Introduction
Ischemic preconditioning (IPC) is induced by
exposure to brief durations of transient ischema, resulting in
ischemic tolerance to a subsequent longer or lethal period of
transient ischemia (1). Kitagawa
et al (2) initially
introduced the concept of ischemic tolerance in the brain, and
demonstrated that 2 min of transient cerebral ischemia, 1–2 days
prior to a subsequent longer or lethal period of transient cerebral
ischemia, exerted protective effects against ischemic injury in the
gerbil hippocampal CA1 region. Further studies have demonstrated
the protective effects of IPC in other animal models, including
global and focal cerebral ischemia (1,3–5).
Ischemic tolerance has also been demonstrated in human clinical
practice; less severe strokes occured in patients that suffered
from a prior transient ischemic attack within a short period of
time (6,7). IPC-induced neuroprotection is
considered a promising target for the development of a potential
therapeutic strategy; however, the basic mechanisms underlying
IPC-induced neuroprotection remain to be elucidated (1).
Platelet-derived growth factor (PDGF) is a
polypeptide that acts as a potent mitogen in several cell types,
which consists of two homodimers (AA and BB) and one heterodimer
(AB) (8,9). The biological activities of PDGF are
mediated through binding to PDGF receptors (PDGFR), designated α
and β. The α subunit can bind to either the -A or -B chain, whereas
the β subunit can bind only to the-B chain (10–12).
PDGF-BB is widely expressed in the central nervous system (13) and is upregulated in neurons
following cerebral ischemia in animal models (14–17).
Suppression of PDGF-BB mRNA expression enhances
N-methyl-D-aspartate (NMDA)-induced excitotoxicity in the neonatal
rat brain (18). Conversely, the
intraventricular administration of PDGF-BB markedly promotes
neuronal survival following cerebral ischemia in rats (19). These findings suggest that PDGF-BB
may serve a crucial role in the protection of neurons against
cerebral ischemic insults.
To the best of our knowledge, the expression pattern
of endogenous PDGF-BB in the IPC-mediated hippocampus, following
subsequent transient cerebral ischemia, has not been studied.
Therefore, the present study aimed to investigate the effects of
IPC on cellular localization and alterations in endogenous PDGF-BB
in the IPC-induced hippocampus. The hippocampus is an important
structure for the study of neuronal damage and its mechanism
following transient cerebral ischemia, and the gerbil is considered
a good animal for studying transient cerebral ischemia (20–23).
Materials and methods
Experimental groups and ischemic
surgery
As previously described (20), 102 male gerbils were obtained from
the Experimental Animal Center, Kangwon National University,
Chuncheon, South Korea. The gerbils were 6 months old with a body
weight of 65–75 g. The animals were housed in a conventional state
under adequate temperature (23°C) and humidity (60%) control with a
12-h light/dark cycle, and provided with free access to water and
food. The gerbils were used according to guidelines that are in
compliance with the current international laws and policies
(24). The present study was
approved by the Institutional Animal Care and Use Committee at
Kangwon National University (Chuncheon, South Korea; approval no.
KW-160802-1).
The gerbils were divided into four groups (n=7 at
each time point: 0, 1, 2 and 5 days in each group): i)
Sham-operated group, both common carotid arteries were exposed;
however, the gerbils were not exposed to ischemia (sham-operation);
ii) ischemia-operated group was exposed to 5 min of transient
cerebral ischemia (lethal transient cerebral ischemia, LTCI); iii)
IPC + sham-operated group was subjected to 2 min sublethal
transient ischemia prior to sham-operation; and iv) IPC +
ischemia-operated group was subjected to 2 min of sublethal
ischemia 1 day prior to 5 min of transient ischemia. The IPC
paradigm has been proven to be effective at protecting neurons
against ischemic damage in this ischemic model (25). The gerbils in the ischemia-operated
and the IPC + ischemia-operated groups were given recovery times of
1, 2 and 5 days, since pyramidal neurons in the hippocampal CA1
region survive until 3 days and begin to die 4–5 days
post-LTCI.
Surgery for ischemic insults
IPC and LTCI were developed according to our
previously described method (20).
Briefly, the experimental animals were anesthetized with a mixture
of 2.5% isoflurane in 33% oxygen and 67% nitrous oxide. Ischemia
was induced by occluding the bilateral common carotid arteries with
non-traumatic aneurysm clips (Yasargil FE 723K; Aesculap AG,
Tuttlingen, Germany). After 2 or 5 min of occlusion, the aneurysm
clips were removed from the arteries. The body temperature under
free-regulating or normothermic (37±0.5°C) conditions was monitored
using a rectal temperature probe (TR-100; Fine Science Tools,
Foster City, Inc., CA, USA) and was maintained using a thermometric
blanket prior to, during and after surgery until the animals
completely recovered from anesthesia. Thereafter, the gerbils were
maintained in a thermal incubator (temperature, 23°C; humidity,
60%; Mirae Medical Industry, Seoul, South Korea) to maintain the
body temperature of animals until they were sacrificed at 1, 2 and
5 days following ischemia.
Spontaneous locomotor activity
In order to elucidate increased hyperactivity
following ischemia-reperfusion (I-R), spontaneous locomotor
activity was measured, according to a previously published
procedure (26). Briefly, gerbils
(n=7 at each time point in each group) were maintained in a
Plexiglas cage (25×20×12 cm), located inside a soundproof chamber.
Locomotor activity was recorded using a Photobeam Activity
system-Home Cage (San Diego Instruments, San Diego, CA, USA).
Spontaneous locomotor activity was monitored during 1 h, a total of
24 h after I-R and, simultaneously, the number of times each animal
stood on its hind legs and the time (in sec) spent exhibiting
grooming behavior were recorded. Each animal was observed
continuously via a 4×8 photobeam. Scores were generated from live
observations, and video sequences were used for subsequent
re-analysis.
Tissue processing for histology
According to our previously published procedure
(27), the gerbils were deeply
anesthetized with pentobarbital sodium (40 mg/kg, i.p; JW
Pharmaceutical Co., Ltd., Seoul, South Korea,) and were
transcardially perfused with 4% paraformaldehyde. The brains were
removed and the tissues cryoprotected by infiltration with 30%
sucrose overnight at 4°C. Subsequently, their brains were serially
sectioned into 30 µm coronal sections using a cryostat (Leica
Microsystems GmbH, Wetzlar, Germany).
