Introduction
Epilepsy is the most common brain disorder affecting
>50 million people worldwide (1). Despite advances in therapies for
epilepsy, a third of patients with epilepsy are non-responsive or
respond poorly to anti-epilepsy drugs (AEDs) and subsequently
develop drug-resistant epilepsy (DRE) (2). There are a number of theories as to
how this develops, including the involvement of P-glycoprotein
(P-gp) overexpression, which is an efflux pump that is anchored at
the blood-brain barrier (3,4).
This has been confirmed by brain tissues collected during surgery
for DRE (5). P-gp is a multidrug
resistance protein and is encoded by MDR1A/B gene. It is
primarily distributed at the luminal surface of capillary
endothelial cells. However, P-gp can also be expressed in glial
cells and neurons. Experiments have demonstrated that P-gp pumps
out AEDs from neuronal cells to reduce the concentration of AEDs in
brain tissue (6). Inhibiting P-gp
expression may improve the efficacy of AEDs in DRE (7,8).
Therefore, P-gp is postulated to be a novel clinical target for the
treatment of multidrug refractory epilepsy (9). However, the exact underlying
mechanisms of P-gp overexpression and its regulatory pathways in
DRE are not fully understood.
The immunity and inflammatory processes affecting
P-gp expression in epilepsy have been established (10–12).
Seizures may trigger multidimensional local inflammatory reactions
in brain (13) that are involved
in the generation and propagation of epileptic activity.
High-mobility group box 1 (HMGB1), a ubiquitous chromatin
component, is passively released by stressed or necrotic cells, and
is overexpressed in epileptic brains. The interaction between HMGB1
and Toll-like receptor 4 (TLR4) stimulates the innate immune system
and inflammation in tissues (14,15).
Our previous study demonstrated that P-gp may be downregulated by
inhibition of TLR4 or HMGB1 during kainic acid (KA)-induced
seizures in rat brains (16).
Receptor for advanced glycation end-product (RAGE), another
important ligand of HMGB1, has key role in innate immune activation
and seizures (17). However, it is
unknown whether RAGE activation contributes to the upregulation of
P-gp following seizures. Nuclear factor-κB (NF-κB) is a downstream
signaling molecule of RAGE, and is a ubiquitous transcription
factor that serves a role in the regulation of immune response and
inflammation (18). Our previous
study suggested that silencing of inhibitor of NF-κB kinase subunit
β (IKKβ), which regulates the activity of NF-κB, reduced the
expression levels of P-gp and reduced seizures (19). HMGB1 binds to RAGE to stimulate the
biological effects of NF-κB. RAGE/NF-κB is known to be a regulator
of inflammation (20,21). The present study investigated
whether HMGB1 binds to RAGE, leading to activation of NF-κB and
enhanced transcription of MDR1A/B and P-gp expression during
brain seizures. The results of the present study may provide a
molecular mechanism and a novel target for the treatment of
DRE.
Materials and methods
Animals
Male Sprague-Dawley (SD) rats (age, 6–7 weeks;
weight, 200–250 g) were purchased from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China) and were housed under controlled
standard conditions at 65±5% humidity and 21±2°C, a 12 h light/dark
cycle and with food and water available. The rats were housed under
these conditions for one week prior to treatment. Experimental
procedures were approved by the Animal Ethics Committee of Nanjing
Medical University, according to international standards.
Experimental groups
Animals were divided randomly into 6 groups: Sham
group (treated with saline as a control, n=21); EP group (treated
with pilocarpine; n=78); small interfering RNA (siRNA) group
(treated with pilocarpine plus siRNA; n=24);
pyrrolidinedithiocarbamic acid (PDTC) group (treated only with
PDTC; n=12); EP + PDTC group (treated with pilocarpine plus PDTC;
n=12); scrambled group (treated with pilocarpine plus scrambled
siRNA; n=4).
Surgery for intracerebroventricular
(ICV) injection
The purpose of the operation was to inject siRNA. SD
rats were anesthetized with 10% chloral hydrate (3 ml/kg) and
placed on stereotactic apparatus. A stainless steel single guide
cannula (PlasticsOne Inc., Roanoke, VA, USA) was implanted into the
right lateral ventricle with coordinates of 0.8 mm posterior to the
bregma, 1.5 mm lateral to the midline and 4.5 mm depth to the
surface of the skull (22). The
cannula was fixed with dental cement (Heraeus Kulzer GmbH,
Wehrheim, Germany).
Pilocarpine-induced status epilepticus
(SE)
Rats were administrated with lithium chloride (127
mg/kg; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) by
intraperitoneal (IP) injection 24 h before pilocarpine treatment.
Methyl scopolamine (1 mg/kg, IP; Sigma-Aldrich) was delivered 30
min before pilocarpine to reduce the peripheral effects and to
enhance survival. A single dose of pilocarpine (30 mg/kg, IP)
(Sigma-Aldrich) was administered. Subsequently, rats received
repeated injections of pilocarpine (10 mg/kg, IP) every 30 min
until they developed convulsive seizures. The maximum number of
pilocarpine injections was 5 per animal (23). Diazepam (10 mg/kg; Sigma-Aldrich)
was utilized to terminate seizure activity 90 min after the onset
of SE. Severity of the convulsions were evaluated by Racine's
classification and denoted the following stages: 0, behavioral
arrest; 1, face clonus; 2, head nodding; 3, forelimb clonus; 4,
forelimb clonus and rearing, 5, forelimb clonus with rearing and
falling (24). Animals classified
as less than Racine's stage 4 were excluded from the experiment.
The sham groups were injected with normal saline instead of
pilocarpine.
siRNA micro-injection and PDTC
administration
siRNAs targeting HMGB1 and RAGE mRNA were designed
and synthesized by Shanghai GenePhama Co., Ltd. (Shanghai, China).
siRNA sequences are listed in Table
I. As described previously, the rats of the siRNA group were
treated with 10 µl siRNA (5 µmol/l) (25) by a microinjection pump via a
pre-implanted cannula 30 min prior to pilocarpine injection.
Following each injection, the needle was left in place for 5 min to
allow for drug diffusion. The sham group was treated with 10 µl
saline. The Scrambled group was administrated with 10 µl scrambled
siRNA (5 µmol/l) and served as the negative control.
 | Table I.siRNA sequences. |
Table I.
siRNA sequences.
| siRNA | Sequence
(5′-3′) |
|---|
| HMGB1-F |
GGAAGACGAAGAUGAAGAATT |
| HMGB1-R |
UUCUUCAUCUUCGUCUUCCTT |
| RAGE-F |
GCCGGAAAUUGUGAAUCCUTT |
| RAGE-R |
AGGAUUCACAAUUUCCGGCTT |
| Negative
control-F |
UUCUCCGAACGUGUCACGUTT |
| Negative
control-R |
ACGUGACACGUUCGGAGAATT |
Rats in the EP + PDTC group were treated with PDTC
(100 mg/kg, IP; BioVision, Inc., Milpitas, CA, USA) at 14.5 h and
15 min prior to the first pilocarpine injection and at 90 min after
onset of SE. A further two injections of PDTC at the same dose was
administrated 5 h after termination of SE and 23.5 h after the
first injection of pilocarpine (26). The sham group was treated with the
same volume of saline and the PDTC group was treated IP with 100
mg/kg PDTC alone.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the hippocampus of the
rats using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's protocol.
