Introduction
Hepatic insulin resistance is a critical component
in the development of type 2 diabetes. Excess adiposity and a
high-fat diet is associated with lipid accumulation in the liver,
resulting in increased circulating free fatty acids (FFAs), and the
subsequent production of pro-inflammatory cytokines, including
tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and
monocyte chemoattractant protein-1 (MCP-1), which are important in
the development of hepatic insulin resistance (1,2).
The c-Jun amino-terminal kinases (JNKs),
particularly JNK1, have been considered to be a critical molecular
link between hepatic insulin resistance and inflammation, and a
potential target for therapeutics (3). JNK1 can be activated by inflammatory
cytokines and FFA, resulting in the phosphorylation of insulin
receptor substrates (IRSs) at serine and threonine residues in the
liver. This inhibits downstream insulin signaling, including the
inactivation of phosphatidylinositol 3-kinase (PI3K) and protein
kinase B (PKB/AKT). In the liver, AKT phosphorylates a number of
substrates, including forkhead box O1 (FOXO1), controlling the
transcription of genes encoding gluconeogenic enzymes, and glycogen
synthase kinase (GSK)-3α/β, regulating glycogen synthesis (4,5).
Erythropoietin (EPO), a kidney cytokine regulating
hematopoiesis, has been widely used for the treatment of anemia in
patients with chronic kidney disease (6). EPO has also been shown to exhibit
anti-inflammatory effects in previous studies. It was demonstrated
that EPO attenuated ischemia-induced inflammation by reducing the
levels of TNF-α, IL-6 and MCP-1 in the ischemic brain and heart in
an experimental model of autoimmune myocarditis (7,8). EPO
treatment has also been shown to decrease serum levels of TNF-α and
IL-6, and reduce insulin resistance in non-diabetic patients on
maintenance hemodialysis (9). Our
previous study in mice suggested that EPO treatment improved
glucose intolerance by inhibiting inflammation in the liver
(10). This led to the hypothesis
that hepatic insulin signaling may be regulated by EPO treatment
in vitro through suppression of the inflammatory response;
however, the mechanism remains to be fully elucidated.
The aim of the present study was to evaluate the
anti-inflammatory activity of EPO involved in the modulation of
insulin sensitivity in PA-induced HepG2 cells, which may assist in
clarifying the pharmacological function of EPO in hepatic insulin
resistance. The data revealed that EPO inhibited the gene
expression of TNF-α, IL-1β and MCP-1 and the phosphorylation of
JNK1, which may be closely associated with the improvement of
hepatic insulin signaling and glucose metabolism by EPO.
Materials and methods
Reagents
Palmitic acid (PA) was purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). Recombinant human EPO was from
Shenyang Sunshine Pharmaceutical Company (Shenyang, China). TRIzol
and primers were from Invitrogen; Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). The reverse transcription kit was purchased
from Takara Biotechnology Co., Ltd. (Dalian, China). Antibodies
against phosphorylated (p)-insulin receptor (IR)-β (Tyr1361), p85
subunit of PI3K (PI3K-p85), p-AKT (Ser473), AKT, FOXO1, p-FOXO1
(Ser256), GSK-3β, p-GSK-3β (Ser9), p-IRS-1 (Ser307), IRS-1, IRS-2,
p-JNK (Thr183/Tyr185), JNK, and the PI3K inhibitors, LY294002 and
wortmannin, were from Cell Signaling Technology, Inc. (Danvers, MA,
USA). Anti-p-IRS-2 (Ser731) was purchased from AnaSpec (Fremont,
CA, USA). Anti-EPO receptor (EPOR), anti-phosphoenolpyruvate
carboxykinase (PEPCK), total (t-)IR-β, and anti-β-actin antibodies
were from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The
PI3K inhibitors were reconstituted fresh in DMSO prior to use and
were protected from light exposure during the experiments.
Cell culture
The HepG2 cells were obtained from the Chinese
Academy of Sciences, cultured in Dulbecco's modified Eagle's medium
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc.). Insulin resistance was induced by the addition of 0.25
mmol/l PA to the medium for 24 h (11). The solution of PA was prepared as
previously described (12). To
examine the effect of EPO, the cells were incubated in a 37°C, 5%
CO2 incubator for 12 h in serum-free medium and treated
with 5 or 10 U/ml EPO for 24 h. The PI3K inhibitors, wortmannin or
LY294002, were added 1 h prior to EPO exposure and were present
throughout EPO administration. The cells were washed twice with PBS
following incubation with 100 nmol/l insulin (Sigma-Aldrich; Merck
KGaA) for 15 min. The protein and mRNA were then harvested
immediately.