Cresyl violet (CV) staining and
Fluoro-Jade B (F-J B) histofluorescence
To investigate neuronal damage in the hippocampus
following I-R, CV and F-J B histofluorescence staining were
performed, as previously described (28). Briefly, for CV staining, the
sections were stained with 1.0% (w/v) CV acetate (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) and dehydrated. The section were
then mounted with Canada balsam (Kanto Chemical Co., Inc., Tokyo,
Japan). For F-J B histofluorescence, the sections were immersed in
a 0.0004% F-J B (Histochem, Inc., Jefferson, AR, USA) staining
solution. After washing, the sections were examined using an
epifluorescent microscope (Zeiss Carl, Göttingen, Germany) with
blue (450–490 nm) excitation light and a barrier filter.
Immunohistochemistry for neuronal
nuclei (NeuN) and PDGF-BB
For immunohistochemical staining, the sections were
analyzed according to our previously described procedure (28). The brain sections were blocked with
10% normal goat serum (cat. no. S-1000; Vector Laboratories Inc.,
Burlingame, CA, USA) in 0.05 M PBS followed by staining with
primary mouse anti-NeuN (a neuron-specific soluble nuclear antigen;
cat. no. 574597; 1:1,000; EMD Millipore, Billerica, MA, USA) and
rabbit anti-PDGF-BB (cat. no. ab21234; 1:200; Abcam, Cambridge, MA,
USA) overnight at 4°C. The sections were then incubated with
secondary antibodies (cat. no. I-1000; 1:250; Vector Laboratories
Inc.) and were developed using Vectastain ABC (Vector Laboratories
Inc.). The sections were visualized with 3,3′-diaminobenzidine
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in 0.1 M Tris-HCl
buffer. In order to establish the specificity of the
immunostaining, a negative control test was carried out with
pre-immune serum, instead of primary antibody. The negative control
resulted in the absence of immunoreactivity in any structures.
Western blot analysis
To examine alterations in the protein expression
levels of PDGF-BB in the hippocampal CA1 region, western blotting
was conducted, according to our previously published procedure
(28). Briefly, tissues (n=7 at
each time point) were homogenized, and the protein concentrations
were determined in the supernatants using a Micro Bicinchonininc
Acid Protein Assay kit with bovine serum albumin as the standard
(Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Aliquots containing 20 µg total protein were boiled and loaded onto
a 12.5% polyacryamide gel. After electrophoresis, the gels were
transferred to nitrocellulose transfer membranes (Pall Corporation,
Port Washington, NY, USA). To reduce background staining, the
membranes were incubated with 5% non-fat dry milk for 30 min at 4°C
prior to incubation with rabbit anti-PDGF-BB (cat. no. ab178409;
1:1,000; Abcam) for 2 hrs at 4°C. Membranes were subsequently
exposed to peroxidase-conjugated goat anti-rabbit immunoglobulin G
(cat. no. A0545; 1:5,000; Sigma-Aldrich; Merck KgaA). Antibodies
were visualized using an enhanced chemiluminescence kit (Pierce;
Thermo Fisher Scientific, Inc.). Antibodies against β-actin were
used as a loading control (cat. no. ab8227; 1:2,000; Abcam).
Data analysis
For cell counts, as previously described (20), sections were selected according to
anatomical landmarks corresponding to anterior-posterior, from −1.4
to −1.8 mm of gerbil brain atlas. The number of NeuN-immunoreactive
and F-J B-positive cells was counted in a 200×200 µm square,
applied at the approximate center of the CA1 region, including the
stratum pyramidale. Cell counts were analyzed as a percentage, with
the sham group designated as 100%.
In order to analyze PDGF-BB immunoreactivity, we
used our previously published method (28). Briefly, cellular immunoreactivity
of PDGF-BB was graded in the hippocampal CA1 and CA3 regions.
Digital images were captured using an AxioM1 light microscope
(Zeiss Carl) equipped with a digital camera (Axiocam; Zeiss Carl)
connected to a PC monitor. Semi-quantification of the
immunoreactivity of PDGF-BB was evaluated using digital image
analysis software (MetaMorph 4.01; Molecular Devices, LLC,
Sunnyvale, CA, USA). The staining intensity of PDGF-BB was
evaluated on the basis of optical density (OD), which was obtained
following transformation of the mean gray level using the formula:
OD = log (256/ mean gray level). The background OD was subtracted
from areas adjacent to the measured area. After the background
density was subtracted, a ratio of the OD of an image file was
calibrated in Adobe Photoshop 8.0 (Adobe Systems, San Jose, CA,
USA) and was analyzed as a percentage, with the sham-operated group
designated as 100% in NIH Image 1.59 (National Institutes of
Health, Bethesda, MD, USA).
Protein expression levels were analyzed, according
to our previously published method (29). Briefly, western blots were scanned
and semi-quantification was conducted using Scion Image software
version 2.0 (Scion Corp., Frederick, MD, USA), which was used to
determine relative OD (ROD): A ratio of the ROD was calibrated as
%, with the sham-operated group designated as 100 %.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. A multiple-sample comparison was applied to test the
differences between groups and days. The differences between groups
on the same day were assessed using one-way analysis of variance
(ANOVA) and Tukey's post hoc test. For analysis of time-dependent
differences between the groups, two-way ANOVA was used with a
Bonferroni post hoc test. P<0.05 was considered to indicate a
statistically significant difference. All experiments were repeated
twice.