The concentration and purity of the RNA was measured
spectrophotometrically at 260 and 280 nm. Total RNA (500 ng) was
subjected to DNase I digestion (Takara Bio, Inc., Otsu, Japan), and
was subsequently utilized for cDNA synthesis using the PrimeScript™
RT Master Mix (Takara Bio, Inc.). qPCR was then performed using the
SYBR® Premix Ex Taq™ kit (Takara Bio, Inc.) with 1 µl 0.5 µM
forward and reverse primers (final concentration, 0.5 µm; Sangon
Biotech Co., Ltd., Shanghai, China; Table II) and 8 µl diluted cDNA on a 7300
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Cycling conditions were as follows: Initial predenaturation
step at 95°C for 30 sec, followed by 40 cycles of denaturation at
95°C for 5 sec and annealing at 60°C for 31 sec. The relative mRNA
expression of the target gene was normalized to GAPDH and was
calculated using the 2−ΔΔCq method (27).
| Table II.Primers used for reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Primers used for reverse
transcription-quantitative polymerase chain reaction.
| Primer | Sequence
(5′-3′) | Product size
(bp) |
|---|
| HMGB1-F |
CTGATGCAGCTTATACGAAG | 20 |
| HMGB1-R |
TCAGGTAAGGAGCAGAACAT | 20 |
| RAGE-F |
GAATCCTCCCCAATGGTTCA | 20 |
| RAGE-R |
GCCCGACACCGGAAAGT | 17 |
| MDR1A-F |
GAGTGAAAAGGTCGTCCAGGAAGCG | 25 |
| MDR1A-R |
TCTCGCATGGTCACAGTTCATGAGC | 25 |
| MDR1B-F |
CCCAAAGTGACACTGGTGCCTCTG | 24 |
| MDR1B-R |
GCCTGGAGCCCATAGCCCCTTTA | 24 |
| GAPDH-F |
ATGACTCTACCCACGGCAAG | 20 |
| GAPDH-R |
TACTCAGCACCAGCATCACC | 20 |
Western blot analysis
Total protein was extracted from the hippocampus of
rats using a protein extraction kit (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China). Samples were homogenized and centrifuged at
14,000 × g for 15 min, and the supernatant was harvested and
diluted to 6.7 µg/µl in SDS/bromophenol blue loading buffer, prior
to heating for 5 min at 100°C. Samples were loaded onto 10% gels,
separated by SDS-PAGE and transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes
were blocked in 5% bovine serum albumin (Sigma-Aldrich; Merck KGaA)
at room temperature for 2 h and subsequently incubated at 4°C
overnight with the following primary antibodies: Rabbit anti-HMGB1
(1:1,000; catalog no. ab18256; Abcam, Cambridge, UK); rabbit
anti-RAGE (1:1,000; catalog no. ab3611; Abcam); mouse anti-P-gp
(1:50, catalog no. 517310; Calbiochem; EMD Millipore); rabbit
anti-phosphorylated-NF-κB p65 (p-p65) (1:1,000, catalog no. 3033;
Cell Signaling Technology, Inc., Danvers, MA, USA). Following
incubation with horseradish peroxidase-conjugated goat anti-rabbit
secondary antibodies (1:5,000; catalog no. ZB230; ZSGB-BIO,
Beijing, China) or anti-mouse secondary antibodies (1:5,000;
catalog no. ZB2305; ZSGB-BIO) for 1.5 h at room temperature,
proteins were detected using electrochemiluminescence (catalog no.
WBKLS0500; EMD Millipore) according to the manufacturer's
instructions, with Quantity One software (version 4.62; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The optical density of each
sample was measured using Scion Image software (version 1.47; Scion
Corporation, Frederick, MD, USA). The optical density of each
sample was normalized to the mouse anti-β-actin (1:500; catalog no.
BM0627; Wuhan Boster Biological Technology, Ltd., Wuhan, China)
loading control.
Immunofluorescence
Rat brains were fixed in 4% paraformaldehyde
following trans-cardiac perfusion overnight at 4°C, and
subsequently soaked in 20 and 30% sucrose for 2 to 3 days,
respectively. The brains were then embedded into optimal cutting
temperature compound (catalog no. C1400; Applygen Technologies,
Inc., Beijing, China). Sections (8-µm thick) were prepared using a
vibrating slicer and staining was performed using a standard
procedure (28). Briefly, sections
were blocked in 0.01 mol/l serum (either rabbit or mouse according
to the secondary antibody used; Wuhan Boster Biological Technology,
Ltd.) at room temperature for 2 h. The redundant serum was removed
then sections were incubated with the following primary antibodies:
Rabbit anti-HMGB1 (1:500; catalog no. ab18256; Abcam), rabbit
anti-RAGE (1:200; catalog no. ab3611; Abcam) and mouse anti-P-gp
(1:50; catalog no. 517310; EMD Millipore) at 4°C overnight. Goat
anti-rabbit FITC-conjugated IgG (1:1,000 PBS dilution; catalog no.
BA1105; Wuhan Boster Biological Technology, Ltd.) or goat
anti-mouse cyanine 3-conjugated IgG (1:1,000 PBS dilution; catalog
no. BA1101; Wuhan Boster Biological Technology, Ltd.) secondary
antibodies were added at room temperature for 1 h, followed by DAPI
(catalog no. AR1177; Wuhan Boster Biological Technology, Ltd.)
nuclear stain. Sections were visualized using a Leica fluorescence
microscope (DM4000; Leica Microsystems GmbH, Wetzlar, Germany) and
the Leica QWin version 3 software (Leica Microsystems GmbH) was
used for analysis. Different cell types were identified by
analyzing cell morphology.
Statistical analysis
Data was analyzed using SPSS software version 13.0
(SPSS, Inc., Chicago, IL, USA) and are presented as the mean ±
standard deviation. Significant differences were analyzed using a
one-tailed Student's t-test, or by one-way analysis of variance
followed by the Least Significant Difference post-hoc test for
multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Seizure behavior
The concentration of pilocarpine (35.50±9.12 mg/kg)
utilized for inducing seizures was equal to that of previous
experiments (26). Following
pilocarpine administration, hypersalivation, piloerection, licking,
chromodacryorrhea, swallowing and chewing occurred immediately.
