using the Griess reagent (Beyotime Institute of
Biotechnology). A standard curve of nitric oxide was determined
using known concentrations of nitrates, and the sample NO
concentrations were calculated using the standard curve. Each assay
was conducted in duplicate, and the results are expressed as
nitrite nM/mg protein in the cells.
Statistical analysis
The cell viability, cell apoptosis ratio and nitrite
concentration results are expressed as the mean ± standard
deviation. The statistical analysis was performed using one-way
analysis of variance followed by the Tukey's post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference. Analyses were performed using the SPSS statistical
software package (version, 20.0; IBM SPSS, Armonk, NY, USA).
Results
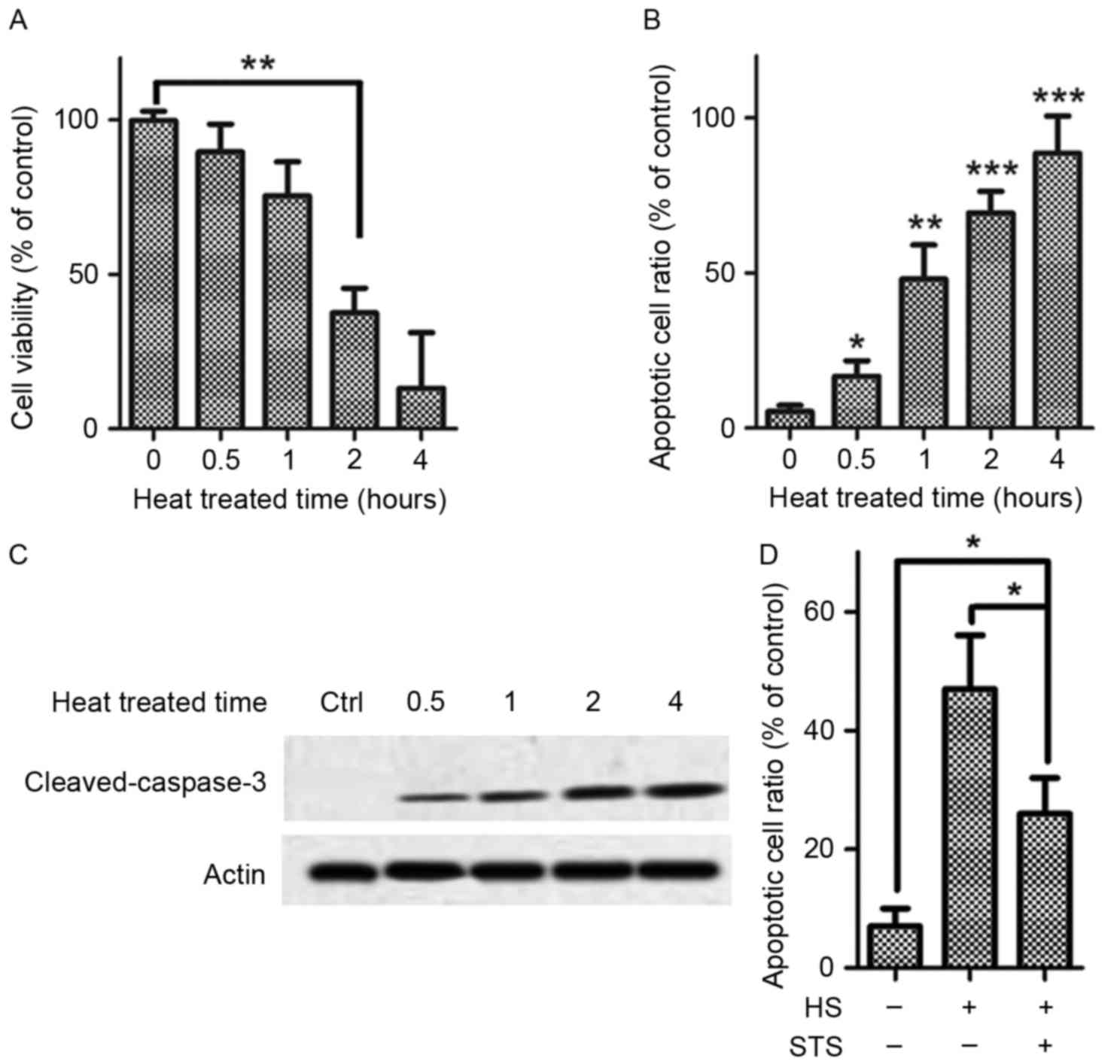
High temperature stimulates apoptosis
of HUVECs
Exposure to high temperature at 42°C for various
time periods (0.5, 1, 2 or 4 h) resulted in a drastic
time-dependent decrease in HUVEC viability. A significant decrease
in HUVEC viability was first observed by 2 h compared with baseline
determined via a CCK-8 assay (Fig.