Analysis of glycogen content
The glycogen levels were measured in cells incubated
at 37°C for 3 h in the presence of 1 nmol/l insulin, using a
glycogen assay kit (BioVision, Inc., Milpitas, CA, USA) as
previously described (11).
Briefly, 106 HepG2 cells were homogenized with 200 µl
dH2O on ice, and the homogenates were boiled for 5 min
to inactivate enzymes, followed by centrifugation of the boiled
samples at 18,000 × g for 5 min at room temperature to remove
insoluble material. The supernatants were ready for use in assays
following hydrolyzing by adding hydrolyzing enzyme mix to standards
and samples, mixing and incubating for 30 min at room temperature.
The optical density values of samples were measured at 570 nm.
Western blot analysis
The HepG2 cells were washed twice with cold PBS and
lysed in ice-cold cell lysis buffer supplemented with protease
inhibitor cocktail (Roche Diagnostics, Basel, Switzerland), and the
protein concentration was determined using the bicinchoninic acid
method. The protein lysates were dissolved in loading buffer and
boiled for 5 min. The 30-µg samples of proteins were separated by
SDS-PAGE on a 10% gel, transferred onto a PVDF membrane and blocked
with 7.5% non-fat milk. The membrane was then incubated with
appropriate primary antibodies overnight at 4°C: Rabbit
anti-phosphorylated (p)-IR-β (Tyr1361; cat. no. 3023; 1:2,000);
rabbit anti-PI3K-p85 (cat. no. 4292; 1:2,000); rabbit anti-p-AKT
(Ser473; cat. no. 9271; 1:2,000); rabbit anti-tAKT (cat. no. 9272;
1:2,000); rabbit anti-t-FOXO1 (cat. no. 2880; 1:1,000); rabbit
anti-p-FOXO1 (Ser256; cat. no. 9461; 1:1,000); rabbit anti-GSK-3β
(cat. no. 9315; 1:1,000); rabbit anti-p-GSK-3β (Ser9; cat. no.
9336; 1:1,000); rabbit anti-p-IRS-1 (Ser307; cat. no. 2381;
1:1,000); rabbit anti-t-IRS-1 (cat. no. 2382; 1:1,000); rabbit
anti-t-IRS-2 (cat. no. 3089; 1:1,000); rabbit anti-p-JNK
(Thr183/Tyr185; cat. no. 9251; 1:1,000); rabbit anti-t-JNK (cat.
no. 9252; 1:1,000); rabbit anti-p-IRS-2 (Ser731; cat. no. AS-28122;
1:1,000); rabbit anti-EPOR (cat. no. sc-695; 1:4,000); rabbit
anti-PEPCK (cat. no. sc-32879; 1:5,000); goat anti-t-IR-β (cat. no.
sc-31367; 1:2,000); and mouse anti-β-actin (cat. no. sc-47778;
1:4,000). Subsequently, the membrane was incubated with appropriate
secondary antibodies (all Santa Cruz Biotechnology, Inc.) for 1 h
at room temperature: Bovine anti-goat immunoglobulin
(Ig)G-horseradish peroxidase (HRP) (cat. no. sc-2350; 1:8,000);
bovine anti-mouse IgG-HRP (cat. no. sc-2371; 1:10,000); and bovine
anti-rabbit IgG-HRP (cat. no. sc-2370; 1:8,000). The proteins were
visualized using enhanced chemiluminescence (EMD Millipore,
Billerica, MA, USA) and quantified using densitometry (Quantity One
software; version 462; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted using the standard TRIzol
RNA isolation method (Invitrogen; Thermo Fisher Scientific, Inc.).
cDNA was synthesized from 2 µg total RNA using a PrimeScript RT
reagent kit (Takara Bio, Inc., Otsu, Japan). cDNA was stored at
−80°C until qPCR analysis, which was performed in an Applied
Biosystems 7500 Real-Time PCR system, using SYBR Premix Ex Taq
(Takara Bio, Inc.). The reaction mixture (20 µl) consisted of 2 µl
cDNA, 10 µl 2X SYBR Premix Ex Taq™, 0.4 µl each of forward and
reverse primers, and 0.4 µl 50X ROX Reference dye. The PCR
conditions were as follows: 95°C for 30 sec, followed by 40 cycles
at 95°C for 5 sec and 60°C for 34 sec. The specific primers used
are presented in Table I. The
results were normalized against the gene expression of β-actin
using the 2−ΔΔCq method (13).