Subsequently, clonic movements of forelimbs, head bobbing and motor
limbic seizures were observed. The average latency of first
seizures (29) and from first
pilocarpine injection to SE were 35.02±24.43 and 37.93±24.78 min,
respectively. Induction success refers to the percentage of rats
who had seizures, however, the level of which may not be suitable
for further experiments; this also includes rats who had seizures
however, they succumbed within 90 min following SE. Model success
refers to the percentage of rats whose seizure level reached the
experimental requirements; these rats survived the 90 min period
following SE. The rates of induction and model success in rats in
the present study were 91.28 and 82.05%, respectively. The rate of
failure, the percentage of rats that were not model successful, was
8.21% and the rate of total mortality, which included
anesthesia-associated mortality, rats succumbing during the
induction of SE and mortality prior to the scheduled time of
specimen collection, was 18.46%.
Expression of HMGB1, RAGE, p-p65 and
P-gp following SE
At 6, 12, 24, 48 and 72 h, and 5 and 7 days
post-termination of seizures, HMGB1, RAGE, p-p65 and P-gp were
measured in the hippocampus of rat brains by western blotting.
Compared with the Sham group, the expression levels of HMGB1 were
significantly enhanced following 48 h (P<0.05), 72 h
(P<0.0001) and 5 days (P<0.01) SE termination, as were the
expression levels of RAGE (P<0.01 and P<0.05) and P-gp
(P<0.05 and P<0.0001) at 72 h and 5 days respectively, and
p-p65 was significantly enhanced following 72 h (P<0.01;
Fig. 1). However, the expression
levels of HMGB1 were reduced at 24 h and 7 days compared with 12 h
and 5 days SE termination, respectively (P<0.05; Fig. 1). The expression levels of RAGE
(P<0.05) and p-p65 (P<0.01) were reduced following 7 days
compared with 72 h, as were the expression levels of p-p65 and P-gp
at 7 days compared with 5 days termination (P<0.05; Fig. 1). The greatest expression of HMGB1,
RAGE and p-p65 was observed at 72 h, and declined at later time
points (Fig. 1). However,
expression levels of P-gp continued to increase at 5 days, which
were subsequently reduced by 7 days (Fig. 1).
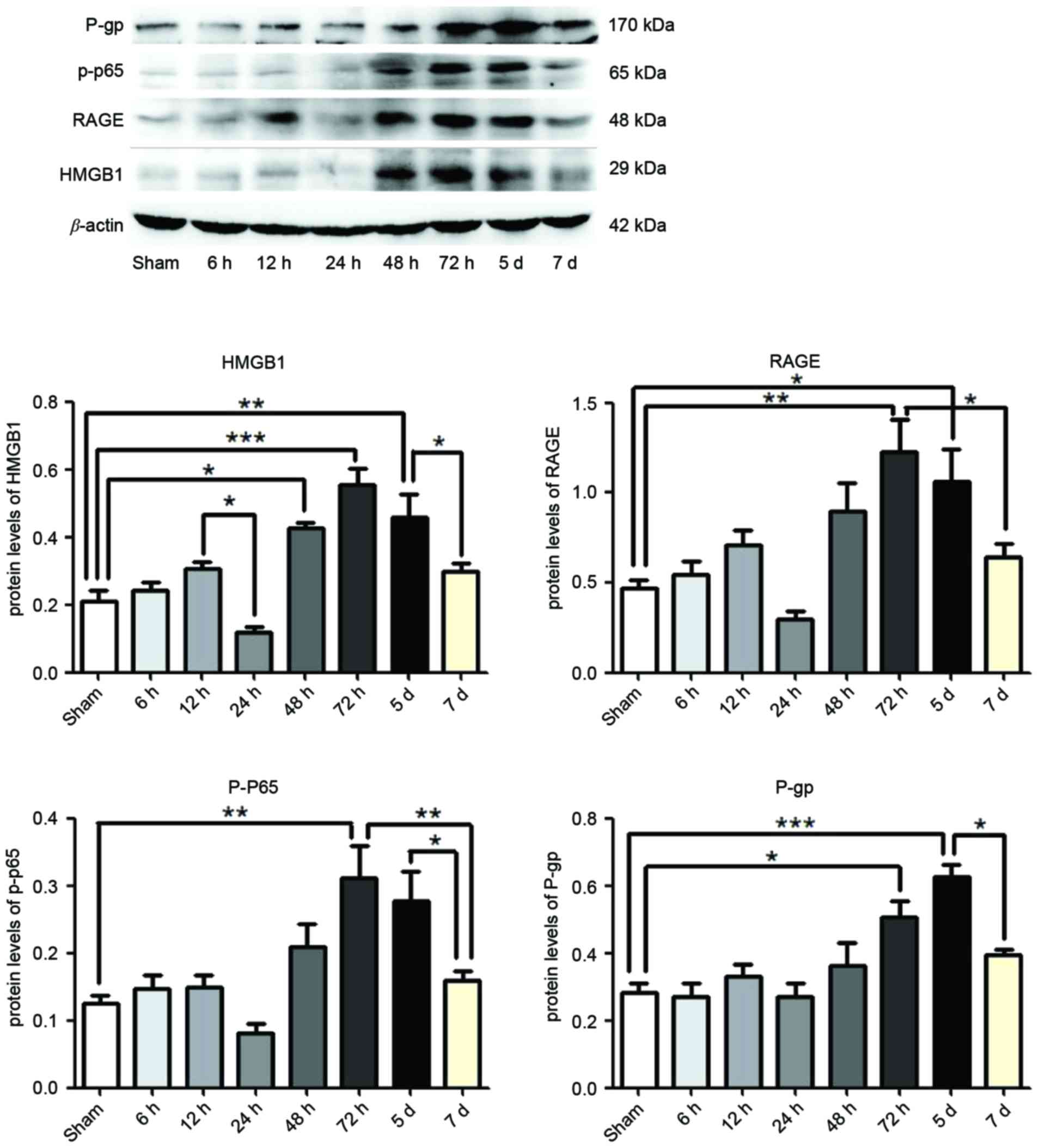 | Figure 1.Protein expression levels of HMGB1,
p-p65, RAGE and P-gp in the hippocampus following status
epilepticus. Proteins were measured at 6, 12, 24, 48, 72 h, 5 and 7
days after termination of seizures, by western blotting. β-actin
served as the loading control. Densitometry analysis was performed
and data are presented as mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.0001. HMGB1, high-mobility group box 1;
RAGE, receptor for advanced glycation end-product; p-p65,
phosphorylated-NF-κB p65; P-gp, P-glycoprotein. |
HMGB1 siRNA pre-treatment inhibits
seizure-induced overexpression of p-p65, RAGE and P-gp
At 30 min prior to IP pilocarpine treatment, HMGB1
siRNA was administered to rats (ICV injection). At 3 days after SE
termination, HMGB1, RAGE, p-p65 and P-gp expression levels were
measured by western blotting. Protein levels of HMGB1 (P<0.01),
RAGE (P<0.01), P-gp (P<0.01) and p-p65 (P<0.0001) in the
EP group were significantly greater compared with expression in the
sham group (Fig. 2A).