Results
Spontaneous motor activity
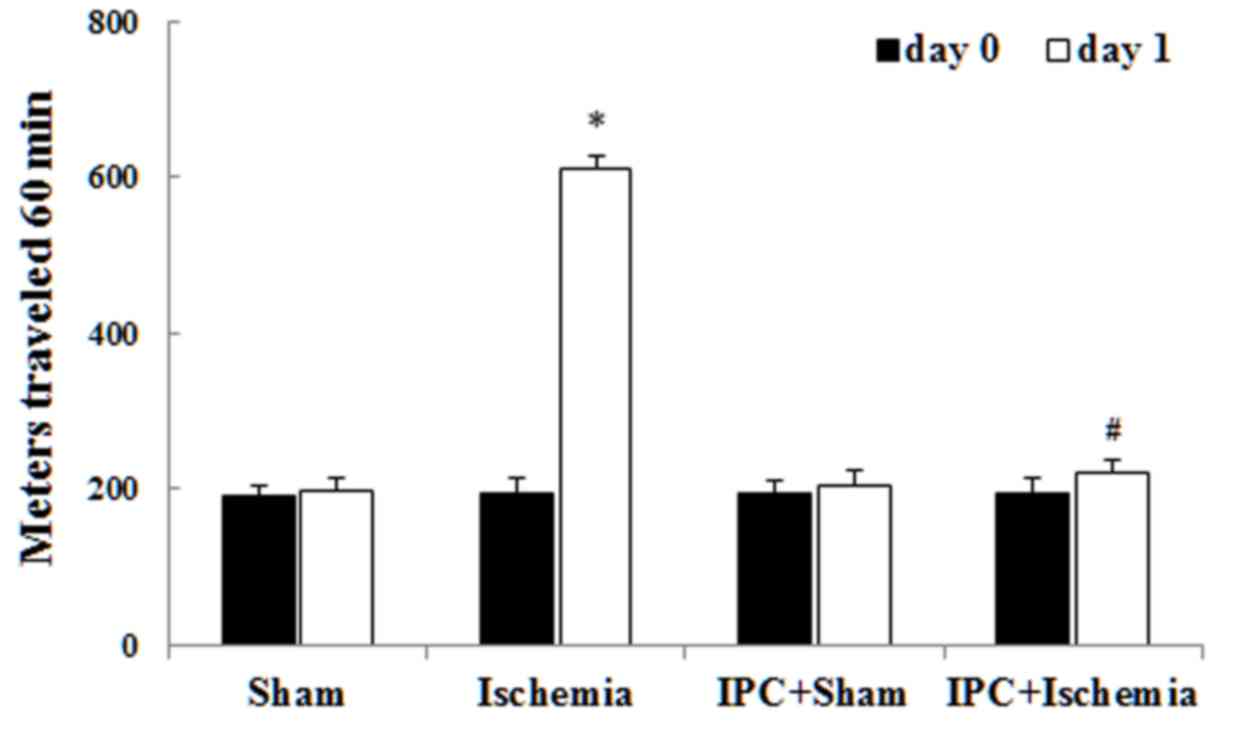
Spontaneous motor activity was detected to
investigate the alterations in motor behavior at day 0 (before
ischemia) and day 1 (1 day after ischemia; Fig. 1) (30,31).
Similar levels of locomotor activity were observed in all
experimental groups on day 0. In the ischemia-operated group,
locomotor activity was evidently increased compared with that in
the sham-operated group 1 day after LTCI. In the IPC +
ischemia-operated group, locomotor activity was not significantly
increased compared with that in the IPC + sham-operated group 1 day
after LTCI. Previous studies have demonstrated that hippocampal
neuronal damage leads to locomotor hyperactivity (29,30).
CV+ cells
CV staining was used to determine the distribution
of all cells in the hippocampus. In all experimental groups on day
0 after LTCI, CV+ cells were easily observed in all subregions of
the hippocampus (Fig. 2Aa-Ah). In
particular, pyramidal neurons in the stratum pyramidale were
identified by their larger and pyramid-like or round shape
(Fig. 2Ab, Ad, Af and Ah). In the
ischemia-operated group, CV+ pyramidal neurons were markedly
decreased in number in the CA1 region, but not in the CA2/3 region,
5 days after LTCI compared with in the sham-operated group
(Fig. 2Ak, Al); at a high
magnification, damaged CA1 pyramidal neurons were shrunken and
contained dark and polygonal nuclei (Fig. 2Al). In the IPC + sham-operated
group, the distribution pattern of CV+ cells in the CA1 region was
similar to that in the sham-operated group (Fig. 2Am and An). In the IPC +
ischemia-operated group, the morphology of CV+ pyramidal neurons in
the CA1 region was similar to that in the IPC + sham-operated group
(Fig. 2Ao and Ap).
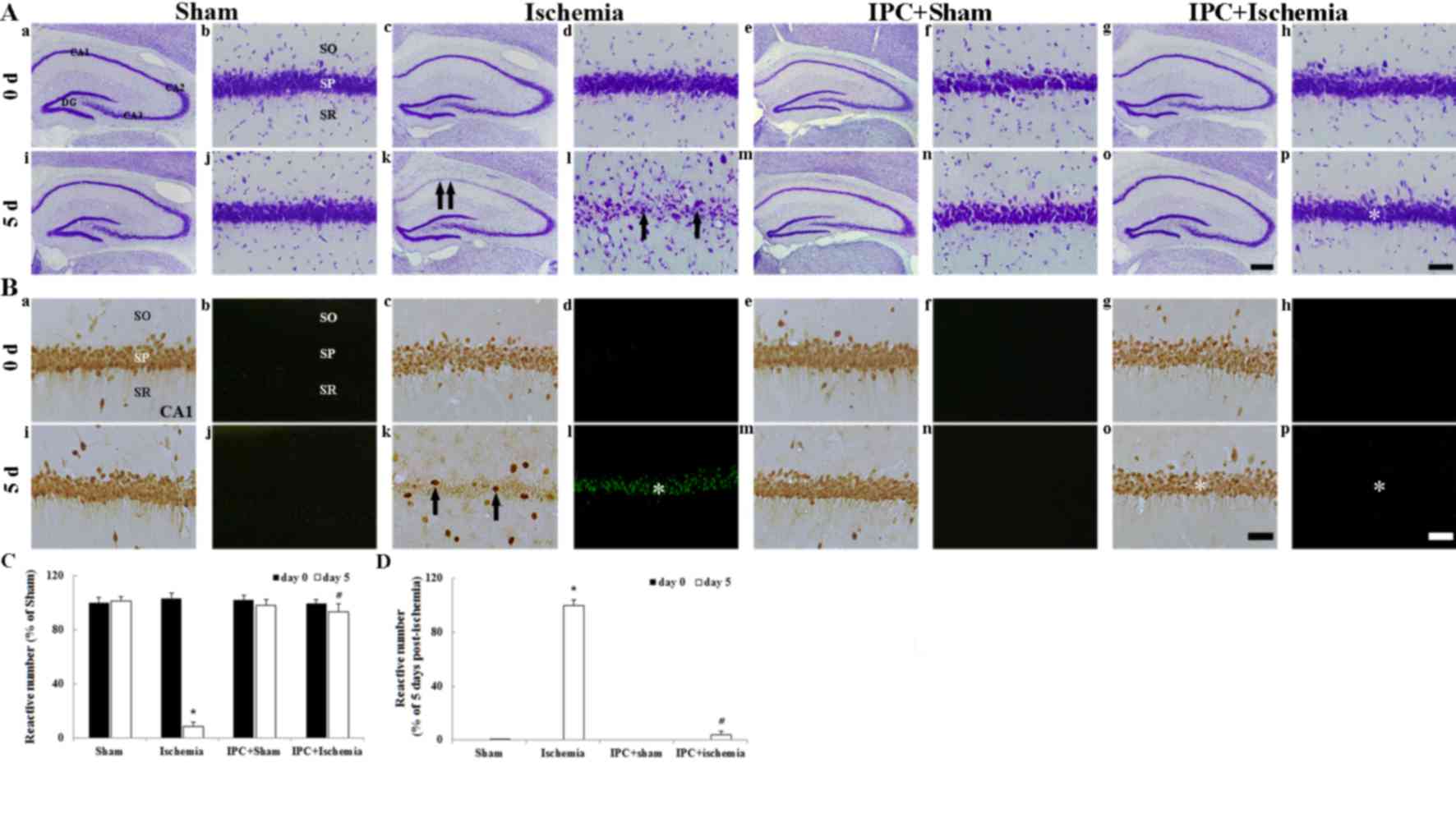 | Figure 2.(A) CV, (B) NeuN and F-J B staining
were conducted. (Aa-Ap) CV staining, (Ba, Bc, Be, Bg, Bi, Bk, Bm
and Bo) NeuN immunohistochemistry and (Bb, Bd, Bf, Bh, Bj, Bl, Bn
and Bp) F-J B histofluorescence staining in the hippocampus of
sham-operated, ischemia-operated, IPC + sham-operated and IPC +
ischemia-operated groups. In the ischemia-operated group,
CV+ cells (arrows in Ak and Al) were damaged in the SP
of the CA1 region 5 days after lethal transient cerebral ischemia.
