Introduction
Malignant mesothelioma (MM) is a rare and aggressive
tumor; its occurrence is associated with substantial exposure to
airborne asbestos fibers (1).
Approximately 70% of the reported cases of MM occur in the pleura,
with a further ~30% occurring in the peritoneum and 1–2% in the
pericardium and the tunica vaginalis testis (2). The incidence of MM is increasing
worldwide owing to an increase in asbestos exposure earlier in
life; however, owing to the long latency period, MM is often
diagnosed late in life (3). MM is
mostly treated with chemotherapy over the course of the disease
(3).
Platinum compounds, such as cisplatin
(cis-dichlorodiammineplatinum; CDDP), are currently regarded
as first-line treatments for patients with MM, although their
efficacy remains limited (4).
These drugs inhibit DNA synthesis through the formation of
intrastrand cross-links and the formation of DNA adducts. A
majority of patients eventually relapse with the emergence of
CDDP-resistant cell populations, despite the initial response to
therapy. Previously published results from a phase III trial in
patients with MM indicated that the combination therapy of CDDP
with pemetrexed improved response rates (41.3 vs. 16.7%), median
survival time (12.1 vs. 9.3 months) and median time to progressive
disease (5.7 vs. 3.9 months) compared with CDDP alone (5). A number of strategies, including
various drug combinations, have been used to prevent or delay
resistance to chemotherapeutic agents; however, it currently
appears inevitable that drug-resistant variants will emerge during
disease progression. It has been suggested that functional defects
in apoptosis signaling are important for the development of
chemoresistance in MM (6).
Therefore, the development of effective chemosensitizing agents
that represent distinct and complementary modes of killing cells
may have important clinical implications. Sulforaphane (SFN) is an
isothiocyanate compound derived from glucoraphanin, which is
present in cruciferous vegetables, and has previously been
demonstrated to have the ability to suppress malignant cancer
phenotypes, including cell proliferation, angiogenesis and
metastasis (7). SFN has also been
reported to sensitize cancer cells to imatinib- and tumor necrosis
factor-related apoptosis inducing ligand-induced apoptosis, via a
reactive oxygen species (ROS)-dependent pathway (8,9),
suggesting potential therapeutic value as an adjunct to current
cancer therapies.
Autophagy is an evolutionarily conserved catabolic
process in which damaged or dysfunctional cellular components are
fused with a lysosome for degradation and recycled for use in the
metabolic and biosynthetic pathways. Autophagy is considered an
adaptive response that promotes survival against cellular stresses;
in other cases, however, prolonged activation of autophagy appears
to promote cell death, termed autophagic cell death (10). As a way to deal with cellular
stresses, such as glucose deprivation, hypoxia, oxidative stress
and endoplasmic reticulum stress, cancer cells utilize the
autophagic pathway, which enables continued growth by maintaining
cellular energy production (11).
Previous research has indicated that the inhibition of autophagy
may potentiate cancer cell death and enhance the efficacy of
anticancer drugs on cells that use autophagy to survive (12,13).
The present study aimed to identify the
chemosensitizing effect of SFN and to understand the implication of
autophagy in MM cell resistance to CDDP therapy. The effects of SFN
on enhancing the anticancer role of CDDP in H-28 MM cells were
evaluated, as well as alterations to apoptosis and autophagy.
Potential roles of autophagy as a factor for MM cell survival are
discussed.
Materials and methods
Reagents and cell culture
SFN, CDDP,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
Rhodamine 123, bafilomycin A1, N-acetylcysteine (NAC), propidium
iodide (PI), DAPI, 2′,7′-dichlorodihydrofluorescein diacetate
(DCF-DA) and the β-actin antibody (catalog no. A2228) were obtained
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Antibodies
against B-cell lymphoma 2 (Bcl-2; catalog no. 2820), Bcl-2
associated X (Bax; catalog no. 5023), cyclin D1 (catalog no. 2978),
cyclin B1 (catalog no. 12231), phosphorylated (p)-cyclin dependent
kinase 2 (p-Cdc2Tyr15; catalog no. 4539), Akt (catalog no. 9272),
p-Akt (catalog no. 9271), mTOR (catalog no. 2972), p-mTOR (catalog
no. 2971), microtubule-associated protein 1 light chain 3B (LC3B;
catalog no. 3868), PARP (catalog no. 9542), cleaved PARP (catalog
no. 9541), procaspase-3 (catalog no. 9665), and cleaved caspase-3
(catalog no. 9664) were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). Antibodies against p53 (catalog no.
sc-126), p21WAF1/CIP1 (catalog no. sc-6246), goat anti-rabbit
IgG-HRP (catalog no. sc-2004), goat anti-mouse IgG-HRP (catalog no.
sc-2005), and the Enhanced Chemiluminescence (ECL) kit were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA).
The human malignant mesothelioma cell line H-28 and
the human mesothelial cell line MeT-5A were obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA). H-28
cells were maintained at 37°C in RPMI-1640 medium (catalog no.
SH30027.01; GE Healthcare Life Sciences, Chalfont, UK) supplemented
with 10% fetal bovine serum (FBS; catalog no. SH30084.03; GE
Healthcare Life Sciences), 100 U/ml penicillin and 100 µg/ml
streptomycin. MeT-5A cells were maintained in M-199 medium
(Welgene, Inc., Daegu, Korea) supplemented with 3.3 nM epidermal
growth factor (catalog no. E9644; Sigma-Aldrich), 5% FBS, 100 U/ml
penicillin and 100 µg/ml streptomycin. Cells were grown to 70%
confluence in a submerged monolayer culture for 24 h prior to
treatment.
Cell viability assay
Cell viability was measured using the MTT assay.
Cells (5×103 cells/well) were seeded into 96-well microtiter plates
and incubated with the vehicle [0.1% dimethylsulfoxide (DMSO) in
medium], SFN (0, 10, 20, 30, 40, 50, 60, 80 and 100 µM), CDDP (0,
40, 80, 160, 240, 480, 640 and 960 µM), or a combination of SFN and
CDDP (20 and 40 µM, respectively) for 48 h at 37°C, and then
exposed to MTT (final concentration, 0.1 mg/ml) for an additional 4
h at 37°C. Absorbance values were measured at a wavelength of 560
nm with a GloMax-Multi Microplate Multimode Reader (Promega
Corporation, Madison, WI, USA). The number of viable cells was
determined by comparing results with the vehicle-treated control
cells. The two-compound combination effect was evaluated by
calculating the combination index (CI), as previously described
(14); where CI<1 indicates a
synergistic effect, CI=1 indicates an additive effect and CI>1
indicates an antagonistic effect.
DAPI staining
Nuclear condensation and fragmentation were observed
by DAPI staining. Cells (1×105 cells/well) were seeded into a
6-well culture plate and incubated with SFN (20 µM) and CDDP (40
µM), alone or in combination, for 48 h at 37°C. The cells were
trypsinized, pelleted by centrifugation at 500 × g for 7 min at
4°C, and fixed in 100% methanol at room temperature for 20 min.
After centrifugation, the pellet was resuspended in DAPI (2 µg/ml)
for 10 min in the dark and washed with 1X PBS. Cells were spread on
slides and the coverslip was then mounted using mounting medium
(catalog no. 08381; Polysciences, Inc., Warrington, PA, USA).
Apoptotic cells were observed with a FluoView FV10i confocal
fluorescence microscope (Olympus Corporation, Tokyo, Japan). Five
random fields were analyzed on each slide.