1A). The apoptosis cell ratio for the HUVECs that were exposure
to the high temperature (42°C) increased significantly by 30 min,
and significant increases in the apoptosis ratio were also found at
2 and 4 h (Fig. 1B). Caspase-3,
the most important effector caspase for both extrinsic and
intrinsic apoptosis pathways and engages subsequent activation of
caspase-3, was detected, including its cleavage in heat-treated
HUVECs. Cleaved caspase-3 accumulated in a time-dependent manner
(Fig. 1C). The results above
suggested that high temperatures suppress HUVEC viability and exert
an apoptosis-inducing effect on HUVECs. Treatment with STS (2
µg/ml) significantly suppressed apoptosis of the HUVECs that were
exposed to high temperature (42°C; Fig. 1D). This result suggests that STS is
a potential suppressor of heat stress-induced apoptosis.
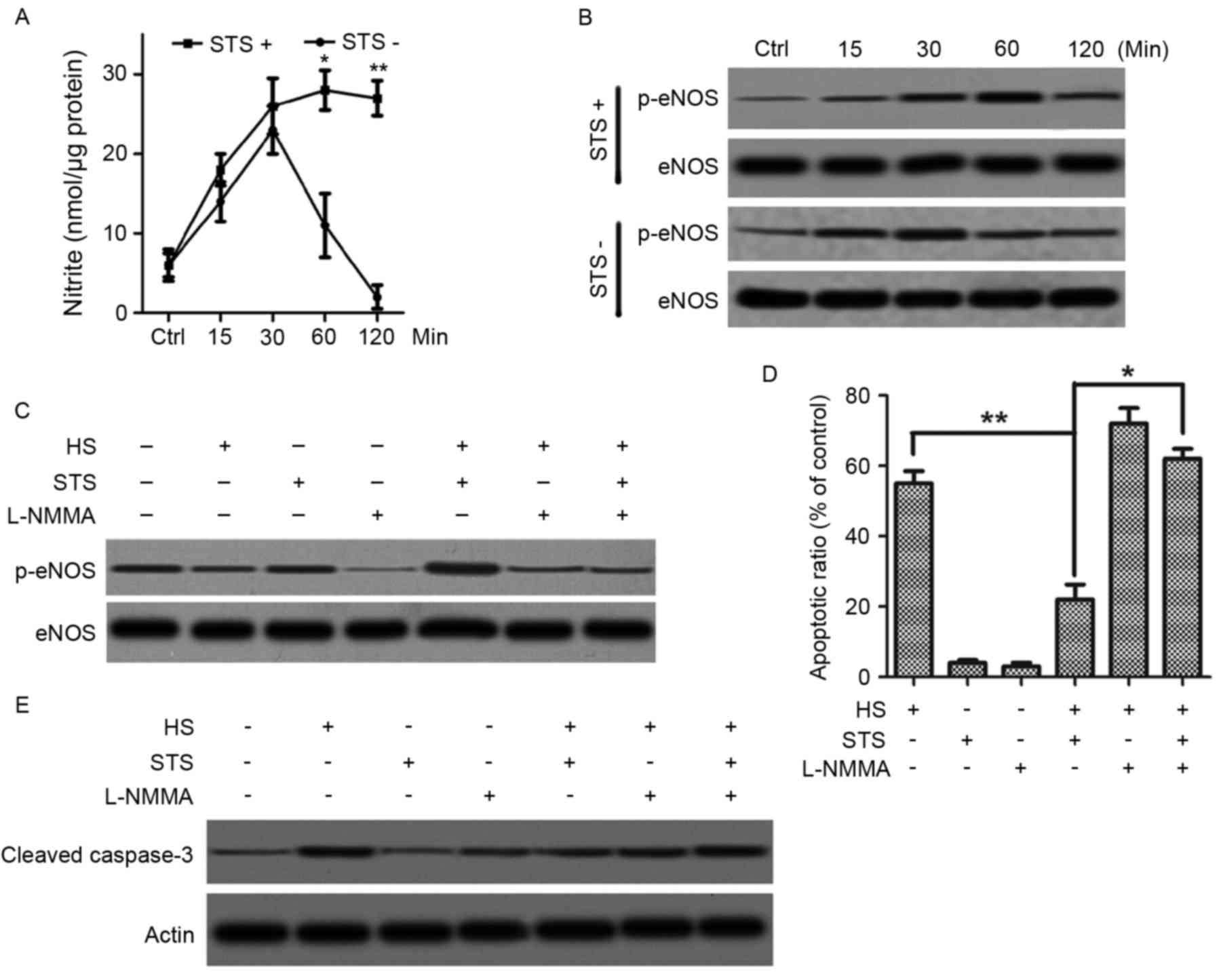
STS inhibits heat stress-induced
apoptosis of HUVECs by upregulating phosphorylated eNOS
NO is a crucial factor in vascular homeostasis and
is endogenously produced by the terminal guanidino nitrogen of
L-arginine of eNOS. To explore the mechanism of the effect of STS
on heat stress-induced apoptosis of HUVECs, NO production was
assessed via the production of nitrite. HUVECs were exposed to a
high temperature at 42°C with or without treatment of STS (2 µg/ml)
for various time periods (15, 30 min, 1 or 2 h). Without treatment
with STS, nitrite production in the HUVECs under heat stress
significantly increased at 0.5 h and decreased sharply at 1 and 2
h, and nitrite production was under the baseline level at 2 h
(Fig. 2A). However, for treatment
with STS, the production of nitrite progressively increased and