Pre-treatment with HMGB1 siRNA significantly reduced the expression
levels of all proteins compared with the EP and scrambled groups
(P<0.01). The mRNA expression levels of HMGB1, RAGE, MDR1A and
MDR1B supported the western blotting data (Fig. 2B). Immunofluorescence staining of
tissue sections demonstrated that HMGB1 was localized to the nuclei
and cytoplasm of neurons (Fig.
2C). P-gp was primarily localized to blood vessels. HMGB1 and
P-gp were expressed in low levels in the Sham group; however, a
greater number of cells expressed HMGB1 and P-gp in the hippocampal
CA3 region of the EP group. Compared with the EP group, the number
of cells expressing HMGB1 and P-gp was reduced in the group treated
with HMGB1 siRNA (Fig. 2C).
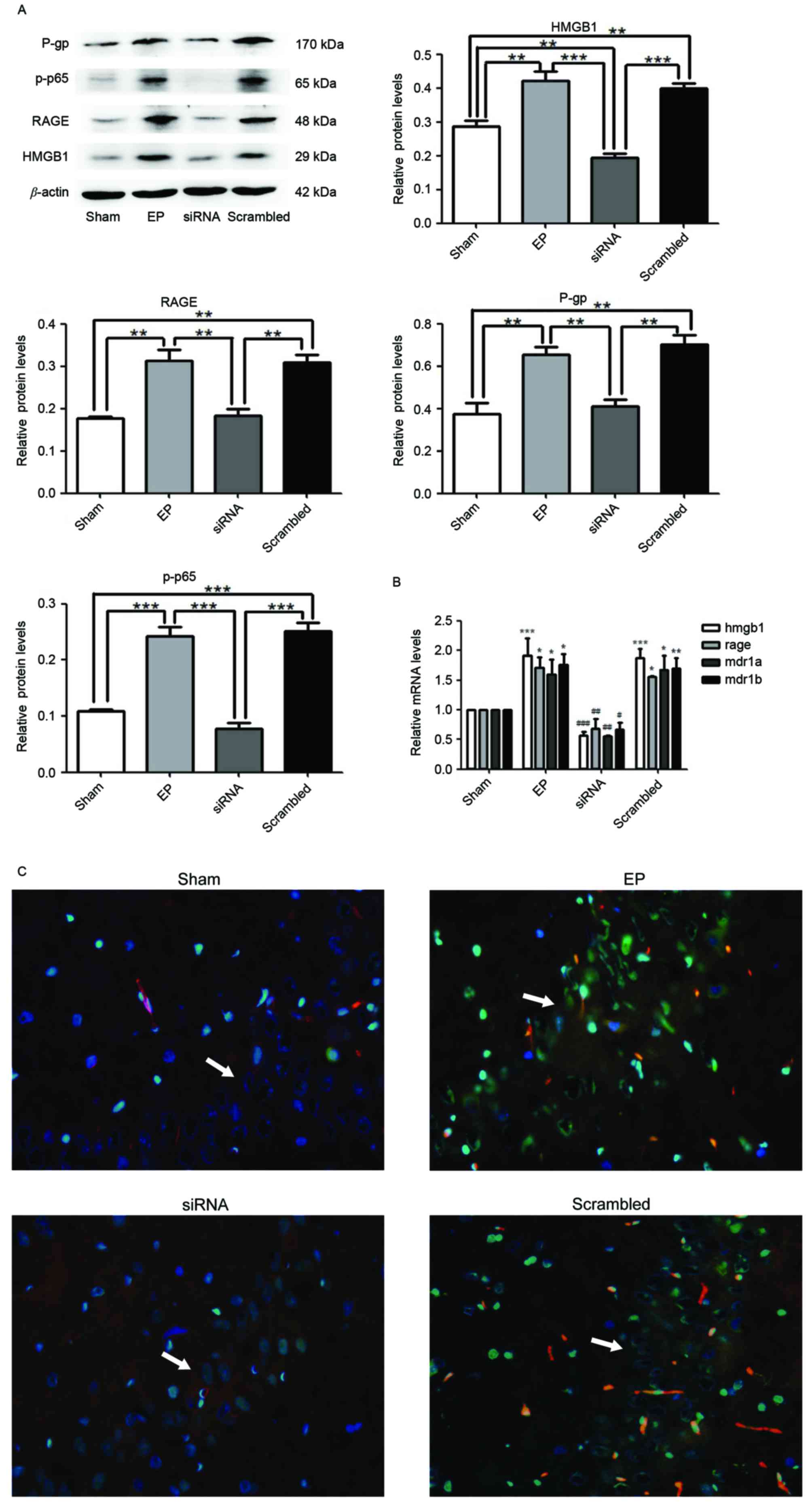 | Figure 2.HMGB1 siRNA reduces the expression
levels of RAGE, p-p65 and P-gp. (A) Protein samples were analyzed
72 h following status epilepticus termination. Densitometry
analysis was performed and data are presented as mean ± standard
deviation. *P<0.05, **P<0.01, ***P<0.0001. (B) HMGB1, RAGE
and MDR1A and MDR1B mRNA expression levels were significantly
reduced following treatment with HMGB1 siRNA. *P<0.05,
**P<0.01 and ***P<0.0001 vs. Sham; #P<0.05,
##P<0.01 and ###P<0.0001 vs. EP. (C)
The localization of HMGB1 and P-gp in the hippocampal CA3 region
(indicated by white arrows) of rat brains, as determined by
immunofluorescence staining. Blue=nuclei, green=HMGB1 and red=P-gp.
Images were captured using ×400 magnification. HMGB1 and P-gp were
significantly reduced in the siRNA group compared with the EP and
scrambled groups. HMGB1 was localized to the nuclei and cytoplasm.
P-gp was primarily localized to blood vessels. P-gp,
P-glycoprotein; p-p65, phosphorylated-NF-κB p65; RAGE, receptor for
advanced glycation end-product; HMGB1, high-mobility group box 1
gene; MDR1A and MDR1B, P-glycoprotein genes; EP,
pilocarpine-induced epilepsy group. |
RAGE knockdown attenuates
seizure-induced upregulation of p-p65 and P-gp
Protein expression levels of RAGE, p-p65 and P-gp
were significantly reduced in the siRNA group compared with the EP
and scrambled group (P<0.01; Fig.