 | Table I.Specific primers pairs used in reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Specific primers pairs used in reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| TNF-α |
CCTCTCTCTAATCAGCCCTCTG |
GAGGACCTGGGAGTAGATGAG |
| IL-1β |
AGCTACGAATCTCCGACCAC |
CGTTATCCCATGTGTCGAAGAA |
| MCP-1 |
CAGCCAGATGCAATCAATGCC |
TGGAATCCTGAACCCACTTCT |
| β-actin |
ACGGGGTCACCCACACTGTGC |
CTAGAAGCATTTGCGGTGGACGATG |
Statistical analysis
Data were analyzed using SPSS software (version
13.0; SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean
± standard error of the mean. Statistical significance was
determined using one-way analysis of variance followed by Dunnett's
or the LSD post-hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
EPO ameliorates insulin signaling
through the phosphorylation of IRS-1 and IRS-2 serine residues, but
not IR, in PA-induced HepG2 cells
The serine phosphorylation of IRS inhibits insulin
signaling through the inactivation of tyrosine phosphorylation of
IRS, and results in insulin resistance (14,15).
The results of the present study suggested that the protein
expression of insulin-stimulated p-IRS-2 (Ser731) was reduced by
treatment with 5 and 10 U/ml EPO in the PA-induced HepG2 cells,
whereas the protein expression of p-IRS-1 (Ser307) was inhibited
only in the 10 U/ml EPO treatment group (Fig. 1A and B). However, the protein
levels of t-IRS-2, IRS-1, p-IR-β (Tyr1361) and t-IR-β were not
altered by EPO treatment (Fig.
1C). EPO mediates biological effects and downstream signaling
primarily through binding to its receptor, EPOR. Therefore, the
protein expression of EPOR (Fig.
1D) was detected in the HepG2 cells. The result showed that
EPOR was expressed in the HepG2 cells, suggesting an EPO-mediated
regulatory effect on glucose metabolism.
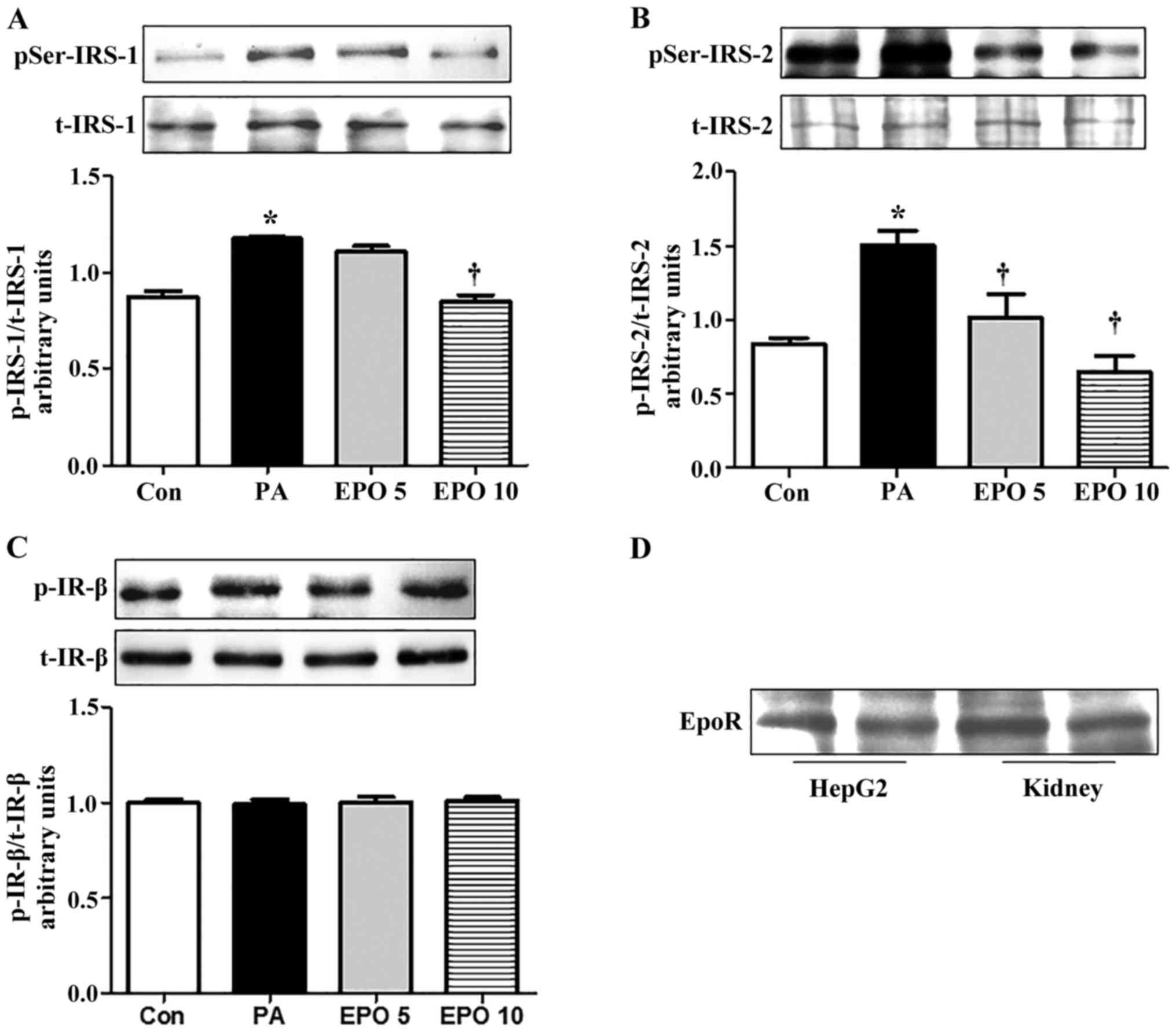 | Figure 1.EPO treatment inhibits serine
phosphorylation of IRS-1 and IRS-2, but not IR, in PA-induced HepG2