reached steady state at 1 h, which was higher than the level of
nitrite for no STS treatment (Fig.
2A). Because eNOS phosphorylation is a key event of NO
production, the authors next determined the change in eNOS
phosphorylation due to STS treatment in HUVECs exposed to heat
stress. The time course of eNOS phosphorylation in high
temperature-exposed cells was determined by immunoprecipitating
with eNOS antibody and immunoblotting with phosphorylated eNOS at
Ser 1177. Exposure to a high temperature at 42°C for various time
periods (15, 30 min 1 and 2 h) resulted in increased eNOS
phosphorylation at 15 and 30 min and sharp decreases in eNOS
phosphorylation at 1 and 2 h compared with baseline (Fig. 2B). However, treatment with 5 µg/ml
STS resulted in a continual increase in eNOS phosphorylation from
15 min to 1 h, which was sustained at 2 h (Fig. 2B). To confirm whether the
STS-induced NO production increase and eNOS phosphorylation were
associated with apoptosis suppression, the eNOS inhibitor L-NMMA
was applied. HUVECs were pretreated with 30 µM L-NMMA for 30 min
followed by exposure to a temperature of 42°C with or without 2
µg/ml STS treatment for 1 h. Pretreatment with L-NMMA resulted in
suppressed nitrite production even after treatment with STS
(Fig. 2C). In addition,
pre-treatment with L-NMMA stimulated apoptosis, which was triggered
by the high temperature even in the presence of STS (Fig. 2D) and also resulted an increase in
caspase-3 cleavage (Fig. 2E).
These results suggested that STS suppressed HUVEC apoptosis that is
otherwise induced by exposure to high temperatures via a mechanism
that promotes NO production by increasing the phosphorylation of
eNOS at Ser 1177.