3A). Similarly, in the group treated with RAGE siRNA, mRNA
expression of RAGE, MDR1A and MDR1B were significantly reduced
compared with the EP and scrambled groups (Fig. 3B). Immunofluorescence staining
demonstrated that RAGE and P-gp were upregulated in the EP group
compared with the sham group. Pre-treatment with RAGE siRNA
resulted in reduced numbers of P-gp positive cells compared with
the scrambled group (Fig. 3C).
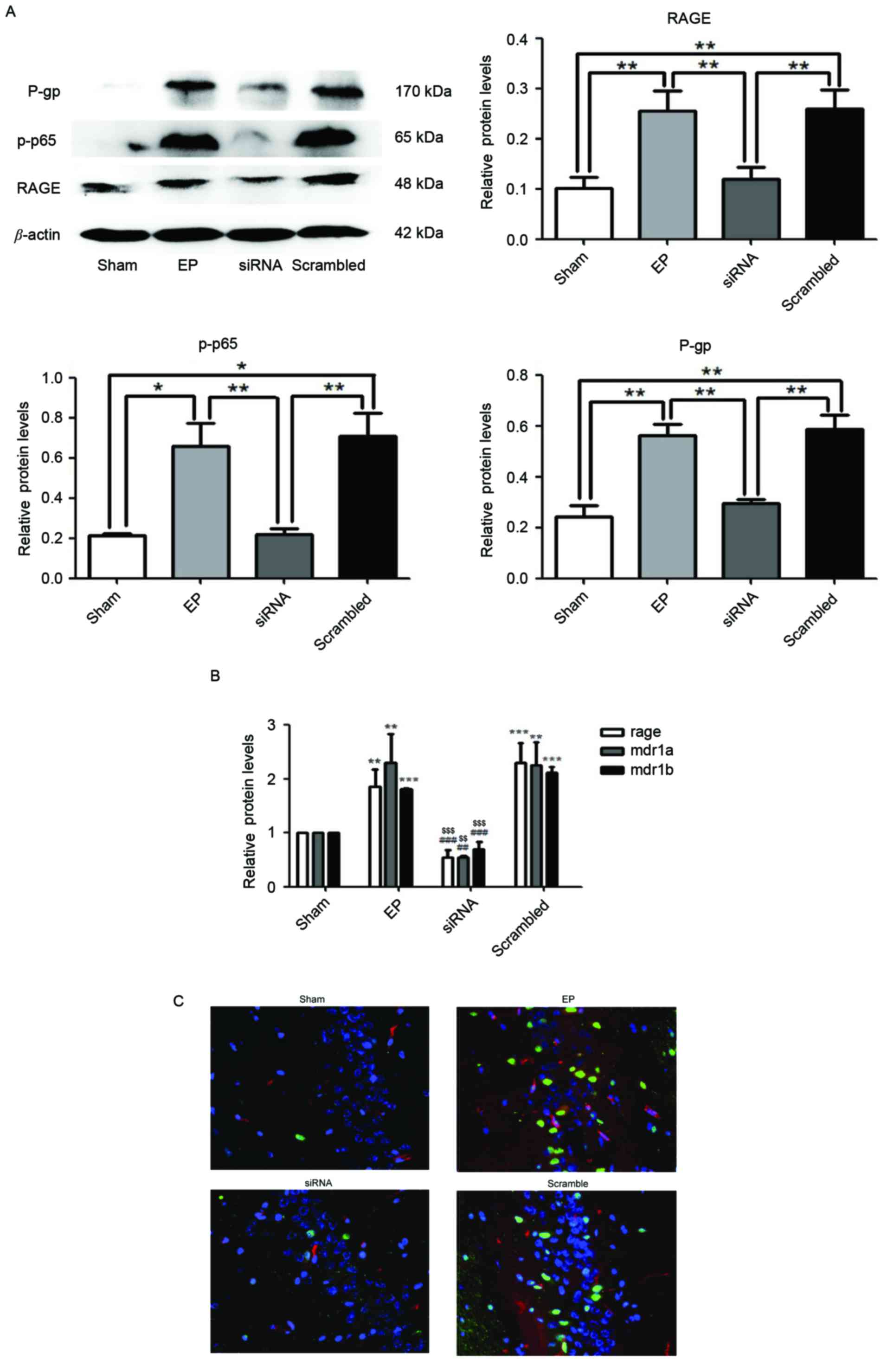 | Figure 3.RAGE knockdown reduces
seizure-induced p-p65 and P-gp overexpression. Specimens were
collected 72 h following status epilepticus termination. (A) RAGE,
p-p65 and P-gp were significantly reduced in the siRNA group
compared with the EP and scrambled groups, as determined by western
blotting. Densitometry analysis was performed and data are
presented as the mean ± standard deviation. *P<0.05 and
**P<0.01. (B) mRNA expression levels of RAGE and MDR1A/B in the
siRNA group were significantly reduced compared with the EP and
Scrambled group. **P<0.01 and ***P<0.0001 vs. sham;
##P<0.01 and ###P<0.0001 vs. EP;
$$P<0.01 and $$$P<0.0001 vs. Scrambled.