Cell cycle analysis
Cell cycle distribution was measured by quantitation
of DNA content in the PI-stained cells. Trypsinized cells (~106
cell/ml) were pelleted by centrifugation at 500 × g for 7 min at
4°C, fixed in 70% ice-cold ethanol overnight at −20°C and treated
with DNase-free RNase A (150 µg/ml) and PI (20 µg/ml) for 1 h at
4°C. Data from 10,000 single-cell events were collected by a
MACSQuant Analyzer flow cytometer and analyzed using MACSQuantify™
software version 2.5 (Miltenyi Biotec GmbH, Bergisch Gladbach,
Germany).
Apoptosis assay
Apoptotic cell distribution was determined with a
Muse Annexin V and Dead Cell Assay kit (catalog no. MCH100105;
Merck KGaA, Darmstadt, Germany), according to the manufacturer's
protocol. This kit includes a fluorescent dye phycoerythrin (PE)
conjugated to Annexin V to detect phosphatidylserine on the
external membrane of apoptotic cells and 7-amino-actinomycin D as a
dead cell marker. Briefly, cells (1×105 cells/well) were seeded
into a 6-well culture plate and incubated with SFN (20 µM) and CDDP
(40 µM), alone or in combination, for 48 h at 37°C. The cells were
trypsinized and collected into culture medium, mixed with the Muse
Annexin V & Dead Cell reagent, and analyzed using a Muse Cell
Analyzer (Merck KGaA).
Western blot analysis
A total of 1×105 cells/well were seeded into 6-well
culture plate and incubated with or without NAC (5 mM) or
bafilomycin A1 (50 nM) for 2 h at 37°C prior to co-treatment with
SFN (20 µM) and CDDP (40 µM) for a further 48 h. Cells were lysed
in radioimmunoprecipitation assay buffer [1X PBS, 1% NP-40, 0.5%
sodium deoxycholate, 0.1% SDS, 10 µg/ml
phenylmethanesulfonylfluoride and a protease inhibitor cocktail
tablet (Boehringer Mannheim, Germany)] for 30 min on ice, and
subsequently pelleted by centrifugation 10,000 × g for 10 min at
4°C. The protein concentration was determined using a bicinchoninic
acid assay kit (catalog no. 23225; Pierce™; Thermo
Scientific, Rockford, IL, USA), according to the manufacturer's
protocol. Cell lysates containing 40 µg protein were separated on
NuPAGE 4–12% Bis-Tris polyacrylamide gels (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and electrophoretically
transferred to Immuno-Blot polyvinylidene fluoride membranes
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membrane was
incubated overnight at 4°C with primary antibodies diluted to 1:500
in casein blocking buffer (catalog no. B6429; Sigma-Aldrich),
followed by incubation for 2 h at room temperature with a secondary
antibody coupled to horseradish peroxidase diluted 1:5,000 in
casein blocking buffer. Signals were visualized using an ECL
detection kit and X-ray film. Blots were stripped using a stripping
buffer (100 mM β-mercaptoethanol, 2% SDS, and 62.5 mM Tris-HCl, pH
6.7) and re-probed with an anti-β-actin antibody to serve as the
loading control.
Measurement of intracellular ROS
levels
Intracellular ROS levels were evaluated by measuring
DCF-DA (Sigma-Aldrich) fluorescence intensity. Briefly, cells
(1×105 cells/well) were seeded into a 6-well culture plate and
incubated with or without 5 mM NAC for 2 h at 37°C prior to
co-treatment with 20 µM SFN and 40 µM CDDP for a further 48 h. The
cells were trypsinized, pelleted by centrifugation at 500 × g for 7
min at 4°C, and resuspended in serum-free RPMI-1640 medium
containing 10 µM DCF-DA for 30 min at 37°C in the dark. Following
incubation, cells were washed twice with 1X PBS, trypsinized,
resuspended in 1X PBS, and immediately analyzed with a MACSQuant
Analyzer flow cytometer and MACSQuantify™ software
version 2.5 (Miltenyi Biotec GmbH). DCF fluorescence was detected
using a 530 nm bandpass filter, and each measurement was based on
the mean fluorescence intensity of 10,000 cells.
Mitochondrial membrane potential (ΔΨm)
analysis
Cells (5×104 cells/well) were seeded onto 6-well
plates and incubated with or without NAC (5 mM) for 2 h at 37°C
prior to co-treatment with 20 µM SFN and 40 µM CDDP for a further
48 h. The cells were trypsinized, harvested by centrifugation at
500 × g for 7 min at 4°C, washed twice with PBS, and stained with
serum-free RPMI-1640 medium containing Rhodamine 123 (final
concentration, 30 nM) at 37°C for 30 min. Following incubation,
cells were washed twice with 1X PBS, trypsinized and resuspended in
1X PBS. Fluorescence intensity was measured and analyzed using a
MACSQuant analyzer flow cytometer and MACSQuantify™
software version 2.5 (Miltenyi Biotec GmbH).
Measurement of reduced glutathione
(GSH)/oxidized glutathione (GSSG) ratio
Total glutathione (that is, total combined GSH and
GSSG expression) and GSSG levels were measured using a GSH/GSSG-Glo
assay (Promega Corporation), according to the manufacturer's
protocol. Briefly, cells (5×103 cells/well) were seeded into a
96-well culture plate and treated with the vehicle (0.1% DMSO in
RMPI-1640 medium) or NAC (5 mM) for 2 h at 37°C prior to
co-treatment with 20 µM SFN and 40 µM CDDP for a further 48 h. The
media was subsequently removed and replaced with total glutathione
or GSSG lysis reagent, for total glutathione or GSSG measurement,
respectively. The GSH probe, luciferin-NT, was converted to
luciferin, which is coupled to a firefly luciferase. The GSH/GSSG
ratio was calculated from the luminescence measurements using a
GloMax-Multi Microplate Multimode Reader (Promega Corporation). The
GSSG value was subtracted from the total glutathione to calculate
the level of GSH.
Flow cytometry analysis of
lysosomes
Cells (1×105 cells/well) were seeded into a 6-well
culture plate and incubated with or without 5 mM NAC for 2 h at
37°C prior to co-treatment with 20 µM SFN and 40 µM CDDP for a
further 48 h. Changes in the activation of the
autophagosome/lysosome pathway were quantitatively determined by
staining the acidic vacuoles inside the cells with LysoTracker Red
DND-99 (25 nmol/l; Molecular Probes; Thermo Fisher Scientific,
Inc.) for 20 min at 37°C. Following incubation, cells were washed
twice with 1X PBS, trypsinized and resuspeded in 1X PBS. The
fluorescence intensity of the cells was analyzed on channel APC-A
using a MACSQuant analyzer flow cytometer and
MACSQuantify™ software version 2.5 (Miltenyi Biotec
GmbH).