In addition, at this time point, few NeuN+ (arrows in
Bk) and numerous F-J B+ (asterisk in Bl) cells were
detected in the SP. However, the distribution of CV+
(asterisk in Ap), NeuN+ (asterisk in Bo) and F-J
B+ (asterisk in Bp) cells in the IPC + ischemia-operated
group was similar to that in the sham-operated group. Scale bar:
(Aa, Ac, Ae, Ag, Ai, Ak, Am and Ao) 800 µm; (Ab, Ad, Af, Ah, Aj,
Al, An, Ap and Ba- Bp) 50 µm. Quantitative analyses of (C)
NeuN+ and (D) F-J B+ cells in all groups.
Data are presented as the mean ± standard error of the mean.
*P<0.05 vs. the sham-operated group; #P<0.05 vs.
the ischemia-operated group. SO, stratum oriens; SR, stratum
radiatum; SP, stratum pyramidale; IPC, ischemic preconditioning;
CV, cresyl violet; NeuN, neuronal nuclei; F-J B, Fluoro-Jade B. |
NeuN+ and F-J B+
cells
IPC-mediated neuroprotection in the CA1 region was
detected using anti-NeuN immunohistochemistry and F-J B
histofluorescence staining (Fig.
2B). In all experimental groups on day 0 after LTCI, pyramidal
neurons in the CA1 region were well stained with NeuN (Fig. 2Ba, Bc, Be, Bg and C) and no F-J
B+ cells were observed (Fig. 2Bb, Bd, Bf, Bh and D). In the
ischemia-operated group, a significant loss of NeuN+
neurons, alongside numerous F-J B+ cells, was observed
in the stratum pyramidale of the CA1 region 5 days after LTCI
(Fig. 2Bk, C and D).
At day 5 following LTCI, in the IPC + sham-operated
group, the pattern of NeuN+ CA1 pyramidal neurons was
similar to that in the sham-operated group (Fig. 2Bm and C) and no F-J B+
cells were detected (Fig. 2Bn and
D). The distribution of NeuN+ and F-J B+
cells in the stratum pyramidale of the IPC + ischemia-operated
group was similar to that in the IPC + sham-operated group
(Fig. 2Bo, Bp, C and D).
PDGF-BB immunoreactivity: CA1
region
PDGF-BB immunoreactivity in the CA1 region was
detected by immunohistochemistry (Fig.
3). PDGF-BB immunoreactivity was easily detected in pyramidal
neurons in the stratum pyramidale of the CA1 region in the
sham-operated groups (Fig. 3A, E, I, M
and Q). In the ischemia-operated group, PDGF-BB
immunoreactivity began to be decreased in the CA1 pyramidal neurons
from 1 day after LTCI and was hardly observed in CA1 pyramidal
neurons 5 days after LTCI (Fig. 3B, F,
J, N and Q).
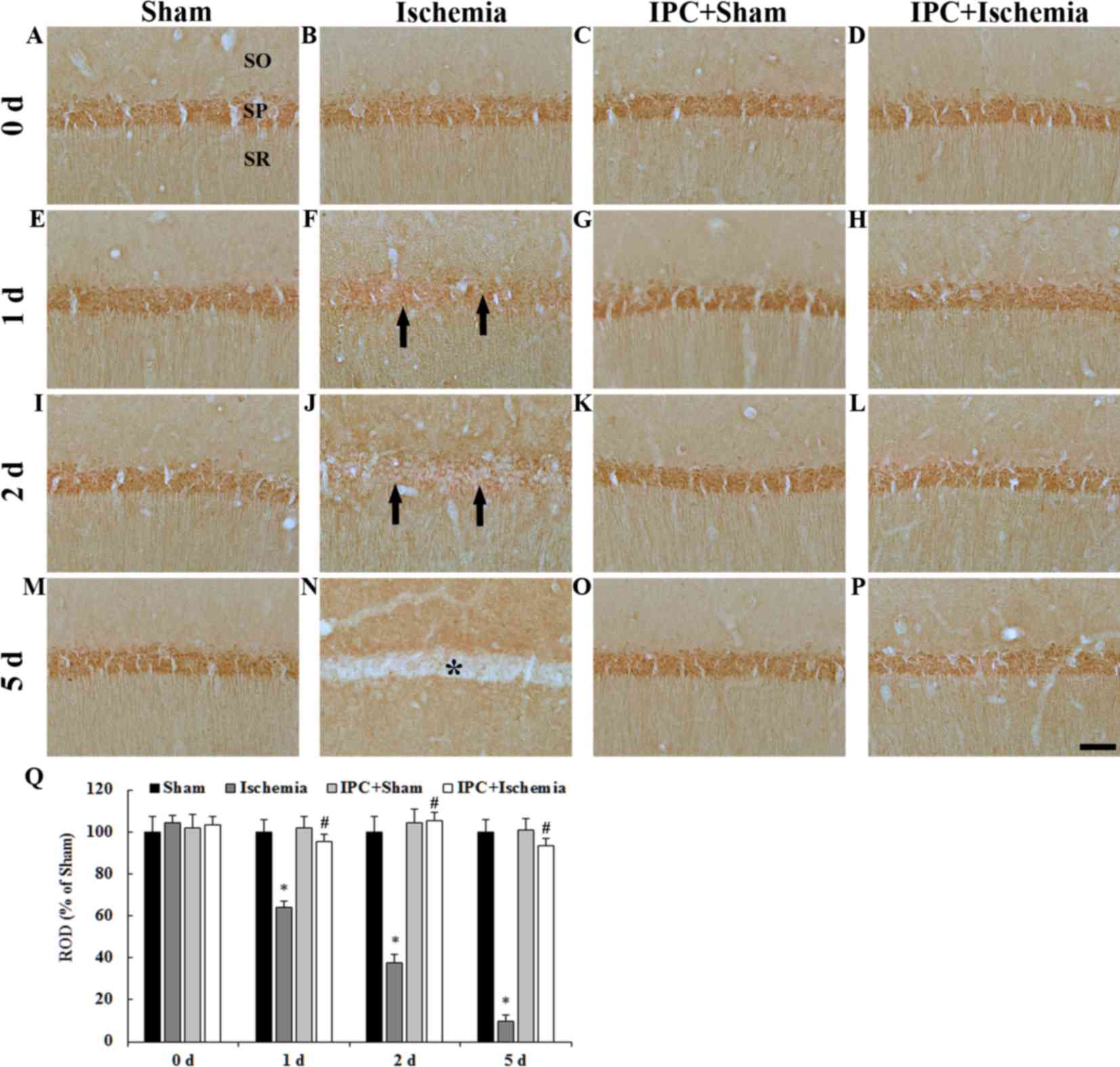 | Figure 3.(A-P) PDGF-BB immunohistochemistry in
the CA1 region of the (A, E, I and M) sham-operated, (B, F, J and
N) ischemia-operated, (C, G, K and O) IPC + sham-operated and (D,
H, L and P) IPC + ischemia-operated groups. In the
ischemia-operated group, PDGF-BB immunoreactivity was markedly
decreased in the SP (arrows in F, J) at 1 day post-ischemia.