(C) Localization of RAGE and P-gp in hippocampal region CA3 of rat
brains. Blue=nuclei, green=RAGE and red=P-gp. Images were captured
using ×400 magnification. Compared with the EP and scramble groups,
the expression levels of RAGE and P-gp in the siRNA group were
significantly reduced. RAGE was primarily localized to the nuclei
of neurons and P-gp was primarily localized to blood vessels. RAGE,
receptor for advanced glycation end-product; p-p65,
phosphorylated-NF-κB p65; P-gp, P-glycoprotein; MDR1A and MDR1B,
P-glycoprotein gene; RAGE, receptor for advanced glycation
end-product; EP, pilocarpine-induced epilepsy group. |
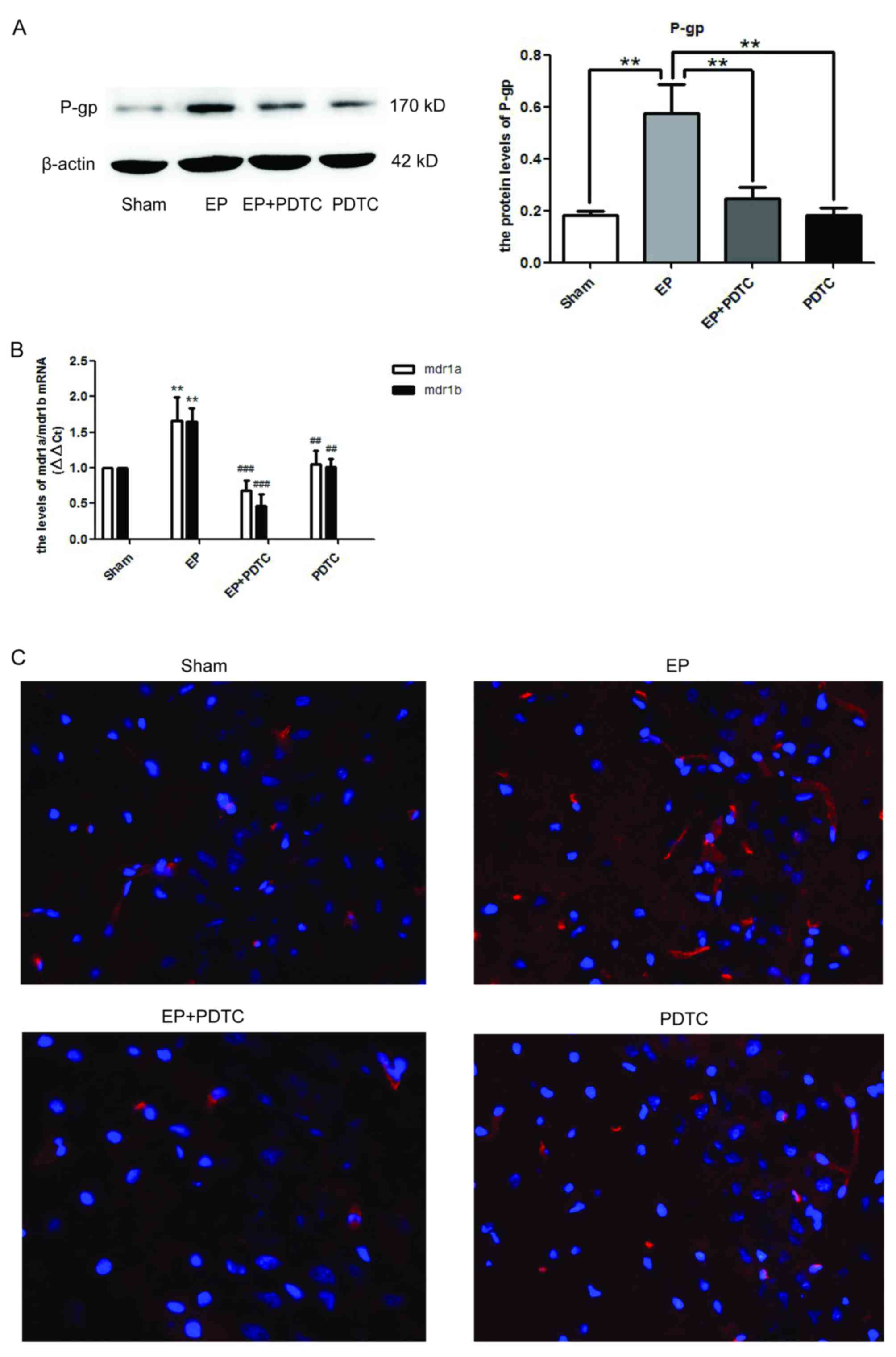
Blocking NF-κB activity using PDTC
reduces P-gp expression in rat brains following seizures
To determine whether NF-κB may regulate the
expression of P-gp, PDTC, a specific inhibitor of NF-κB was
utilized. As demonstrated in Fig.
4A, 72 h after SE termination, the protein expression levels of
P-gp in the EP + PDTC group were significantly reduced compared
with the EP group (P<0.01). This was supported by the mRNA
expression levels of MDR1A/B (Fig.
4B), and the immunofluorescence staining of P-gp (Fig. 4C).
Discussion
The possibility that inflammatory processes in the
brain contribute to seizures and the establishment of a chronic
epileptic foci, is becoming increasingly recognized.
Pro-inflammatory cytokines, including interleukin-1β, and danger
signals, including HMGB1 and S100β, are overexpressed in human and
experimental epileptogenic tissues (30–32).
They have proconvulsant activity in various seizure models,
possibly by reducing the seizure threshold via functional
interactions with classical neurotransmitter systems (33). This demonstrates potential novel
targets for therapeutic intervention to epilepsy.
HMGB1 is a nuclear protein that has cytokine-like
functions following its extracellular release. It is either
passively released from cells undergoing injury or actively
secreted by cells under stressful conditions (34). The hyperacetylated form of HMGB1
regulates the transcription of various pro-inflammatory cytokines
via binding to TLR4 and RAGE (35,36).
Experimental models of epilepsy suggest that
HMGB1-TLR4 signaling may contribute to seizures in humans, and
could potentially be targeted to produce anticonvulsant effects in
patients currently resistant to drugs (14). In addition, our previous study
demonstrated that the susceptibility to seizures may be reduced by
inhibition of HMGB1 and TRL4 in KA-induced epilepsy (37).
The present study revealed that the expression
levels of HMGB1 were enhanced following SE up to 72 h, which were
subsequently reduced again. However, there was a marked decrease in
HMGB1 expression at 24 h. Luo et al (38) reported a similar effect in a
KA-induced seizure mouse model. Wang et al (39) revealed that HMGB1 was released by
macrophages 12 h post-stimulation and this resulted in an
intracellular decrease in HMGB1 stores. In addition, HMGB1
secretion was reduced 24 h post-stimulation and resynthesized. It
was speculated that brain cells may have similar mechanisms.
Proliferated neuroglia cells were demonstrated to be involved in
enhanced secretion of HMGB1 24 h post-stimulation (39).
RAGE is a transmembrane receptor of the
immunoglobulin superfamily and interacts with ligands following
activation of pro-inflammatory and immune responses (40). A previous study suggested that
overexpression of RAGE in temporal epilepsy contributes to
hyperexcitability in acute and chronic seizures. A potential
underlying mechanism may be that HMGB1 activates RAGE, resulting in
inflammation in epileptic brains (17).
It is established that HMGB1/RAGE have a role in
inflammatory responses (41,42),
and their interaction results in downstream activation of the
pro-inflammatory transcription factor, NF-κB (43). NF-κB is a key protein that is
regulated by RAGE signaling (44).
Upon activation, NF-κB translocates to the nucleus and subsequently
binds to DNA sequences to stimulate transcription of target genes,
including various cytokines, adhesion molecules and the gene
encoding RAGE itself (45). In
present study, NF-κB expression levels were reduced following
knockdown of HMGB1 and RAGE, which suggested that HMGB1/RAGE
regulates the transcription of the gene encoding NF-κB.
Our previous study demonstrated that following
knockdown of the gene encoding IKKβ, which phosphorylates an
inhibitory protein of NF-κB to enable its nuclear migration,
expression of the P-gp protein was reduced in a KA-induced
epileptic brain. This result concurred with the present study, as
inhibition of NF-κB by PDTC following SE termination resulted in
reduced expression levels of P-gp. Therefore, it is possible that
NF-κB regulates MDR1A/B gene transcription.