Statistical analysis
Statistical comparisons were performed by one-way
analysis of variance followed by Tukey's post hoc correction for
multiple comparisons, using SPSS version 17.0 (SPSS, Inc., Chicago,
IL, USA). Data were expressed as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Synergistic growth-inhibiting and
apoptosis-promoting effects of combination SFN/CDDP treatment
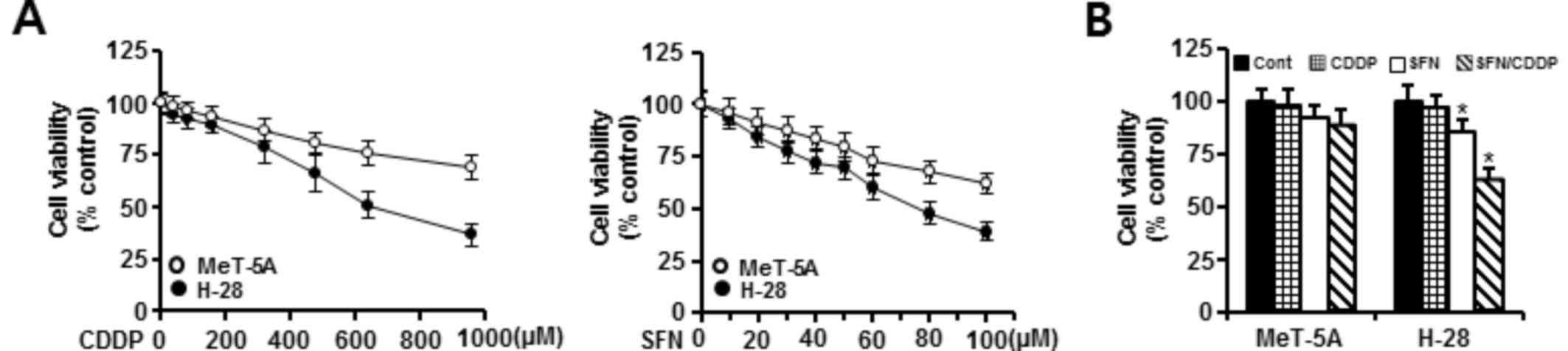
The cytotoxic effects of SFN and CDDP exposure were
assessed by treating H-28 MM cells and MeT-5A normal human
mesothelial cells with increasing concentrations of SFN (10–100 µM)
or CDDP (40–960 µM) for 48 h and analyzed by MTT assay. As
demonstrated in Fig. 1A, treatment
with SFN or CDDP alone inhibited the growth of MeT-5A and H-28
cells in a concentration-dependent manner. MeT-5A cells exhibited
lower cytotoxicity to the two compounds when compared with H-28
cells. Concentrations of 20 and 40 µM for the combination of SFN
and CDDP, respectively, were chosen based on the concentrations
showing a high cell viability (>90%) in MeT-5A cells when
treated with SFN (92%) or CDDP (95%) alone. Combination treatment
of 20 µM SFN and 40 µM CDDP (SFN/CDDP) led to a significant
decrease in the viability (62.5%) of H-28 cells compared with 20 µM
SFN (85.3%) or 40 µM CDDP (96.7%) treatment alone. However, the
combination of 20 µM SFN and 40 µM CDDP did not significantly
inhibit the viability (88.3%) of MeT-5A cells compared with 20 µM
SFN (91.6%) or 40 µM CDDP (98.5%) alone (Fig. 1B). Based on the CI value, the
cytotoxic effect of SFN and CDDP in combination on H-28 cells was
synergistic (CI=0.454).
To further elucidate whether the growth inhibition
of H-28 cells by SFN/CDDP treatment was related to apoptotic cell
death, the proapoptotic effects of the two compounds was examined
by analyzing nuclear phenotypes and apoptotic cells using DAPI and
Annexin V-phycoerythrin (PE) staining, respectively. The proportion
of adherent cells with pyknotic and fragmented nuclei increased
(Fig. 2A), and the proportion of
Annexin V-PE-positive cells undergoing apoptosis increased to
33.05% (Fig. 2B) in combination
treated cells relative to SFN (12.62%) or CDDP (3.01%) treatment
alone. In addition, cell cycle analysis in the PI-stained cells
indicated an increase in the sub-G0/G1 peak,
which was indicative of apoptosis (Fig. 2C). The proportion of cells in the
G2/M phase increased, while the percentage of cells at
G0/G1 and S phases decreased, indicating a
G2/M phase-transition delay in the cell cycle.
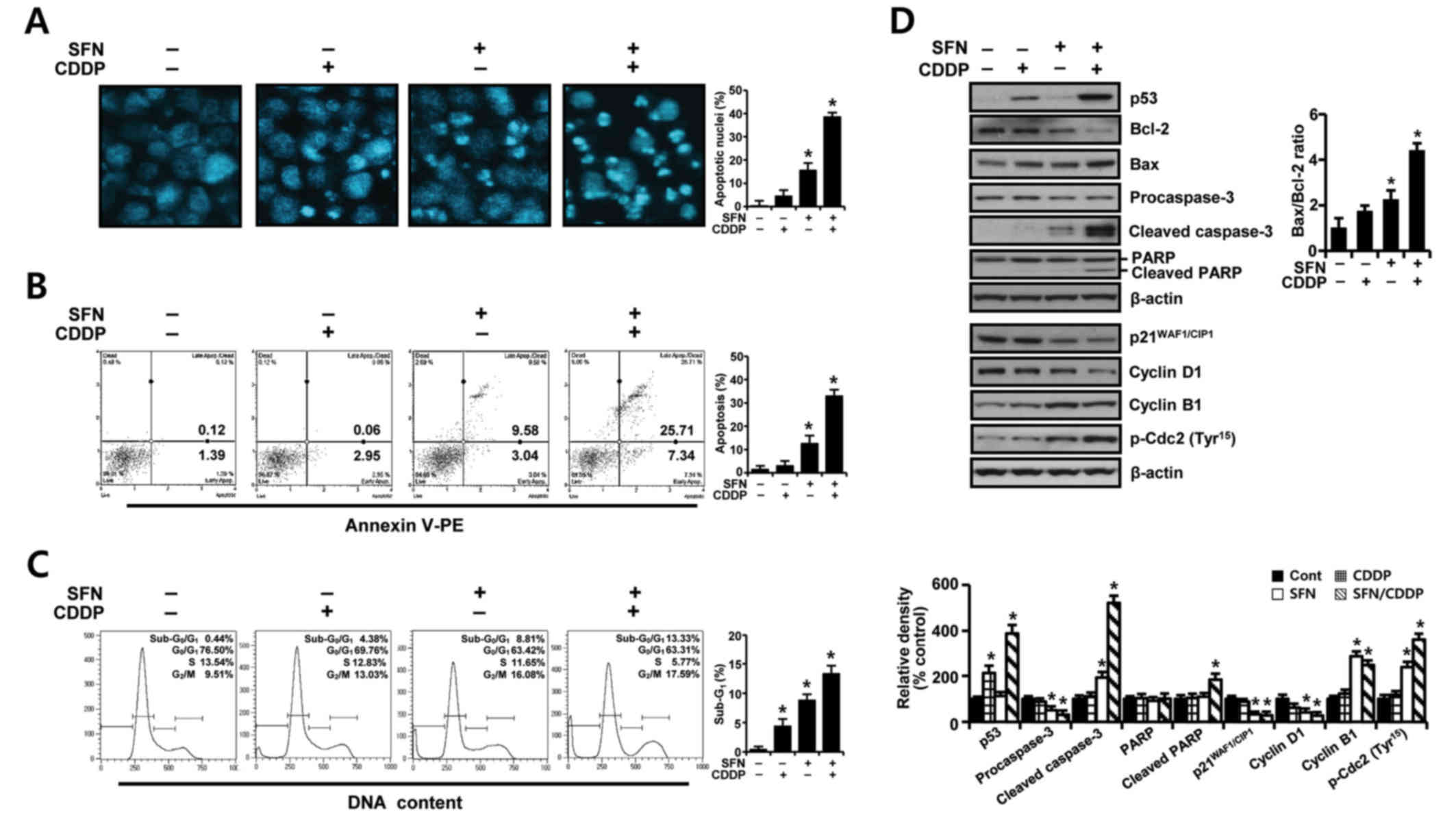 | Figure 2.Apoptosis-promoting and
growth-inhibiting effects of SFN and CDDP in H-28 malignant
mesothelioma cells. H-28 cells were treated with SFN (20 µM) and
CDDP (40 µM), alone or in combination, for 48 h. (A) Nuclear
morphology was assessed by nuclear staining with DAPI
(magnification ×40). (B) The number of apoptotic cells following
Annexin V-PE staining was analyzed using a Muse Cell Analyzer. (C)
Cell distribution in sub-G0/G1,
G0/G1, S and G2/M phases was
analyzed by flow cytometry following staining with propidium iodide
(20 µg/ml). (D) Expression levels of apoptosis- and cell
cycle-regulating proteins were measured by western blot analysis.