PDGF-BB immunoreactivity was barely detected in pyramidal neurons
(asterisk in N) of the CA1 region 5 days after LTCI. In the IPC +
sham- and IPC + ischemia-operated groups, PDGF-BB immunoreactivity
was similar to that in the sham-operated group. Scale bar, 50 µm.
(Q) Quantitative analysis of PDGF-BB immunoreactivity in the SP of
all groups. The ratio of ROD was calibrated as a %, with the
sham-operated group designated as 100%. Data are presented as the
mean ± standard error of the mean. *P<0.05 vs. the sham-operated
group; #P<0.05 vs. the ischemia-operated group. SO,
stratum oriens; SR, stratum radiatum; SP, stratum pyramidale; IPC,
ischemic preconditioning; PDGF-BB, platelet-derived growth
factor-BB; ROD, relative optical density. |
In the IPC + sham-operated group, PDGF-BB
immunoreactivity in CA1 pyramidal neurons was similar to that in
the sham-operated group (Fig. 3C, G,
K, O and Q). In the IPC + ischemia-operated group, PDGF-BB
immunoreactivity in CA1 pyramidal neurons was not altered after
LTCI compared with in the IPC + sham-operated group (Fig. 3D, H, L, P and Q).
PDGF-BB immunoreactivity: CA3
region
PDGF-BB immunoreactivity in the CA3 region was
detected by immunohistochemistry (Fig.
4). In the CA3 region of the sham-operated group, PDGF-BB
immunoreactivity was detected in neurons of the stratum pyramidale
(Fig. 4A, E, I, M and Q). In the
ischemia-operated group, PDGF-BB immunoreactivity was not
significantly altered in the stratum pyramidale (Fig. 4B, F, J, N and Q). In the IPC +
sham-operated group, PDGF-BB immunoreactivity in the stratum
pyramidale was slightly increased 0 days after LTCI compared with
in the sham-operated group (Fig.
4C); thereafter, the immunoreactivity was similar to that in
the sham-operated group until 5 days post-ischemia (Fig. 4G, K, O and Q). In the IPC +
ischemia-operated group, Changes in PDGF-BB immunoreactivity in the
stratum pyramidale was similar to that in the IPC + sham-operated
group (Fig. 4D, H, L, P and
Q).
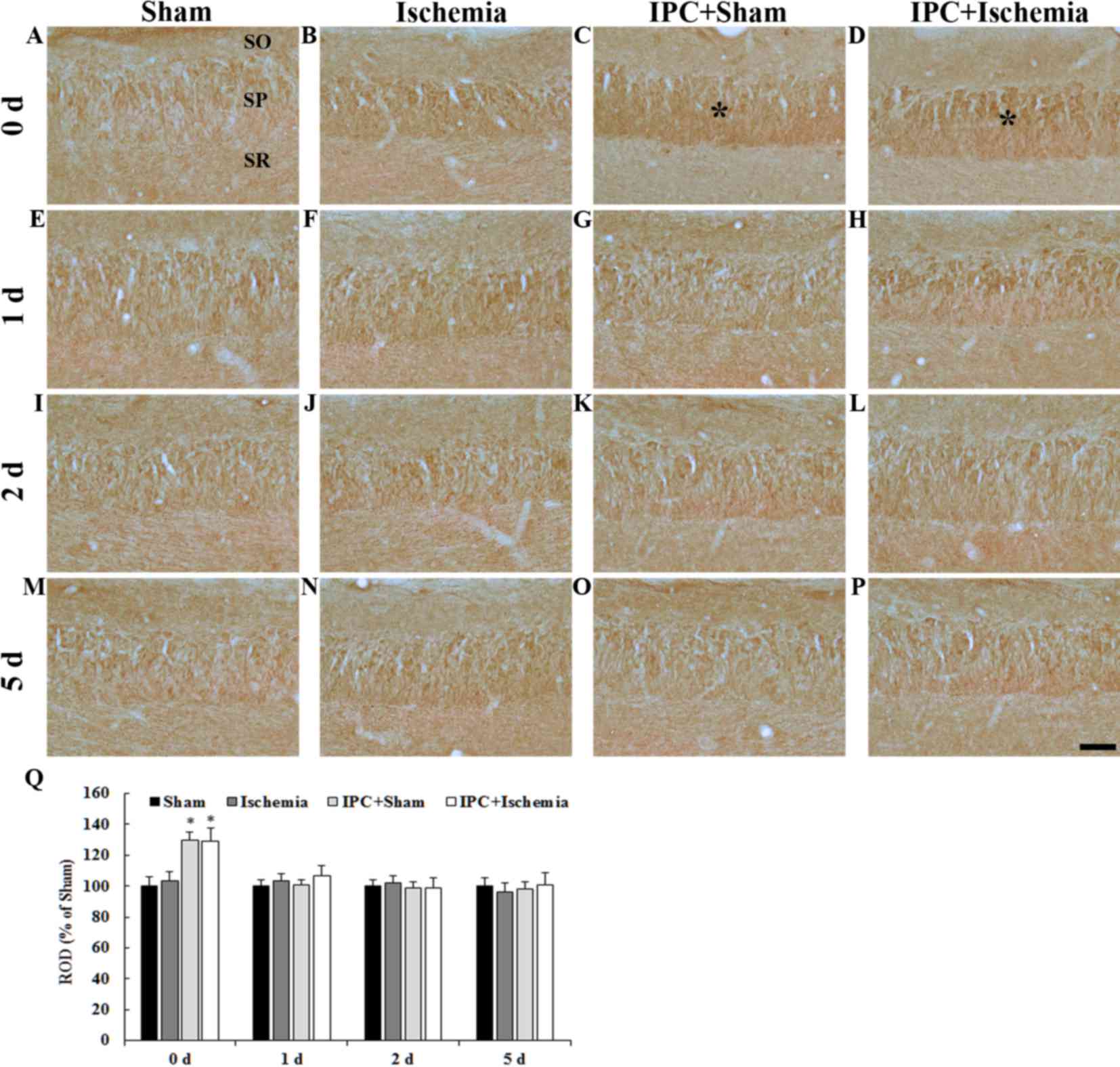 | Figure 4.(A-P) PDGF-BB immunohistochemistry in
the CA3 region of the (A, E, I and M) sham-operated, (B, F, J and
N) ischemia-operated, (C, G, K and O) IPC + sham-operated and (D,
H, L and P) IPC + ischemia-operated groups. PDGF-BB
immunoreactivity was increased in pyramidal neurons of the SP
(asterisks in C and D) at day 0 in the IPC + sham-operated and IPC
+ ischemia-operated groups. Scale bar=50 µm. (Q) Quantitative
analysis of PDGF-BB immunoreactivity in the SP of all groups. The
ratio of the ROD was calibrated as a %, with the sham-operated
group designated as 100%. Data are presented as the mean ± standard
error of the mean. *P<0.05 vs. the sham-operated group. SO,
stratum oriens; SR, stratum radiatum; SP, stratum pyramidale; IPC,
ischemic preconditioning; PDGF-BB, platelet-derived growth
factor-BB; ROD, relative optical density. |
Protein expression levels of
PDGF-BB
Western blot analysis indicated that the alterations
in PDGF-BB protein expression in the CA1 region post-LTCI were
similar to those observed by immunohistochemistry (Fig. 5). In the ischemia-operated group,
PDGF-BB protein levels were significantly decreased 2 days after
LTCI and the levels were lowest 5 days post-LTCI. In the IPC +
sham-operated group, PDGF-BB protein expression was significantly
increased compared with in the sham-operated group. In the IPC +
ischemia-operated group, PDGF-BB protein levels were not
significantly altered following LTCI (Fig. 5).
Discussion
Transient brain ischemia leads to selective
damage/death of pyramidal neurons in the hippocampal CA1 region
several days after I-R. This neuronal death is commonly referred to
as ‘delayed neuronal death’, since it occurs very slowly for 4–5
days following 5 min of transient brain ischemia, which is a lethal
type of ischemia for CA1 pyramidal neurons (27). Conversely, pyramidal neurons in the