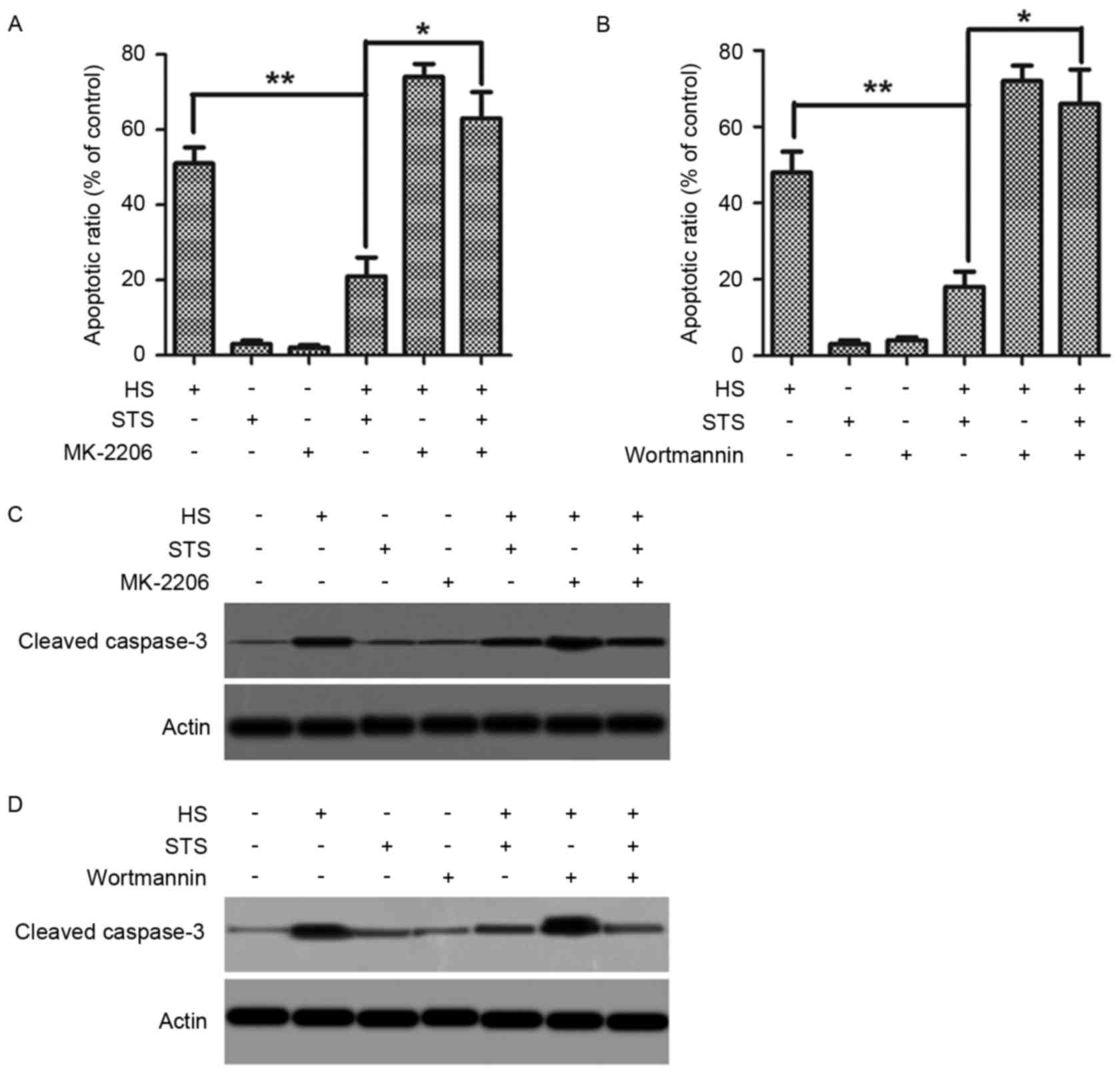
STS promotes phosphorylation of eNOS
in heat-treated HUVECs via the PI3K/AKT pathway
Following this, the authors examined the time course
of AKT phosphorylation in high temperature-exposed HUVECs with or
without STS treatment by immunoblotting cell lysates with an
antibody directed against phosphorylated AKT at Ser 473. HUVECs
were exposed to a high temperature of 42°C for various time periods
(15, 30 min, 1 or 2 h) with or without 2 µg/ml STS treatment.
Without STS treatment, phosphorylated AKT levels at 15 min and 30
min were higher compared to baseline and lower at 1 and 2 h
compared to baseline. However, STS treatment resulted in a
continual increase in AKT phosphorylation in a time-dependent
manner. It appeared that AKT phosphorylation stabilized by 1 h
(Fig. 3A). The AKT inhibitor
MK-2206 was applied to confirm whether eNOS phosphorylation was
regulated by AKT phosphorylation. HUVECs were exposed to a
temperature of 42°C for 1 h with or without 2 µg/ml STS after
pretreatment with 5 µM MK-2206. eNOS phosphorylation levels were
determined by immunoprecipitating with the eNOS antibody and
immunoblotting with phosphorylated eNOS at Ser 1177. Pretreatment
with MK-2206 suppressed the increase in eNOS phosphorylation
induced by STS treatment in the high temperature-exposed HUVECs
(Fig. 3B). The nitrite product was
also determined. Pretreatment with MK-2206 significantly suppressed
the STS-induced increase in nitrite production in the high
temperature-exposed HUVECs (Fig.