The Bax/Bcl-2 ratio and relative density of protein bands were
obtained from densitometric analysis of the western blot images
normalized to β-actin. Representative results are presented from
one of three independent experiments; β-actin was used as a loading
control. Error bars indicate the mean ± standard deviation for
three independent experiments. *P<0.05 vs. respective untreated
controls. Bax, Bcl-2 associated X protein; Bcl-2, B-cell lymphoma
2; CDDP, cis-dichlorodiammineplatinum (cisplatin); PARP,
poly(ADP-ribose) polymerase; p-Cdc2; phosphorylated cyclin
dependent kinase 2; PE, phycoerythrin; SFN, sulforaphane. |
To identify possible effector molecules that may be
responsible for the growth-inhibiting effect of SFN/CDDP treatment,
the induction of apoptosis- and cell cycle-regulating molecules was
assessed by western blot analysis. As presented in Fig. 2D, SFN/CDDP induced an upregulation
of p53 protein expression levels and enhanced the cleavage of
procaspase-3 and poly (ADP-ribose) polymerase (PARP), and increased
the Bax/Bcl-2 expression ratio compared with cells treated with
either drug alone or untreated. In addition, SFN/CDDP treatment
increased the levels of p-Cdc2Tyr15 and cyclin B1
protein expression levels, whereas the levels of
p21WAF1/CIP1 and cyclin D1 proteins were decreased
compared with the untreated control.
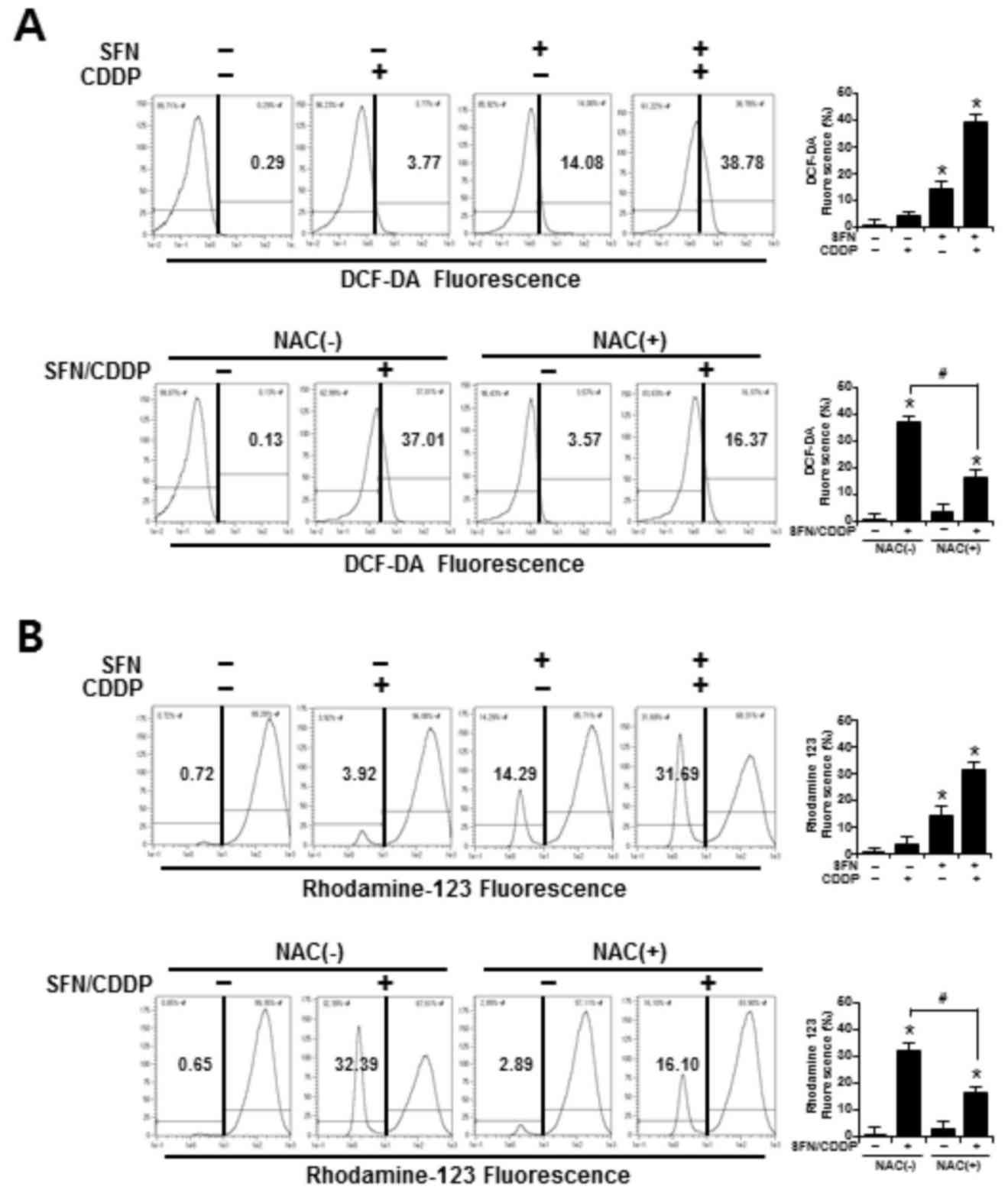
ROS accumulation and ΔΨm
To determine whether the synergistic cytotoxic
effects of SFN/CDDP treatment on H-28 cells were associated with
oxidative mitochondrial damage, intracellular ROS levels were
measured by flow cytometry using an ROS-sensitive fluorophore,
DCF-DA. As presented in the representative histogram in Fig. 3A, cells treated with SFN or CDDP
alone induced a slight increase in ROS accumulation, as indicated
by a shift in the DCF fluorescence to the right; however, combined
treatment with the two compounds increased ROS production to
38.78%. In addition, the integrity of mitochondrial function was
analyzed by flow cytometry using the fluorescent dye Rhodamine 123,
where decreased fluorescence was correlated with a loss of ΔΨm. In
the presence of SFN/CDDP, the number of cells with ΔΨm loss
increased to 31.69% (Fig. 3B).
Pretreatment with the ROS scavenger NAC significantly reduced the
levels of SFN/CDDP-induced ROS expression to 16.37% and lowered the
number of cells with ΔΨm loss to 16.10%, compared with the
NAC-untreated SFN/CDDP-induced control cells (Fig. 3A and B).