In rodents, P-gp is encoded by the MDR1A and
MDR1B genes. P-gp encoded by MDR1A serves a role as
an efflux pump in cells of the liver, intestine, kidney and
blood-brain barrier (46). The
mRNA expression levels of MDR1A and MDR1B in the
present study were not significantly different. In addition to
vascular endothelial cells, nerve cells can express P-gp in the
brain following seizures (47).
However, the immunofluorescence staining in the present study
demonstrated that P-gp was primarily localized to blood vessels,
and there was little expression observed in neurons. This may be
due to the model utilized and the time in which specimens were
observed. van Vliet et al (48) demonstrated that P-gp is observed in
glial-like cells in the rat dentate gyrus 1 week after seizures
induced by electrical kindling. However, Guo et al (49) reported that P-gp was not expressed
in hippocampus neurons 4 weeks after pilocarpine-induced epilepsy
in rats. During immunofluorescence testing, specific markers for
the different cell types were not applied. This was a limitation of
the present study, which could be resolved with specific markers
for blood vessels and neurons in future studies.
In conclusion, the present study demonstrated that
HMGB1 knockdown may reduce the expression levels of MDR1A/B
mRNA and P-gp protein via RAGE/NF-κB inflammatory signaling
pathways. This may partly explain the underlying mechanism of P-gp
overexpression in pilocarpine-induced SE rat brains. Therefore,
targeting the HMGB1/RAGE/NF-κB signaling pathway may be a potential
strategy for the treatment of DRE.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81171222). The
authors would like to thank Dr Zhang Qiaoquan (Department of
Neuropathology, Nanjing Brain Hospital, Jiangsu, China) for their
assistance with the immunofluorescence.
Glossary
Abbreviations
Abbreviations:
|
AED
|
anti-epilepsy drug
|
|
DRE
|
drug-refractory epilepsy
|
|
EP
|
pilocarpine-induced epilepsy
|
|
HMGB1
|
high-mobility group box 1
|
|
NF-κB
|
nuclear factor-κB
|
|
P-gp
|
p-glycoprotein
|
|
p-P65
|
phospho-NF-κB p65
|
|
RAGE
|
receptor for advanced glycation
end-product
|
|
siRNA
|
small interfering RNA
|
References
|
1
|
Ngugi AK, Bottomley C, Kleinschmidt I,
Sander JW and Newton CR: Estimation of the burden of active and
life-time epilepsy: A meta-analytic approach. Epilepsia.
51:883–890. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ni H, Sun Q, Tian T, Feng X and Sun BL:
Long-term expression of metabolism-associated genes in the rat
hippocampus following recurrent neonatal seizures and its
regulation by melatonin. Mol Med Rep. 12:2727–2734. 2015.PubMed/NCBI
|
|
3
|
Stepień KM, Tomaszewska J and Czuczwar SJ:
The multidrug transporter P-glycoprotein in pharmacoresistance to
antiepileptic drugs. Pharmacol Rep. 64:1011–1019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang CB, Kwan P, Zuo Z and Baum L: The
transport of antiepileptic drugs by P-glycoprotein. Adv Drug Deliv
Rev. 64:930–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Avemary J, Salvamoser JD, Peraud A, Rémi
J, Noachtar S, Fricker G and Potschka H: Dynamic regulation of
P-glycoprotein in human brain capillaries. Mol Pharm. 10:3333–3341.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Potschka H and Luna-Munguia H: CNS
transporters and drug delivery in epilepsy. Curr Pharm Des.
20:1534–1542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartels AL, de Klerk OL, Kortekaas R, de
Vries JJ and Leenders KL: 11C-verapamil to assess P-gp function in
human brain during aging, depression and neurodegenerative disease.
Curr Top Med Chem. 10:1775–1784. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Holtman L, van Vliet EA, Edelbroek PM,
Aronica E and Gorter JA: Cox-2 inhibition can lead to adverse
effects in a rat model for temporal lobeepilepsy. Epilepsy Res.
91:49–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hartz AM, Notenboom S and Bauer B:
Signaling to P-glycoprotein-A new therapeutic target to treat
drug-resistant epilepsy? Drug News Perspect. 22:393–397. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vezzani A, French J, Bartfai T and Baram
TZ: The role of inflammation in epilepsy. Nat Rev Neurol. 7:31–40.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kawase A, Norikane S, Okada A, Adachi M,
Kato Y and Iwaki M: Distinct alterations in ATP-Binding cassette
transporter expression in liver, kidney, small intestine, and brain
in adjuvant-induced arthritic rats. J Pharm Sci. 103:2556–2564.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Doorduin J, de Vries EF, Dierckx RA and
Klein HC: P-glycoprotein activity in the blood-brain barrier is
affected by virus-induced neuroinflammation and antipsychotic
treatment. Neuropharmacology. 85:548–553. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rojas A, Jiang JX, Ganesh T, Yang MS,
Lelutiu N, Gueorguieva P and Dingledine R: Cyclooxygenase-2 in
epilepsy. Epilepsia. 55:17–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maroso M, Balosso S, Ravizza T, Liu J,
Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi
AA, et al: Toll-like receptor 4 and high-mobility group box-1 are
involved in ictogenesis and can be targeted to reduce seizures. Nat
Med. 16:413–419. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiavegato A, Zurolo E, Losi G, Aronica E
and Carmignoto G: The inflammatory molecules IL-1β and HMGB1 can
rapidly enhance focal seizure generation in a brain slice model of
temporal lobe epilepsy. Front Cell Neurosci. 8:1552014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Y, Huang XJ, Yu N, Xie Y, Zhang K,
Wen F, Liu H and Di Q: HMGB1 contributes to the expression of
P-glycoprotein in mouse epileptic brain through Toll-like receptor
4 and receptor for advanced glycation end products. PLoS One.
10:e01409182015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iori V, Maroso M, Rizzi M, Iyer AM,
Vertemara R, Carli M, Agresti A, Antonelli A, Bianchi ME, Aronica
E, et al: Receptor for advanced glycation endproducts is
upregulated in temporal lobe epilepsy and contributes to
experimental seizures. Neurobiol Dis. 58:102–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roberts DJ and Goralski KB: A critical
overview of the influence of inflammation and infection on
P-glycoprotein expression and activity in the brain. Expert Opin
Drug Metab Toxicol. 4:1245–1264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu N, Liu H, Zhang YF, Su LY, Liu XH, Li
LC, Hao JB, Huang XJ and Di Q: Effects of brain IKKβ gene silencing
by small interfering RNA on P-Glycoprotein expression and brain
damage in the rat kainic acid-induced seizure model. CNS Neurol
Disord Drug Targets. 13:661–672. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Angelo MF, Aguirre A, Avilés Reyes RX,
Villarreal A, Lukin J, Melendez M, Vanasco V, Barker P, Alvarez S,
Epstein A, et al: The proinflammatory RAGE/NF-κB pathway is
involved in neuronal damage and reactive gliosis in a model of
sleep apnea by intermittent hypoxia. PLoS One. 9:e1079012014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Batkulwar KB, Bansode SB, Patil GV,
Godbole RK, Kazi RS, Chinnathambi S, Shanmugam D and Kulkarni MJ:
Investigation of phosphoproteome in RAGE signaling. Proteomics.