3C). The results suggest that STS upregulates eNOS
phosphorylation via AKT activation. Because both eNOS and AKT are
downstream targets of PI3K, whether STS-induced phosphorylation of
eNOS and Akt are regulated by a PI3Kinase-dependent mechanism was
investigated. HUVECs were exposed to a high temperature of 42°C
with or without 2 µg/ml STS treatment following pretreatment with
the PI3K inhibitor wortmannin (500 nM). eNOS phosphorylation at Ser
1177 and AKT phosphorylation at Ser 473 were detected, as above.
Treatment with wortmannin suppressed the increases in eNOS and AKT
phosphorylation that is induced by STS in high temperature-exposed
HUVECs (Fig. 3D and E). In
addition, treatment with wortmannin suppressed the increased
nitrite levels that are induced by STS in high temperature-exposed
HUVECs (Fig. 3F). The results
suggested that PI3K promotes AKT and eNOS phosphorylation, thus
stimulating NO production in high temperature-exposed HUVECs.
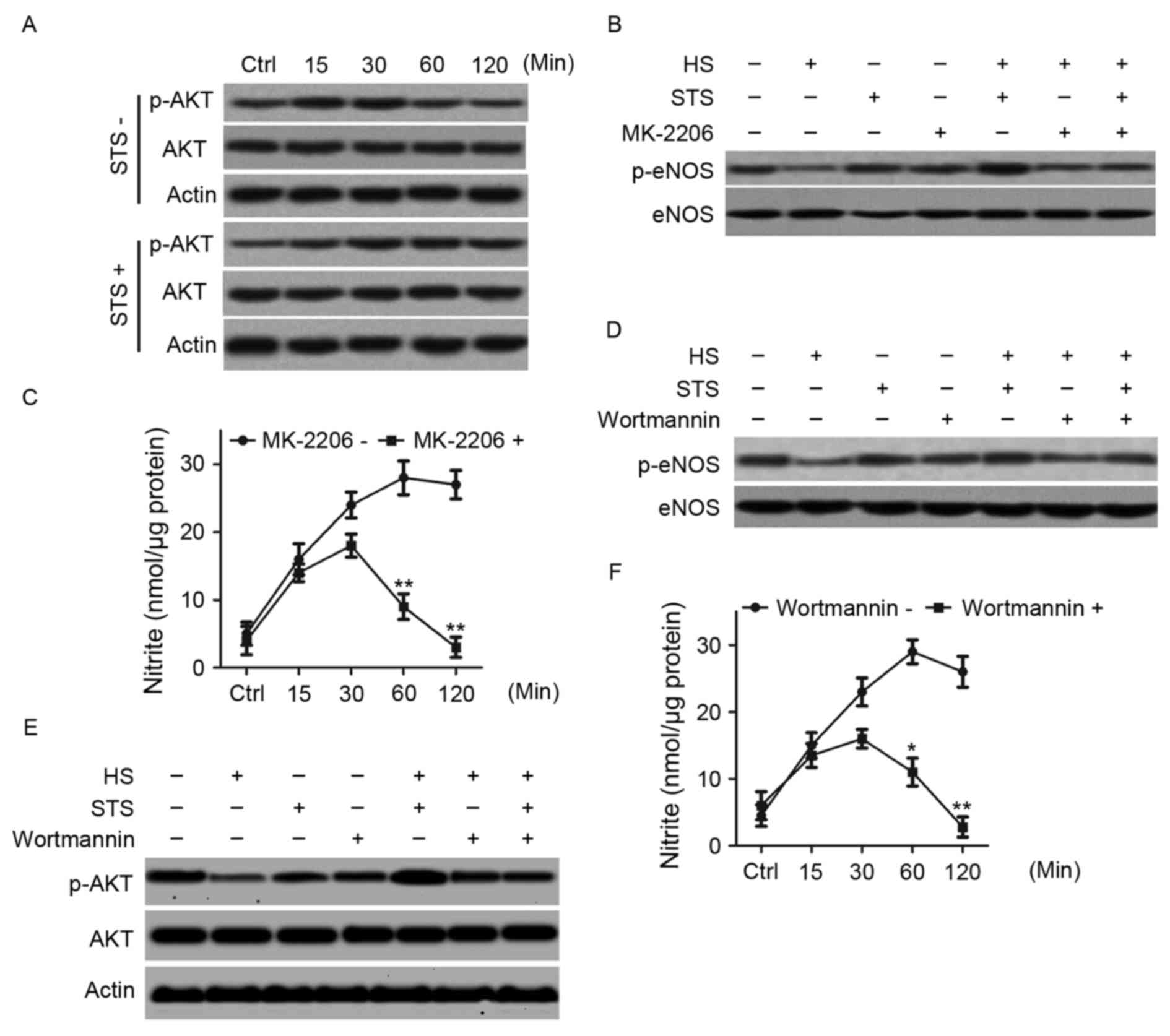 | Figure 3.STS activated the PI3K/AKT pathway to
promote nitric oxide production in HUVECs under heat stress. HUVECs
were exposed to 42°C for various time periods (15, 30 min, 1 or 2
h) with or without 2 µg/ml STS treatment. (A) The total HUVEC
proteins of were used for western blot analysis with antibodies
against AKT, phosphorylated AKT (at Ser 473) and β-actin. HUVECs
were exposed to 42°C for 1 h with or without 5 µM MK-2206
pre-treatment for 30 min and with or without treatment with 2 µg/ml
STS, which was added 30 min in advance. (B) Whole-cell lysates were
immunoprecipitated with an eNOS antibody and immunoblotted with
phosphorylated eNOS (at Ser 1177). (C) The HUVECs were exposed to
42°C for 1 h and STS was added 30 min in advance with or without
pre-treatment with 5 µM MK-2206 for 30 min. Nitrite in whole cell
lysates were determined using nitrite kits for the different time
periods, as indicated (15, 30 min, 1 or 2 h). *P<0.05,
**P<0.01 vs. controls. HUVECs were exposed to 42°C for 1 h with
or without 500 nM wortmannin pre-treatment for 30 min and with or
without 2 µg/ml STS treatment, which was added 30 min in advance.
(D) Whole-cell lysates were collected and analyzed as described in