hippocampal CA3 region are much less vulnerable to ischemic insults
(32). The present study examined
the delayed neuronal death of CA1 pyramidal neurons using CV
histochemistry, NeuN immunohistochemistry and F-J B
histofluorescence.
The results of the present study demonstrated that
CA1 pyramidal neurons in the IPC-induced gerbil hippocampus did not
die following LTCI. IPC, which is induced by exposure to brief
durations of transient ischema, does not induce neuronal
damage/death in ischemic areas and prevents ischemic injury
following a subsequent longer or lethal transient ischemic insult
(33). The first description of
IPC in the brain was reported by Kitagawa et al (34) in a gerbil model, and similar
findings have been reported in rats (35,36)
and mice (37). In addition,
IPC-mediated neuroprotection has been studied in brain slices
(38), as well as in murine cell
culture (39). In the present
study, brief IPC (2 min of transient ischemia) was used to prevent
neuronal death in the hippocampal CA1 region, and this brief IPC
stimulus did not induce neuronal damage, as assessed by CV, NeuN
and F-J B staining, which is very sensitive to acute neuronal
injury (40). The results
indicated that CA1 pyramidal neurons exhibited normal features in
the IPC-induced gerbil brain 5 days after LTCI. To the best of our
knowledge, although IPC provides marked neuroprotection against
ischemic brain injury, its underlying mechanisms require further
elucidation for the development of therapeutic strategies for the
treatment of ischemic stroke, as the molecular mechanisms
underlying IPC-induced ischemic tolerance are not fully understood
(1).
It is well known that endogenous PDGF-BB is
expressed in neurons (13) and its
expression is altered after some brain insults (13,14).
In particular, it has been reported that PDGF-BB may be considered
a potent neuroprotective factor under cerebral ischemic conditions
(14,19,41,42).
Previous studies have demonstrated that the application of
exogenous PDGF-BB protein may prevent neuronal cell death in the
CA1 region after global brain ischemia, and may induce infarct
tolerance against reversible focal brain ischemia (19,42,43).
Albers et al (44) previously demonstrated that NMDA
antagonists may prevent neuronal injury in a gerbil model of
transient cerebral ischemia by inhibiting the excitotoxicity
induced by NMDA receptors. NMDA-induced excitotoxicity triggers
several downstream cascades, including nitrosative and oxidative
stress, mitochondrial dysfunction, and protease and phospholipase
activation, which culminate in cell death (45). This NMDA-induced excitotoxicity is
widely considered to mediate the delayed neuronal death of
hippocampal CA1 pyramidal neurons following transient global
cerebral ischemia (46).
Therefore, inhibition of Ca2+ overload may be a
potential mechanism underlying PDGF-mediated neuroprotection
(47). Furthermore, the
suppression of PDGF-BB mRNA expression has been reported to
increase susceptibility of the brain to NMDA-induced excitotoxicity
or ischemia (18,48,49).
In concordance with these in vivo data, the activation of
PDGFR-β by PDGF-BB strongly inhibited NMDA-induced excitotoxicity
in cultured hippocampal neurons (50,51).
These studies indicated that endogenously synthesized and
exogenously applied PDGF-BB may exert neuroprotective effects, and
that PDGFR-β expression in neurons may principally mediate this
protective effect.
On the basis of these aforementioned studies, the
present study compared alterations in PDGF-BB immunoreactivity in
the CA1 region between the ischemia-operated and IPC +
ischemia-operated groups. PDGF-BB immunoreactivity was
significantly decreased in the CA1 pyramidal neurons following LTCI
and was barely detected in the neurons 5 days after LTCI; however,
IPC maintained PDGF-BB immunoreactivity in the CA1 pyramidal
neurons in the IPC + sham- and IPC + ischemia-operated groups.