cells, and the expression of EPOR in HepG2 cells. All experiments
for analysis of IRS-1, IRS-2, AKT and PEPCK in HepG2 cells were
performed in the presence of 100 nM insulin for 15 min. (A) Western
blot analysis of the protein expression of t-IRS-1 and p-IRS-1
(Ser307). (B) Quantification of protein expression of t-IRS-2 and
p-IRS-2 (Ser731). (C) Western blot analysis of the effects of EPO
on the protein expression of t-IR-β and p-IR-β (Tyr1361). (D)
Western blot analysis of the expression of EPOR in HepG2 cells.
Data are presented as the mean ± standard error of the mean.
*P<0.05, compared with the Con group; †P<0.05,
compared with the PA group. Con, control; PA, palmitic acid; EPO,
erythropoietin; EPO 5, 5 U/ml EPO; EPO 10, 10 U/ml EPO; IRS,
insulin receptor substrate; IR, insulin receptor; EPOR,
erythropoietin receptor; AKT, protein kinase B; PEPCK,
phosphoenolpyruvate carboxykinase; p-, phosphorylated; t-,
total. |
Inhibitors of PI3K inhibit the
EPO-mediated beneficial effects on the hepatic insulin signaling
pathway
The PI3K/AKT signaling pathway, activated by the
tyrosine phosphorylation of IRS, is primarily responsible for the
effect of insulin on gluconeogenesis and glycogen synthesis in
hepatocytes. To understand the mechanisms underlying the effect of
EPO, the present study examined the levels of PI3K-p85, t-AKT,
p-AKT (Ser473), t-FOXO1, p-FOXO1 (Ser256), t-GSK-3β and p-GSK-3β
(Ser9) in the PA-induced HepG2 cells. The results showed that EPO
treatment led to increased protein expression levels of PI3K-p85,
p-AKT (Ser473), p-FOXO1 (Ser256) and p-GSK-3β (Ser9), whereas no
significant differences were found in the levels of t-AKT, t-FOXO1
or t-GSK-3β (Fig. 2A-D). Exposure
of the cells to the specific PI3K signaling pathway inhibitors,
wortmannin (0.5 µmol/l) or LY294002 (10 µmol/l), for 1 h markedly
inhibited the EPO-mediated increase in the protein levels of p-AKT
(Ser473), p-GSK-3β (Ser9) and p-FOXO1 (Ser256) (Fig. 3A-C).