(B). (E) Whole cell lysates were collected and analyzed as
described in (A). (F) The HUVECs were exposed to 42°C for 1 h, and
STS was added 30 min in advance with or without 500 nM wortmannin
pre-treatment for 30 min. Nitrite in whole cell lysates were
determined using nitrite kits for different time periods, as
indicated (15, 30 min, 1, or 2 h). *P<0.05, **P<0.01 vs.
controls. STS, sodium tanshinone IIA sulfonate; PI3K,
phosphatidylinositol-3 kinase; AKT, protein kinase B; HUVECs, human
umbilical vein endothelial cells; eNOS, endothelial nitric oxide
synthase. |
STS suppressed heat stress-stimulated
apoptosis involving PI3K/AKT/eNOS pathway activation
The effects of treatment with the eNOS
phosphorylation inhibitor L-NMMA, the AKT phosphorylation inhibitor
MK-2206, or the PI3K inhibitor wortmannin on heat stress-stimulated
apoptosis were observed to test whether the PI3K/AKT/eNOS pathway
serves a critical role in the apoptosis-suppressing function of
STS. Following pretreatment with or without MK-2206 or wortmannin,
HUVECs were exposed to a high temperature of 42°C with or without
treatment with 2 µg/ml STS, and apoptosis cell ratios were examined
via flow cytometry. Under the heat stress condition, pretreatment
with MK-2206 resulted in an increase in the apoptosis cell ratio
regardless of STS treatment (Fig.
4A). The same phenomenon occurred due to pretreatment with
wortmannin (Fig. 4B). Whole-cell
lysates were used to analyze the cleavage of casepase-3.
Pretreatment with MK-2206 resulted in an increase in cleaved
casepase-3 regardless of the existence of STS (Fig. 4C). Pretreatment with wortmannin
produced the same results (Fig.
4D). These results suggested that STS suppresses the
heat-induced apoptosis of HUVECs though the PI3K/AKT pathway. The
data presented in Fig. 2D and E
demonstrate that the reduction in heat-induced apoptosis also
involved eNOS phosphorylation at Ser 1177. STS-induced eNOS
phosphorylation was regulated by PI3K/AKT, as presented in Fig. 3. Therefore, the authors believe
that STS suppressed heat stress-induced apoptosis by stimulating
the PI3K/AKT/eNOS pathway, which was also critical for the increase
in STS-induced NO production.