These findings indicated that PDGF-BB may be associated with
neuronal damage after ischemic insults and that the regulation of
PDGF-BB expression may be affected by PDGFR-β.
In conclusion, the findings of the present study
indicated that IPC (2 min of transient cerebral ischemia) increased
PDGF-BB expression in the gerbil hippocampal CA1 pyramidal neurons
following 5 min of LTCI. The present study provides evidence
regarding the mechanism underlying IPC-mediated neuroprotection
against transient cerebral ischemic injury.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
NRF-2014R1A1A2056105), and the Bio-Synergy Research Project (grant
no. NRF-2015M3A9C4076322) of the Ministry of Science, ICT and
Future Planning through the National Research Foundation.
References
|
1
|
Gidday JM: Cerebral preconditioning and
ischaemic tolerance. Nat Rev Neurosci. 7:437–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kitagawa K, Matsumoto M, Tagaya M, Hata R,
Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et
al: ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res.
528:21–24. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kirino T, Tsujita Y and Tamura A: Induced
tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood
Flow Metab. 11:299–307. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nishi S, Taki W, Uemura Y, Higashi T,
Kikuchi H, Kudoh H, Satoh M and Nagata K: Ischemic tolerance due to
the induction of HSP70 in a rat ischemic recirculation model. Brain
Res. 615:281–288. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stagliano NE, Perez-Pinzón MA, Moskowitz
MA and Huang PL: Focal ischemic preconditioning induces rapid
tolerance to middle cerebral artery occlusion in mice. J Cereb
Blood Flow Metab. 19:757–761. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weih M, Kallenberg K, Bergk A, Dirnagl U,
Harms L, Wernecke KD and Einhäupl KM: Attenuated stroke severity
after prodromal TIA: A role for ischemic tolerance in the brain?
Stroke. 30:1851–1854. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moncayo J, de Freitas GR, Bogousslavsky J,
Altieri M and van Melle G: Do transient ischemic attacks have a
neuroprotective effect? Neurology. 54:2089–2094. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ross R, Raines EW and Bowen-Pope DF: The
biology of platelet-derived growth factor. Cell. 46:155–169. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heldin CH and Westermark B:
Platelet-derived growth factor: Mechanism of action and possible in
vivo function. Cell Regul. 1:555–566. 1990.PubMed/NCBI
|
|
10
|
Hart CE, Forstrom JW, Kelly JD, Seifert
RA, Smith RA, Ross R, Murray MJ and Bowen-Pope DF: Two classes of
PDGF receptor recognize different isoforms of PDGF. Science.
240:1529–1531. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heldin CH, Bäckström G, Ostman A,
Hammacher A, Rönnstrand L, Rubin K, Nistér M and Westermark B:
Binding of different dimeric forms of PDGF to human fibroblasts:
Evidence for two separate receptor types. EMBO J. 7:1387–1393.
1988.PubMed/NCBI
|
|
12
|
Seifert RA, Hart CE, Phillips PE, Forstrom
JW, Ross R, Murray MJ and Bowen-Pope DF: Two different subunits
associate to create isoform-specific platelet-derived growth factor
receptors. J Biol Chem. 264:8771–8778. 1989.PubMed/NCBI
|
|
13
|
Sasahara M, Fries JW, Raines EW, Gown AM,
Westrum LE, Frosch MP, Bonthron DT, Ross R and Collins T: PDGF
B-chain in neurons of the central nervous system, posterior
pituitary, and in a transgenic model. Cell. 64:217–227. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iihara K, Sasahara M, Hashimoto N, Uemura
Y, Kikuchi H and Hazama F: Ischemia induces the expression of the
platelet-derived growth factor-B chain in neurons and brain
macrophages in vivo. J Cereb Blood Flow Metab. 14:818–824. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iihara K, Sasahara M, Hashimoto N and
Hazama F: Induction of platelet-derived growth factor beta-receptor
in focal ischemia of rat brain. J Cereb Blood Flow Metab.
16:941–949. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krupinski J, Issa R, Bujny T, Slevin M,
Kumar P, Kumar S and Kaluza J: A putative role for platelet-derived
growth factor in angiogenesis and neuroprotection after ischemic
stroke in humans. Stroke. 28:564–573. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Renner O, Tsimpas A, Kostin S, Valable S,
Petit E, Schaper W and Marti HH: Time- and cell type-specific
induction of platelet-derived growth factor receptor-beta during
cerebral ischemia. Brain Res Mol Brain Res. 113:44–51. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Egawa-Tsuzuki T, Ohno M, Tanaka N,
Takeuchi Y, Uramoto H, Faigle R, Funa K, Ishii Y and Sasahara M:
The PDGF B-chain is involved in the ontogenic susceptibility of the
developing rat brain to NMDA toxicity. Exp Neurol. 186:89–98. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Iihara K, Hashimoto N, Tsukahara T, Sakata
M, Yanamoto H and Taniguchi T: Platelet-derived growth factor-BB,
but not -AA, prevents delayed neuronal death after forebrain
ischemia in rats. J Cereb Blood Flow Metab. 17:1097–1106. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bae EJ, Chen BH, Yan BC, Shin BN, Cho JH,
Kim IH, Ahn JH, Lee JC, Tae HJ, Hong S, et al: Delayed hippocampal
neuronal death in young gerbil following transient global cerebral
ischemia is related to higher and longer-term expression of p63 in
the ischemic hippocampus. Neural Regen Res. 10:944–950. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao XY, Wu CF, Yang J, Gao Y, Sun FJ,
Wang DX, Wang CH and Lin BC: Effect of arginine vasopressin on the
cortex edema in the ischemic stroke of Mongolian gerbils.
Neuropeptides. 51:55–62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JC, Kim IH, Park JH, Ahn JH, Cho JH,
Cho GS, Tae HJ, Chen BH, Yan BC, Yoo KY, et al: Ischemic
preconditioning protects hippocampal pyramidal neurons from
transient ischemic injury via the attenuation of oxidative damage
through upregulating heme oxygenase-1. Free Radic Biol Med.
79:78–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JC, Park JH, Kim IH, Cho GS, Ahn JH,
Tae HJ, Choi SY, Cho JH, Kim DW, Kwon YG, et al: Neuroprotection of
ischemic preconditioning is mediated by thioredoxin 2 in the
hippocampal CA1 region following a subsequent transient cerebral
ischemia. Brain Pathol. 27:276–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
National Research Council (U.S.), .