In H-28 cells treated with SFN/CDDP, flow cytometric
analysis revealed a G2/M phase-transition delay and an
increase in the number of cells in the
sub-G0/G1 peak, which was significantly
reduced by NAC pretreatment (P<0.05; Fig. 4A). NAC treatment also decreased the
proportion Annexin V-PE-positive cells (P<0.05; Fig. 4A and B). This was accompanied by a
significant improvement in the GSH/GSSG ratio (P<0.05), and cell
viability increased to 85.53 and 81% in the SFN/CDDP-treated cells
respectively, compared with the NAC-untreated SFN/CDDP-induced
control cells (68.6 and 61.9%, respectively) (P<0.05; Fig. 4C and D). Furthermore, cells exposed
to SFN/CDDP and pretreated with NAC exhibited a decrease in the
expression level of p53 protein, an increase in the level of cyclin
D1 protein, a decrease in the Bax/Bcl-2 ratio, a reduction in the
resultant cleavages of procaspase-3 and PARP, and a slight decrease
in the phosphorylation of Cdc2 on Tyr15, compared with
SFN/CDDP-treated cells that were not exposed to NAC (Fig. 4E). However, there were no
significant differences in the expression levels of
p21WAF1/CIP1 and cyclin B1 proteins between the two
treatment groups.
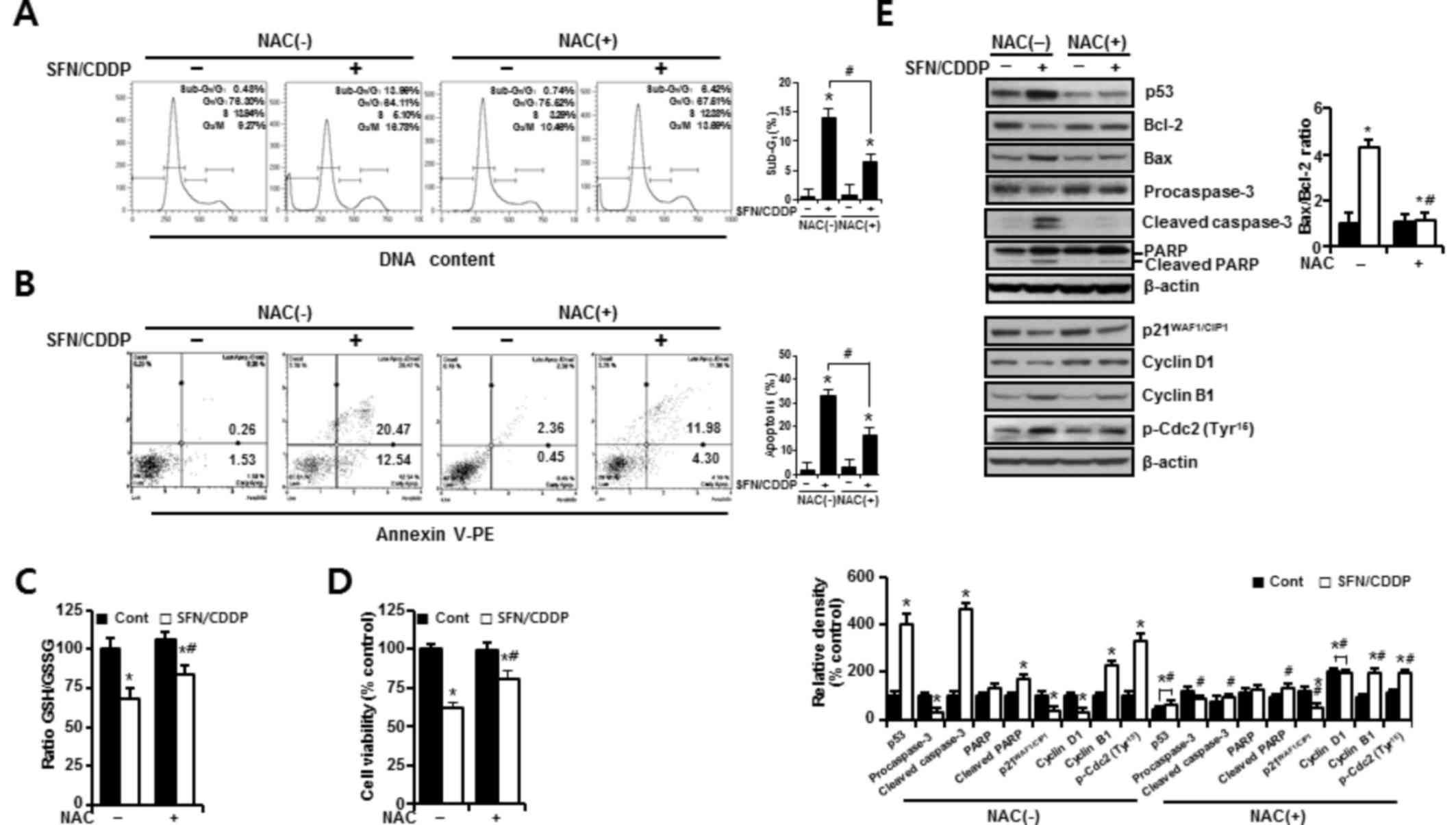 | Figure 4.Protective effects of NAC against the
pro-oxidant activity of SFN/CDDP in H-28 malignant mesothelioma
cells. H-28 cells were pretreated with or without NAC (5 mM) for 2
h prior to co-treatment with SFN (20 µM) and CDDP (40 µM) for a
further 48 h. (A) Cell cycle analysis was determined by propidium
iodide staining. (B) Cellular apoptosis was determined by Annexin
V-PE binding assay. (C) GSH/GSSG ratio was determined by
GSH/GSSG-Glo assay. (D) Cell viability was determined by MTT assay.
(E) Western blot analysis of apoptosis- and cell cycle-regulating
proteins; β-actin was used as a loading control. The Bax/Bcl-2
ratio and relative density of protein bands were obtained from
densitometric analysis of the western blot images normalized to
β-actin. Error bars indicate the mean ± standard deviation for
three independent experiments. *P<0.05 vs. the respective
controls; #P<0.05 vs. the respective NAC(−) group.
Bax, Bcl-2 associated X protein; Bcl-2, B-cell lymphoma 2; CDDP,
cis-dichlorodiammineplatinum (cisplatin); Cont, untreated control;
GSH, reduced glutathione; GSSG, oxidized glutathione; NAC,
N-acetylcysteine; PARP, poly(ADP-ribose) polymerase; p-Cdc2,
phosphorylated-cyclin dependent kinase 2; PE, phycoerythrin; SFN,
sulforaphane. |
ROS-dependent activation of
autophagy
To assess the autophagy-inducing effects of SFN and
CDDP treatment on H-28 cells, cells were treated for 48 h with SFN
and CDDP, alone or in combination, and the acidic vacuoles were
subsequently stained with LysoTracker Red DND-99 and analyzed on
channel APC-A by flow cytometry. As presented in Fig. 5A, treatment with SFN/CDDP or SFN
alone exhibited a significant increase in cells displaying high
LysoTracker Red fluorescence, indicative of autophagy. Compared
with either SFN or CDDP treatments alone, treatment with SFN/CDDP
resulted in higher protein expression levels of the autophagy
marker LC3B-II and a decrease in the phosphorylation of Akt and
mTOR, and these alterations were detected at after 12, 24 and 48 h
treatment (Fig. 5B). These
expression patterns were reversed by NAC pretreatment, suggesting
the involvement of ROS in the activation of autophagy in response
to SFN/CDDP exposure (Fig. 6A and
B). Pretreatment with the autophagy inhibitor bafilomycin A1,
which prevents autophagosome-lysosome fusion, enhanced the
expression levels of cleaved caspase-3 and cleaved PARP, led to a
further increase in the Bax/Bcl-2 ratio (Fig. 6C), and significantly enhanced
SFN/CDDP-induced cytotoxicity (Fig.
6D).