Discussion
The present study of the effects of high temperature
on HUVECs demonstrated that heat stress stimulated a reduction in
cell viability and enhancements in caspase-3 cleavage and apoptosis
in a time-dependent manner. In addition, heat stress induced eNOS
phosphorylation at Ser 1155 in as early as 15 min, peaking at 30
min, and then sharply decreasing after 1 h. However, STS treatment
significantly suppressed the heat stress-induced apoptosis,
maintaining elevated eNOS phosphorylation for up to 2 h, and led to
an increase in NO production. The STS-induced increase in NO
production was ablated by pretreatment with the eNOS inhibitor,
L-NMMA. The present studies also revealed that STS treatment
induces a significant increase in AKT phosphorylation. Further,
inhibition of AKT activation induced significant elevations in
STS-induced eNOS phosphorylation and NO production. Additionally,
inhibition of the PI3Kinase resulted in significant suppression of
the STS-induced increases in AKT phosphorylation, eNOS
phosphorylation and NO production. In addition, these data
demonstrated that inhibition of the PI3K/AKT pathway suppressed the
increase in STS-induced NO production.
The present data suggested that heat stress
suppresses the physiological viability of endothelial cells,
promoting apoptosis and inhibiting the synthesis and release of
vasoactive substances. STS is a potential inhibitor of heat
stress-induced apoptosis, and NO is considered an important
mediator of the effect. A previously published study on endothelial
cells have demonstrated that NO production is regulated by eNOS
activity (18). NOS
phosphorylation at Ser 1177 has been widely considered an important
mechanism for increasing NO production under various stimuli,
including shear stress. The current study of HUVECs under heat
stress demonstrated that high temperatures stimulate
phosphorylation of eNOS at Ser 1177, increasing NO production at an
early stage. However, the mechanism does not apply to long-term
exposure to high temperatures. However, STS treatment promotes eNOS
phosphorylation at Ser1177 and increases NO production in a
sustained manner in HUVECs, even under heat stress conditions.
Furthermore, the present study suggests that inhibition of eNOS
results in suppression of the STS-induced increase in NO
production. Furthermore, the study revealed that inhibition of
eNOS, either by a PI3K inhibitor or an AKT phosphorylation
inhibitor, significantly attenuates STS-induced eNOS activity and
NO production. Therefore, eNOS phosphorylation at Ser 1177 is
likely required for STS-induced eNOS activation and NO production
in HUVECs under heat stress conditions.
AKT, also known as protein kinase B, is an important
oncogene involved in cell survival. AKT is phosphorylated by
tyrosine kinase G-coupled receptors under shear stress, ultimately
downregulating signals (19). The
PI3K/AKT/eNOS phosphorylation signal pathway has been reported to
serve a significant role in NO production. Blocking of the PI3K/AKT
pathway leads to reduced NO release in human endothelial cells. In
addition, eNOS phosphorylation caused by shear stress in cultured
endothelial cells has been reported to be related to PI3K/AKT
signaling (19). Furthermore,
overexpression of a constitutively active AKTMyr mutant
increased eNOS phosphorylation and NO production (20). The present study demonstrated that
STS treatment markedly increases AKT phosphorylation, and a
blockade of PI3K/AKT suppresses STS-induced eNOS phosphorylation,
which indicates that the PI3K/AKT pathway regulates STS-induced
eNOS phosphorylation.
STS is a derivative of tanshinone II A and is used
for the treatment of diabetic nephropathy, acting as an
anti-inflammatory and anti-fiber agent. Some studies have revealed
that STS suppress inflammatory responses (21,22).
Our previous study demonstrated that STS has potential in
systematic inflammation inhibition. The present study suggests that
STS prevents heat stress-induced endothelium injury. This may be
the mechanism responsible for STS-induced systematic
anti-inflammation.
References
|
1
|
Bouchama A and Knochel JP: Heat stroke. N
Engl J Med. 346:1978–1988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Geng Y, Ma Q, Liu YN, Peng N, Yuan FF, Li
XG, Li M, Wu YS, Li BL, Song WB, et al: Heatstroke induces liver
injury via IL-1β and HMGB1-induced pyroptosis. J Hepatol.
63:622–633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang J, Deng P, Jiang Y, Tang Y, Chen B,
Su L and Liu Z: Role of HMGB1 in propofol protection of rat
intestinal epithelial cells injured by heat shock. Cell Biol Int.