Committee for the Update of the Guide for the Care and Use of
Laboratory Animals., Institute for Laboratory Animal Research
(U.S.) and National Academies Press (U.S.): Guide for the care and
use of laboratory animals. National Academies Press; Washington,
D.C.: 2011
|
|
25
|
Nakamura H, Katsumata T, Nishiyama Y,
Otori T, Katsura K and Katayama Y: Effect of ischemic
preconditioning on cerebral blood flow after subsequent lethal
ischemia in gerbils. Life Sci. 78:1713–1719. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohk TG, Yoo KY, Park SM, Shin BN, Kim IH,
Park JH, Ahn HC, Lee YJ, Kim MJ, Kim TY, et al: Neuronal damage
using fluoro-jade B histofluorescence and gliosis in the striatum
after various durations of transient cerebral ischemia in gerbils.
Neurochem Res. 37:826–834. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JC, Park JH, Yan BC, Kim IH, Cho GS,
Jeoung D, Kwon YG, Kim YM, Lee YL, Shin HC and Won MH: Effects of
transient cerebral ischemia on the expression of DNA
methyltransferase 1 in the gerbil hippocampal CA1 region. Neurochem
Res. 38:74–81. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JC, Kim IH, Cho GS, Park JH, Ahn JH,
Yan BC, Kwon HM, Kim YM, Cheon SH, Cho JH, et al: Ischemic
preconditioning-induced neuroprotection against transient cerebral
ischemic damage via attenuating ubiquitin aggregation. J Neurol
Sci. 336:74–82. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee CH, Park JH, Choi JH, Yoo KY, Ryu PD
and Won MH: Heat shock protein 90 and its cochaperone, p23, are
markedly increased in the aged gerbil hippocampus. Exp Gerontol.
46:768–772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bickford P, Heron C, Young DA, Gerhardt GA
and De La Garza R: Impaired acquisition of novel locomotor tasks in
aged and norepinephrine-depleted F344 rats. Neurobiol Aging.
13:475–481. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuroiwa T, Bonnekoh P and Hossmann KA:
Locomotor hyperactivity and hippocampal CA1 injury after transient
forebrain ischemia of gerbils. Neurosci Lett. 122:141–144. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmidt-Kastner R and Freund TF: Selective
vulnerability of the hippocampus in brain ischemia. Neuroscience.
40:599–636. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lehotský J, Burda J, Danielisová V,
Gottlieb M, Kaplán P and Saniová B: Ischemic tolerance: The
mechanisms of neuroprotective strategy. Anat Rec (Hoboken).
292:2002–2012. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kitagawa K, Matsumoto M, Kuwabara K,
Tagaya M, Ohtsuki T, Hata R, Ueda H, Handa N, Kimura K and Kamada
T: ‘Ischemic tolerance’ phenomenon detected in various brain
regions. Brain Res. 561:203–211. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kawahara N, Wang Y, Mukasa A, Furuya K,
Shimizu T, Hamakubo T, Aburatani H, Kodama T and Kirino T:
Genome-wide gene expression analysis for induced ischemic tolerance
and delayed neuronal death following transient global ischemia in
rats. J Cereb Blood Flow Metab. 24:212–223. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Perez-Pinzón MA, Xu GP, Dietrich WD,
Rosenthal M and Sick TJ: Rapid preconditioning protects rats
against ischemic neuronal damage after 3 but not 7 days of
reperfusion following global cerebral ischemia. J Cereb Blood Flow
Metab. 17:175–182. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Atochin DN, Clark J, Demchenko IT,
Moskowitz MA and Huang PL: Rapid cerebral ischemic preconditioning
in mice deficient in endothelial and neuronal nitric oxide
synthases. Stroke. 34:1299–1303. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Raval AP, Dave KR, Mochly-Rosen D, Sick TJ
and Pérez-Pinzón MA: Epsilon PKC is required for the induction of
tolerance by ischemic and NMDA-mediated preconditioning in the
organotypic hippocampal slice. J Neurosci. 23:384–391.
2003.PubMed/NCBI
|
|
39
|
Grabb MC and Choi DW: Ischemic tolerance
in murine cortical cell culture: Critical role for NMDA receptors.
J Neurosci. 19:1657–1662. 1999.PubMed/NCBI
|
|
40
|
Schmued LC and Hopkins KJ: Fluoro-Jade B:
A high affinity fluorescent marker for the localization of neuronal
degeneration. Brain Res. 874:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kaneko M, Sasahara M, Takayama S, Handa J
and Hazama F: Expression of platelet-derived growth factor after
transient forebrain ischemia in the gerbil hippocampus. Acta
Neuropathol. 95:471–478. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kawabe T, Wen TC, Matsuda S, Ishihara K,
Otsuda H and Sakanaka M: Platelet-derived growth factor prevents
ischemia-induced neuronal injuries in vivo. Neurosci Res.
29:335–343. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sakata M, Yanamoto H, Hashimoto N, Iihara
K, Tsukahara T, Taniguchi T and Kikuchi H: Induction of infarct
tolerance by platelet-derived growth factor against reversible
focal ischemia. Brain Res. 784:250–255. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Albers GW, Goldberg MP and Choi DW: Do
NMDA antagonists prevent neuronal injury? Yes. Arch Neurol.
49:418–420. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chinopoulos C and Adam-Vizi V: Calcium,
mitochondria and oxidative stress in neuronal pathology. Novel
aspects of an enduring theme. FEBS J. 273:433–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu
L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, et al: NMDA receptor
subunits have differential roles in mediating excitotoxic neuronal
death both in vitro and in vivo. J Neurosci. 27:2846–2857. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Funa K and Sasahara M: The roles of PDGF
in development and during neurogenesis in the normal and diseased
nervous system. J Neuroimmune Pharmacol. 9:168–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ishii Y, Oya T, Zheng L, Gao Z, Kawaguchi
M, Sabit H, Matsushima T, Tokunaga A, Ishizawa S, Hori E, et al:
Mouse brains deficient in neuronal PDGF receptor-beta develop
normally but are vulnerable to injury. J Neurochem. 98:588–600.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shen J, Ishii Y, Xu G, Dang TC, Hamashima
T, Matsushima T, Yamamoto S, Hattori Y, Takatsuru Y, Nabekura J and
Sasahara M: PDGFR-β as a positive regulator of tissue repair in a
mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab.
32:353–367. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Valenzuela CF, Xiong Z, MacDonald JF,
Weiner JL, Frazier CJ, Dunwiddie TV, Kazlauskas A, Whiting PJ and
Harris RA: Platelet-derived growth factor induces a long-term
inhibition of N-methyl-D-aspartate receptor function. J Biol Chem.
271:16151–16159. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tseng HC and Dichter MA: Platelet-derived
growth factor-BB pretreatment attenuates excitotoxic death in
cultured hippocampal neurons. Neurobiol Dis. 19:77–83. 2005.
View Article : Google Scholar : PubMed/NCBI
|