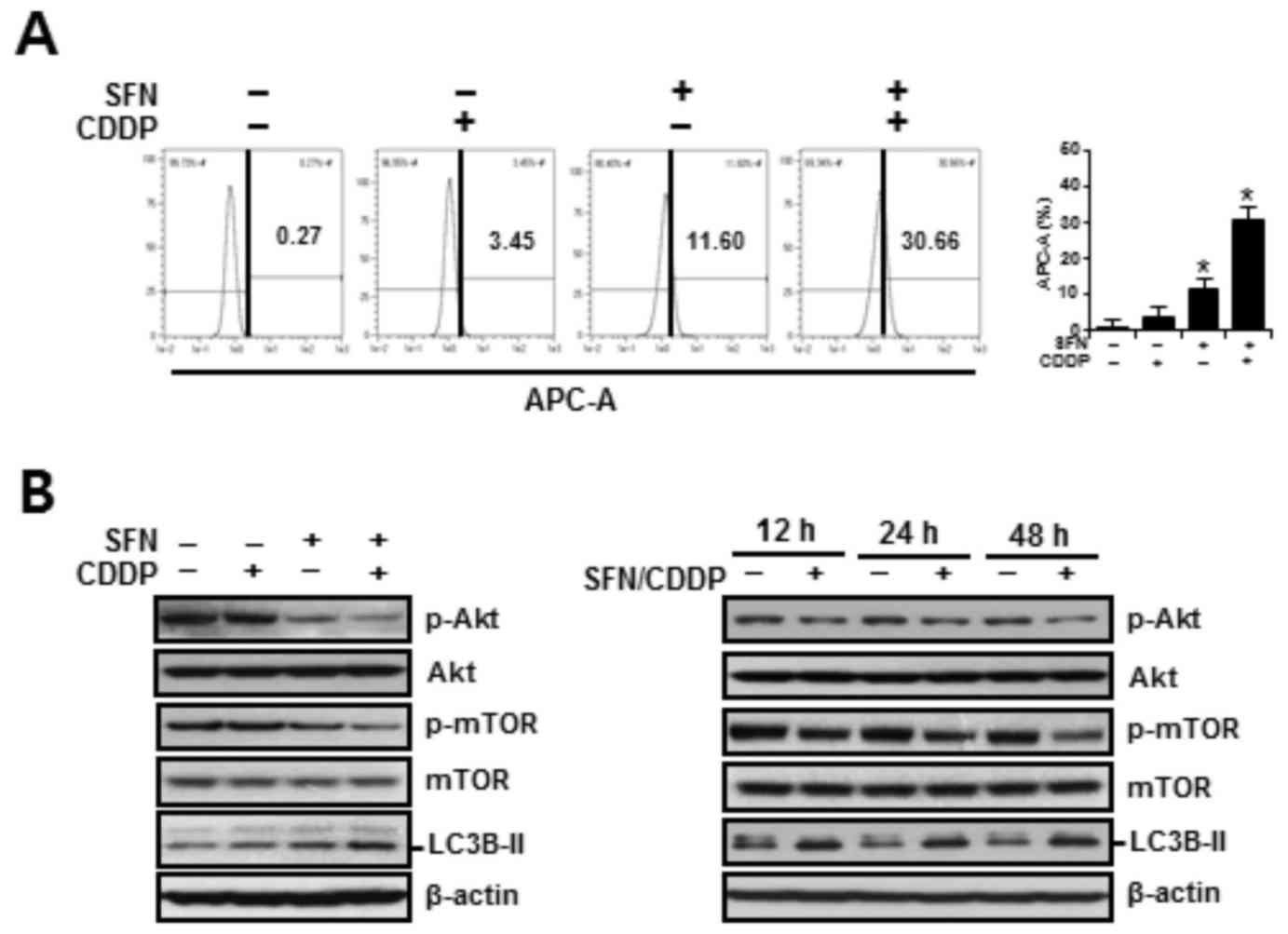 | Figure 5.Autophagy-promoting effects of SFN and
CDDP in H-28 malignant mesothelioma cells. (A) H-28 cells were
treated with SFN (20 µM) and CDDP (40 µM), alone or in combination,
for 48 h. The number of LysoTracker Red(+) cells, as detected by
APC-A fluorescence intensity, were analyzed by flow cytometry. (B)
H-28 cells were treated with SFN (20 µM) and CDDP (40 µM), alone or
in combination, for 48 h or the indicated times (12, 24 and 48 h),
and the levels of autophagy-related proteins were measured by
western blot analysis; β-actin was used as a loading control.
Representative results are presented and error bars indicate the
mean ± standard deviation for three independent experiments.
*P<0.05 vs. the respective controls. APC-A, allophycocyanin
channel; CDDP, cis-dichlorodiammineplatinum (cisplatin);
LC3B-II, microtubule-associated protein 1 light chain 3; βmTOR,
mammalian target of rapamycin; p, phosphorylated; SFN,
sulforaphane. |
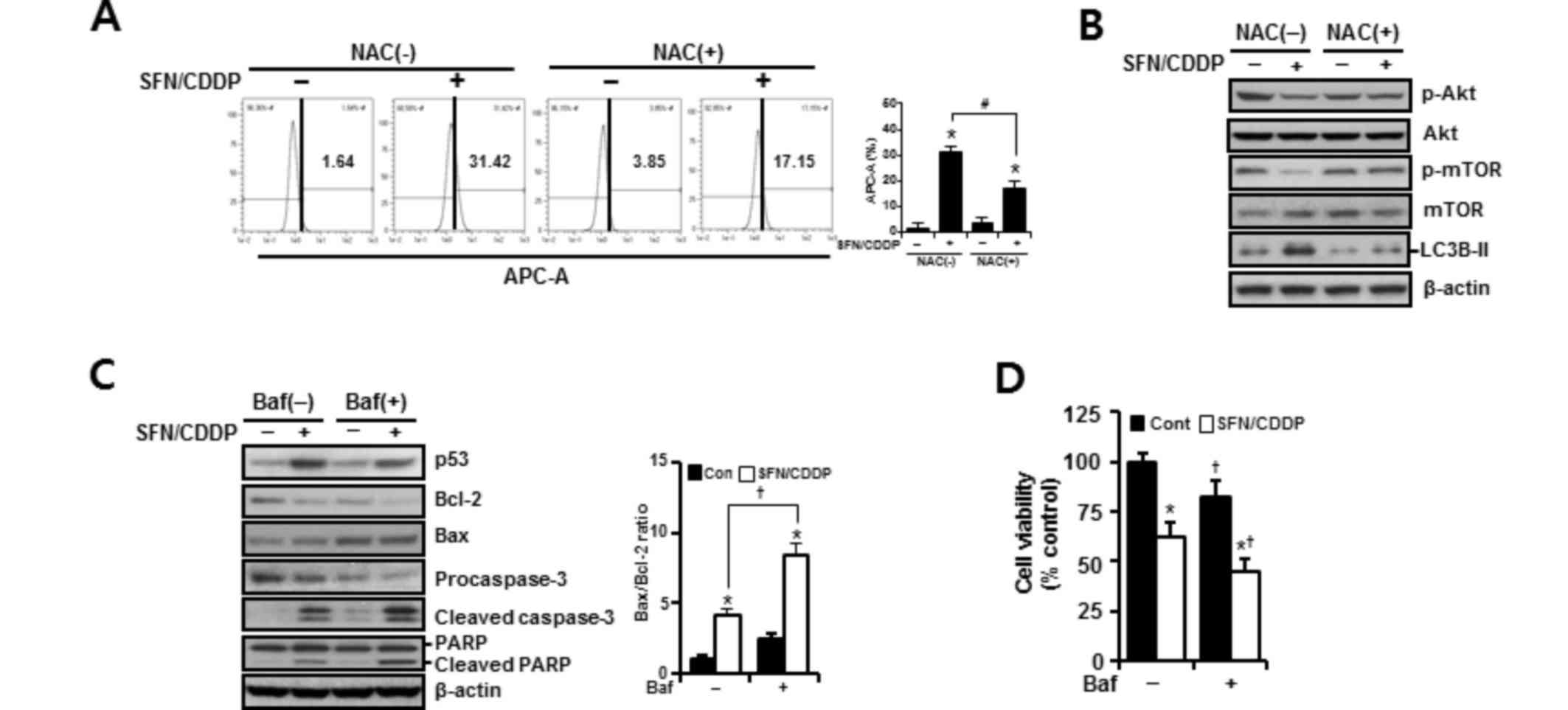 | Figure 6.Effects of NAC and Baf on the
SFN/CDDP-induced autophagy in H-28 malignant mesothelioma cells. (A
and B) H-28 cells were pretreated with or without NAC (5 mM) for 2
h prior to co-treatment with SFN (20 µM) and CDDP (40 µM) for a
further 48 h. (A) The number of LysoTracker Red(+) cells, as
detected by APC-A fluorescence intensity by flow cytometry. (B)
Western blot analysis of autophagy-related proteins; β-actin was
used as a loading control. (C and D) H-28 cells were pretreated
with or without Baf (50 nM) for 2 h prior to co-treatment with SFN
(20 µM) and CDDP (40 µM) for a further 48 h. (C) Western blot
analysis of apoptosis-regulating proteins; β-actin was used as a
loading control. (D) Cell viability was determined by MTT assay.