15:245–259. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Senn C, Hangartner C, Moes S, Guerini D
and Hofbauer KG: Central administration of small interfering RNAs
in rats: A comparison with antisenseoligonucleotides. Eur J
Pharmacol. 522:30–37. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun H, Wu H, Yu X, Zhang G, Zhang R, Zhan
S, Wang H, Bu N, Ma X and Li Y: Angiotensin II and its receptor in
activated microglia enhanced neuronal loss and cognitive impairment
following pilocarpine-induced status epilepticus. Mol Cell
Neurosci. 65:58–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Racine RJ: Modification of seizure
activity by electrical stimulation. II. Motor seizure.
Electroencephalogr Clin Neurophysiol. 32:281–294. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lei C, Lin S, Zhang C, Tao W, Dong W, Hao
Z, Liu M and Wu B: Activation of cerebral recovery by matrix
metalloproteinase-9 after intracerebral hemorrhage. Neuroscience.
230:86–93. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Soerensen J, Pekcec A, Fuest C, Nickel A
and Potschka H: Pyrrolidine dithiocarbamate protects the piriform
cortex in the pilocarpine status epilepticus model. Epilepsy Res.
87:177–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu C, Cui Z, Wang S and Zhang D: CD93 and
GIPC expression and localization during central nervous system
inflammation. Neural Regen Res. 9:1995–2001. 2014.PubMed/NCBI
|
|
29
|
Turski WA, Cavalheiro EA, Schwarz M,
Czuczwar SJ, Kleinrok Z and Turski L: Limbic seizures produced by
pilocarpine in rats: Behavioural, eletroencephalographic and
neuropathological study. Behav Brain Res. 9:315–335. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Choy M, Dubé CM, Patterson K, Barnes SR,
Maras P, Blood AB, Hasso AN, Obenaus A and Baram TZ: A novel
noninvasive predictive epilepsy biomarker with clinical potential.
J Neurosci. 34:8672–8684. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zanotto C, Abib RT, Batassini C,
Tortorelli LS, Biasibetti R, Rodrigues L, Nardin P, Hansen F,
Gottfried C, Leite MC and Gonçalves CA: Non-specific inhibitors of
aquaporin-4 stimulate S100B secretion in acute hippocampal slices
of rats. Brain Res. 1491:14–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kołosowska K, Maciejak P, Szyndler J,
Turzyńska D, Sobolewska A and Płaźnik A: The role of interleukin-1β
in the pentylenetetrazole-induced kindling of seizures, in the rat
hippocampus. Eur J Pharmacol. 731:31–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vezzani A: Epilepsy and inflammation in
the brain: Overview and pathophysiology. Epilepsy Curr. 14 1
Suppl:S3–S7. 2014. View Article : Google Scholar
|
|
34
|
Yang H, Antoine DJ, Andersson U and Tracey
KJ: The many faces of HMGB1: Molecular structure-functional
activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol.
93:865–873. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maroso M, Balosso S, Ravizza T, Liu J,
Bianchi ME and Vezzani A: Interleukin-1 type 1 receptor/Toll-like
receptor signalling in epilepsy: The importance of IL-1beta and
high-mobility group box 1. J Intern Med. 270:319–326. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huebener P, Pradere JP, Hernandez C, Gwak
GY, Caviglia JM, Mu X, Loike JD, Jenkins RE, Antoine DJ and Schwabe
RF: The HMGB1/RAGE axis triggers neutrophil-mediated injury
amplification following necrosis. J Clin Invest. 125:539–550. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang XJ, Hao JB, Di Q, Yu N, Zhang YF,
Wen F and Chen Y: High mobility group protein B1 contributes to the
expression of P-glycoprotein in hippocampus of epileptic rats. J
Nanjing Med University (natural science). 8:1029–1033. 2014.
|
|
38
|
Luo LD, Jin YC, Kim ID and Lee JK:
Glycyrrhizin suppresses HMGB1 inductions in the hippocampus and
subsequent accumulation in serum of a kainic acid-induced seizure
mouse mode. Cell Mol Neurobiol. 34:987–997. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMGB-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ibrahim ZA, Armour CL, Phipps S and Sukkar
MB: RAGE and TLRs: Relatives, friends or neighbours? Mol Immunol.
56:739–744. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xie J, Méndez JD, Méndez-Valenzuela V and
Aguilar-Hernández MM: Cellular signalling of the receptor for
advanced glycation end products (RAGE). Cell Signal. 25:2185–2197.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mazarati A, Maroso M, Iori V, Vezzani A
and Carli M: High-mobility group box-1 impairs memory in mice
through both toll-like receptor 4 and Receptor for Advanced
Glycation End Products. Exp Neurol. 232:143–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dumitriu IE, Baruah P, Valentinis B, Voll
RE, Herrmann M, Nawroth PP, Arnold B, Bianchi ME, Manfredi AA and
Rovere-Querini P: Release of high mobility group box 1 by dendritic
cells controls T cell activation via the receptor for advanced
glycation end products. J Immunol. 174:7506–7515. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kalea AZ, Reiniger N, Yang H, Arriero M,
Schmidt AM and Hudson BI: Alternative splicing of the murine
receptor for advanced glycation end-products (RAGE) gene. FASEB J.
23:1766–1774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bierhaus A, Humpert PM, Morcos M, Wendt T,
Chavakis T, Arnold B, Stern DM and Nawroth PP: Understanding RAGE,
the receptor for advanced glycation end products. J Mol Med (Berl).
83:876–886. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Borst P and Schinkel AH: P-glycoprotein
ABCB1: A major player in drug handling by mammals. J Clin Invest.
123:4131–4133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Aronica E, Sisodiya SM and Gorter JA:
Cerebral expression of drug transporters in epilepsy. Adv Drug
Deliv Rev. 64:919–929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
van Vliet E, Aronica E, Redeker S, Marchi
N, Rizzi M, Vezzani A and Gorter J: Selective and persistent
upregulation of mdr1b mRNA and P-glycoprotein in the
parahippocampal cortex of chronic epileptic rats. Epilepsy Res.
60:203–213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guo Y and Jiang L: Drug transporters are
altered in brain, liver and kidney of rats with chronic epilepsy
induced by lithium-pilocarpine. Neurol Res. 32:106–112. 2010.
View Article : Google Scholar : PubMed/NCBI
|