37:262–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leon LR and Bouchama A: Heat stroke. Compr
Physiol. 5:611–647. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Epstein Y, Roberts WO, Golan R, Heled Y,
Sorkine P and Halpern P: Sepsis, septic shock, and fatal exertional
heat stroke. Curr Sports Med Rep. 14:64–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iba T, Gando S, Murata A, Kushimoto S,
Saitoh D, Eguchi Y, Ohtomo Y, Okamoto K, Koseki K, Mayumi T, et al:
Predicting the severity of systemic inflammatory response syndrome
(SIRS)-associated coagulopathy with hemostatic molecular markers
and vascular endothelial injury markers. J Trauma. 63:1093–1098.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gando S: Systemic inflammatory response
syndrome (SIRS) and endothelial cell injury. Nihon Rinsho.
62:2244–2250. 2004.(In Japanese). PubMed/NCBI
|
|
8
|
Sylman JL, Artzer DT, Rana K and Neeves
KB: A vascular injury model using focal heat-induced activation of
endothelial cells. Integr Biol (Camb). 7:801–814. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hawiger J, Veach RA and Zienkiewicz J: New
paradigms in sepsis: From prevention to protection of failing
microcirculation. J Thromb Haemost. 13:1743–1756. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Krau SD: Heat-related illness: A hot topic
in critical care. Crit Care Nurs Clin North Am. 25:251–262. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bruchim Y, Loeb E, Saragusty J and Aroch
I: Pathological findings in dogs with fatal heatstroke. J Comp
Pathol. 140:97–104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Y, Zhou G, Wang Z, Guo X, Xu Q, Huang
Q and Su L: NF-κB signaling is essential for resistance to heat
stress-induced early stage apoptosis in human umbilical vein
endothelial cells. Sci Rep. 5:135472015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li L, Tan H, Gu Z, Liu Z, Geng Y, Liu Y,
Tong H, Tang Y, Qiu J and Su L: Heat stress induces apoptosis
through a Ca2+-mediated mitochondrial apoptotic pathway
in human umbilical vein endothelial cells. PLoS One. 9:e1110832014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim YM, Bombeck CA and Billiar TR: Nitric
oxide as a bifunctional regulator of apoptosis. Circ Res.
84:253–256. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Napoli C, Paolisso G, Casamassimi A,
Al-Omran M, Barbieri M, Sommese L, Infante T and Ignarro LJ:
Effects of nitric oxide on cell proliferation: Novel insights. J Am
Coll Cardiol. 62:89–95. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takahashi K, Ouyang X, Komatsu K, Nakamura
N, Hattori M, Baba A and Azuma J: Sodium tanshinone IIA sulfonate
derived from Danshen (Salvia miltiorrhiza) attenuates hypertrophy
induced by angiotensin II in cultured neonatal rat cardiac cells.
Biochem Pharmacol. 64:745–749. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang C, Li H, Zhou K, Luo C, Li Y, Xie L
and Hua Y: Sodium tanshinone IIA sulfonate and sodium danshensu
open the placental barrier through down-regulation of placental
P-glycoprotein in mice: Implications in the transplacental digoxin
treatment for fetal heart failure. Int J Cardiol. 176:1331–1333.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cirino G, Fiorucci S and Sessa W C:
Endothelial nitric oxide synthase: The Cinderella of inflammation?
Trends Pharmacol Sci. 24:91–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dimmeler S, Fleming I, Fisslthaler B,
Hermann C, Busse R and Zeiher AM: Activation of nitric oxide
synthase in endothelial cells by Akt-dependent phosphorylation.
Nature. 399:601–605. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gonzalez E, Kou R, Lin AJ, Golan DE and
Michel T: Subcellular targeting and agonist-induced site-specific
phosphorylation of endothelial nitric-oxide synthase. J Biol Chem.
277:39554–39560. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Li W, Zhu S, Jundoria A, Li J,
Yang H, Fan S, Wang P, Tracey KJ, Sama AE and Wang H: Tanshinone
IIA sodium sulfonate facilitates endocytic HMGB1 uptake. Biochem
Pharmacol. 84:1492–1500. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu Q, Wei B, Wei L, Hua K, Yu X, Li H and
Ji H: Sodium tanshinone IIA sulfonate ameliorates ischemia-induced
myocardial inflammation and lipid accumulation in Beagle dogs
through NLRP3 inflammasome. Int J Cardiol. 196:183–192. 2015.
View Article : Google Scholar : PubMed/NCBI
|