Representative results are presented and error bars indicate the
mean ± standard deviation for three independent experiments.
*P<0.05 vs. the respective controls; #P<0.05 vs.
the respective NAC(−) group; †P<0.05 vs. the
respective Baf(−) group. Baf, bafilomycin A1; Bax, Bcl-2 associated
X protein; Bcl-2, B-cell lymphoma 2; CDDP,
cis-dichlorodiammineplatinum (cisplatin); Cont, untreated control;
LC3B-II, microtubule-associated protein 1 light chain 3 β; mTOR,
mammalian target of rapamycin; NAC, N-acetylcysteine; p,
phosphorylated; PARP, poly(ADP-ribose) polymerase; SFN,
sulforaphane. |
Discussion
The present study investigated the effects of SFN
co-treatment on enhancing the anticancer role, along with the
phenomena of apoptosis and autophagy in this process, of CDDP in
H-28 malignant mesothelioma cells, and identified alterations in
apoptotic and autophagic factors. First, SFN/CDDP treatment
produced a significant and synergistic cytotoxic effect, evidenced
by the enhancement of apoptosis and G2/M
phase-transition delay in the cell cycle compared with either
treatment alone. Second, this synergism appeared to be mediated via
proapoptotic and cell-cycle modulators, including p53, Bcl-2, Bax,
phosphatidylinositol-3-kinase (PI3K)/Akt and Cdc2. Third, the
pro-oxidant activity of SFN/CDDP treatment contributed to the
simultaneous induction of apoptosis and autophagy. Fourth,
inhibition of autophagy by bafilomycin A1 augmented
SFN/CDDP-induced cytotoxicity, indicating SFN/CDDP-induced
autophagy is a pro-survival process in H-28 cells.
Cells are continually receiving and integrating a
variety of positive and negative growth signals. Loss of normal
survival signals or an increase in negative growth signals tips the
balance and a cell may undergo programmed cell death (15). Cancer cells may be able to survive
and thrive against cellular stresses by weakening or crippling the
proapoptotic machinery, for example via p53 inactivation and
upregulation of antiapoptotic Bcl-2-related proteins (16). In this regard, the presence of
upregulated p53 expression, increased Bax/Bcl-2 ratio and inhibited
pro-survival PI3K/Akt signaling, which suppresses the actions of
the proapoptotic circuitry, may provide a theoretical basis for the
production of a synergistic apoptosis-promoting effect by SFN/CDDP
treatment in H-28 cells. In addition, the concurrence of
G2/M phase-transition delay with apoptosis observed in
the present study is consistent with a previous report that
revealed reduced viability in ovarian cancer cells treated with SFN
in triple combination with epigallocatechin gallate and CDDP
(17). However, that observation
differs from the results of the present discovery, in which the
increase in cells at the G2/M phase (indicative of an
arrest) was not dependent on the cyclin dependent kinase inhibitor
p21WAF1/CIP1, rather it was associated with an
upregulation of p-Cdc2 expression levels. The phosphorylation of
Cdc2 on Tyr15 has been reported to render the
Cdc2/cyclin B complex inactive, thus keeping the cells in
G2 phase from entering M phase (18).
Oxidative stress-induced damage to macromolecules
has been proposed as a possible mechanism for CDDP-induced
cytotoxicity (19). Previous
research has also demonstrated that SFN treatment increases the
sensitivity of various cancer cells to chemotherapeutic agents, by
inducing a further increase in oxidative stress (8,9).
Excessive ROS production triggers mitochondrial-mediated apoptosis
by increasing the Bax/Bcl-2 ratio, which results in marked ΔΨm loss
and subsequent caspase activation (20). Combining these results with
observations from the present study, which revealed mitochondrial
damage and an increase in apoptosis, indicated that the pro-oxidant
role of SFN is essential in potentiating the cytotoxic effects of
CDDP. NAC is a precursor for cellular biosynthesis of the
antioxidant GSH and stimulates the synthesis of GSH. Exposure to
SFN/CDDP resulted in a significant reduction in the GSH/GSSG ratio
in H-28 cells; however, this was reversed in the presence of NAC.
The replenishment of cellular GSH levels may explain, at least in
part, the effect of NAC on reducing ROS levels. The concurrence of
autophagy with apoptosis in response to SFN/CDDP exposure was
apparently dependent on ROS accumulation. Consistent with our data,
previous studies indicated that ROS may act as upstream molecules
in mediating autophagy and apoptosis (21,22).
Furthermore, elevated ROS levels suppress the PI3K/Akt/mTOR
survival pathway and subsequently induce apoptosis and autophagy,
whereas activation of this survival pathway inhibits several
autophagy-promoting proteins via phosphorylation of mTOR complex 1
(23,24). This rationale is supported by the
present study which demonstrated that SFN/CDDP treatment reduced
the phosphorylation of both Akt and mTOR, induced the upregulation
of LC3B-II expression and caused a significant increase in the
proportion of LysoTracker Red(+) cells. PI3K/Akt/mTOR signaling is
a crucial survival pathway in MM (25). Recently, Echeverry et al
(26) reported that autophagy is
one of the predominant mechanisms of drug resistance to the dual
PI3K/mTOR inhibitors, NVP-BEZ235 and GDC-0980, in MM cells. Bcl-2
has also been reported as a possible effector molecule linked to
both apoptosis and autophagy, as it inhibits apoptosis by
preventing mitochondrial cytochrome c release and suppresses
autophagy by interacting with Beclin-1 (27). In the present study, the
downregulation of Bcl-2 protein expression in H-28 cells exposed to
SFN/CDDP may have contributed to the concurrence of apoptosis and
autophagy; however, further research is required to understand the
significance of this double role of Bcl-2.
A number of previous studies have reported on the
paradoxical functions of autophagy in cancer, in which it can
either promote or inhibit cell survival (11–13).
Cancer cells may take advantage of this mechanism for protection
and to maintain survival against stresses. Evidence of autophagy as
a survival mechanism comes from previous research that indicated
that the activation of autophagy contributes to cancer progression,
therapeutic resistance and survival of dormant cancer cells
(12,28). The present study demonstrated that
autophagic inhibition potentiated the cytotoxic effect of SFN/CDDP
treatment, which supports the cytoprotective role of autophagy.
However, significant levels of apoptotic cell death were observed,
and the activation of autophagy was not able to override H-28
apoptosis induced by SFN/CDDP treatment. Detailed mechanistic
studies under the same conditions are required to unveil the
cytoprotective aspects of autophagy.
In conclusion, SFN/CDDP treatment produced a
synergistic outcome in H-28 cells via activation of the
ROS-dependent mitochondrial apoptotic pathway as a fail-safe
mechanism designed to eliminate damaged cells from the growing
population. Autophagy was additionally activated as a prosurvival
process to SFN/CDDP insults, possibly as an adaptive survival
mechanism. These responses may contribute to the selection of more
malignant clones that escape apoptotic eradication, and may
therefore provide a route for drug-resistant subpopulations to
arise. From this perspective, the present study underlines the
concept that targeting autophagic regulation may be an attractive
therapeutic strategy for the improvement of clinical outcomes for
patients with MM.
Acknowledgements
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF), funded by the Ministry of Education (grant no.
NRF-2015R1D1A3A03020269).
References
|
1
|
Wolff H, Vehmas T, Oksa P, Rantanen J and
Vainio H: Asbestos, asbestosis, and cancer, the helsinki criteria
for diagnosis and attribution 2014: Recommendations. Scand J Work
Environ Health. 41:5–15. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Raptopoulos V: Peritoneal mesothelioma.
Crit Rev Diagn Imaging. 24:293–328. 1985.PubMed/NCBI
|
|
3
|
Scripcariu V, Dajbog E, Radu I, Ferariu D,
Pricop A, Grigoraş M and Dragomir C: Malignant peritoneal
mesothelioma tumours. Evolution, treatment, prognosis: Rev Med Chir
Soc Med Nat Iasi. 111:673–677. 2007.
|
|
4
|
Ray M and Kindler HL: Malignant pleural
mesothelioma: An update on biomarkers and treatment. Chest.
136:888–896. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Villanova F, Procopio A and Rippo MR:
Malignant mesothelioma resistance to apoptosis: Recent discoveries
and their implication for effective therapeutic strategies. Curr
Med Chem. 15:631–641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vogelzang NJ, Rusthoven JJ, Symanowski J,
Denham C, Kaukel E, Ruffie P, Gatzemeier U, Boyer M, Emri S,
Manegold C, et al: Phase III study of pemetrexed in combination
with cisplatin versus cisplatin alone in patients with malignant
pleural mesothelioma. J Clin Oncol. 21:2636–2644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tomczyk J and Olejnik A: Sulforaphane-a
possible agent in prevention and therapy of cancer. Postepy Hig Med
Dosw (Online). 64:590–603. 2010.(In Polish). PubMed/NCBI
|
|
8
|
Lin LC, Yeh CT, Kuo CC, Lee CM, Yen GC,
Wang LS, Wu CH, Yang WC and Wu AT: Sulforaphane potentiates the
efficacy of imatinib against chronic leukemia cancer stem cells
through enhanced abrogation of Wnt/β-catenin function. J Agric Food
Chem. 60:7031–7039. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim H, Kim EH, Eom YW, Kim WH, Kwon TK,
Lee SJ and Choi KS: Sulforaphane sensitizes tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-resistant hepatoma
cells to TRAIL-induced apoptosis through reactive oxygen
species-mediated up-regulation of DR5. Cancer Res. 66:1740–1750.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sharma K, Le N, Alotaibi M and Gewirtz DA:
Cytotoxic autophagy in cancer therapy. Int J Mol Sci.
15:10034–10051. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giuliani CM and Dass CR: Metabolic stress
and cancer: Is autophagy the common denominator and a feasible
target? J Pharm Pharmacol. 66:597–614. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vlahopoulos S, Critselis E, Voutsas IF,
Perez SA, Moschovi M, Baxevanis CN and Chrousos GP: New use for old
drugs? Prospective targets of chloroquines in cancer therapy. Curr
Drug Targets. 15:843–851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jin S and White E: Role of autophagy in
cancer: Management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mehlen P and Thibert C: Dependence
receptors: Between life and death. Cell Mol Life Sci. 61:1854–1866.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen H, Landen CN, Li Y, Alvarez RD and
Tollefsbol TO: Enhancement of cisplatin-mediated apoptosis in
ovarian cancer cells through potentiating G2/M arrest and p21
upregulation by combinatorial epigallocatechin gallate and
sulforaphane. J Oncol. 2013:8729572013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singh SV, Herman-Antosiewicz A, Singh AV,
Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L and Baskaran R:
Sulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Deavall DG, Martin EA, Horner JM and
Roberts R: Drug-induced oxidative stress and toxicity. J Toxicol.
2012:6454602012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deng S, Tang S, Zhang S, Zhang C, Wang C,
Zhou Y, Dai C and Xiao X: Furazolidone induces apoptosis through
activating reactive oxygen species-dependent mitochondrial
signaling pathway and suppressing PI3K/Akt signaling pathway in
HepG2 cells. Food Chem Toxicol. 75:173–186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dunlop EA and Tee AR: mTOR and autophagy:
A dynamic relationship governed by nutrients and energy. Semin Cell
Dev Biol. 36:121–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nazio F and Cecconi F: mTOR, AMBRA1, and
autophagy: An intricate relationship. Cell Cycle. 12:2524–2525.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen B, Li D, Li M, Li S, Peng K, Shi X,
Zhou L, Zhang P, Xu Z, Yin H, et al: Induction of
mitochondria-mediated apoptosis and PI3K/Akt/ mTOR-mediated
autophagy by aflatoxin B2 in hepatocytes of broilers. Oncotarget.
7:84989–84998. 2016.PubMed/NCBI
|
|
24
|
Mi S, Xiang G, Yuwen D, Gao J, Guo W, Wu
X, Wu X, Sun Y, Su Y, Shen Y and Xu Q: Inhibition of autophagy by
andrographolide resensitizes cisplatin-resistant non-small cell
lung carcinoma cells via activation of the Akt/mTOR pathway.
Toxicol Appl Pharmacol. 310:78–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou S, Liu L, Li H, Eilers G, Kuang Y,
Shi S, Yan Z, Li X, Corson JM, Meng F, et al: Multipoint targeting
of the PI3K/mTOR pathway in mesothelioma. Br J Cancer.
110:2479–2488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Echeverry N, Ziltener G, Barbone D, Weder
W, Stahel PA, Broaddus VC and Felley-Bosco E: Inhibition of
autophagy sensitizes malignant pleural mesothelioma cells to dual
PI3K/mTOR inhibitors. Cell Death Dis. 6:e17572015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao GX, Pan H, Ouyang DY and He XH: The
critical molecular interconnections in regulating apoptosis and
autophagy. Ann Med. 47:305–315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare
S, Kondo S, Kondo Y, Yu Y, Mills GB, et al: The tumor suppressor
gene ARHI regulates autophagy and tumor dormancy in human ovarian
cancer cells. J Clin Invest. 118:3917–3929. 2008.PubMed/NCBI
|