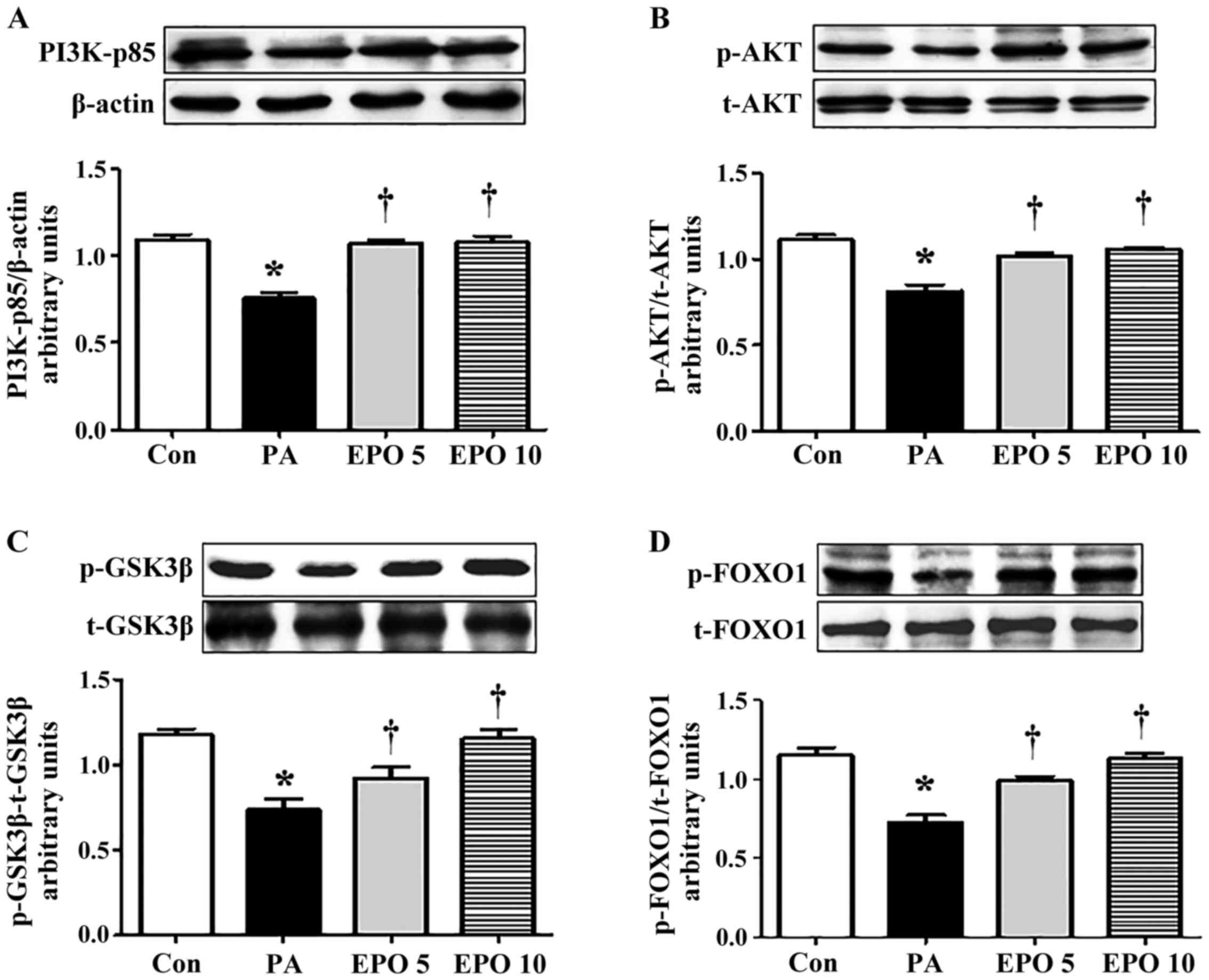 | Figure 2.Effects of EPO treatment on
phosphorylation of PI3K/AKT/FOXO1 and GSK-3β in PA-induced HepG2
cells. The levels of PI3K-p85, t-AKT, p-AKT, t-FOXO1, p-FOXO1,
t-GSK-3β and p-GSK-3β in HepG2 cells were determined using western
blot analysis. The relative levels of PI3K-p85, AKT, FOXO1 and
GSK-3β were analyzed using densitometric analysis with Quantity One
software. Western blot and quantitative analyses of the protein
levels of (A) PI3K-p85, (B) p-AKT/AKT ratio, (C) p-FOXO1/FOXO1
ratio and (D) p-GSK-3β/GSK-3β ratio. Data shown are representative
images and expressed as the mean ± standard error of the mean.
*P<0.05, compared with the Con group; †P<0.05,
compared with the PA group. Con, control; PA, palmitic acid; EPO,
erythropoietin; EPO 5: 5 U/ml EPO treatment; EPO 10: 10 U/ml EPO
treatment; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase
B; FOXO1, forkhead box O1; GSK-3, glycogen synthase kinase 3; p-,
phosphorylated; t-, total. |
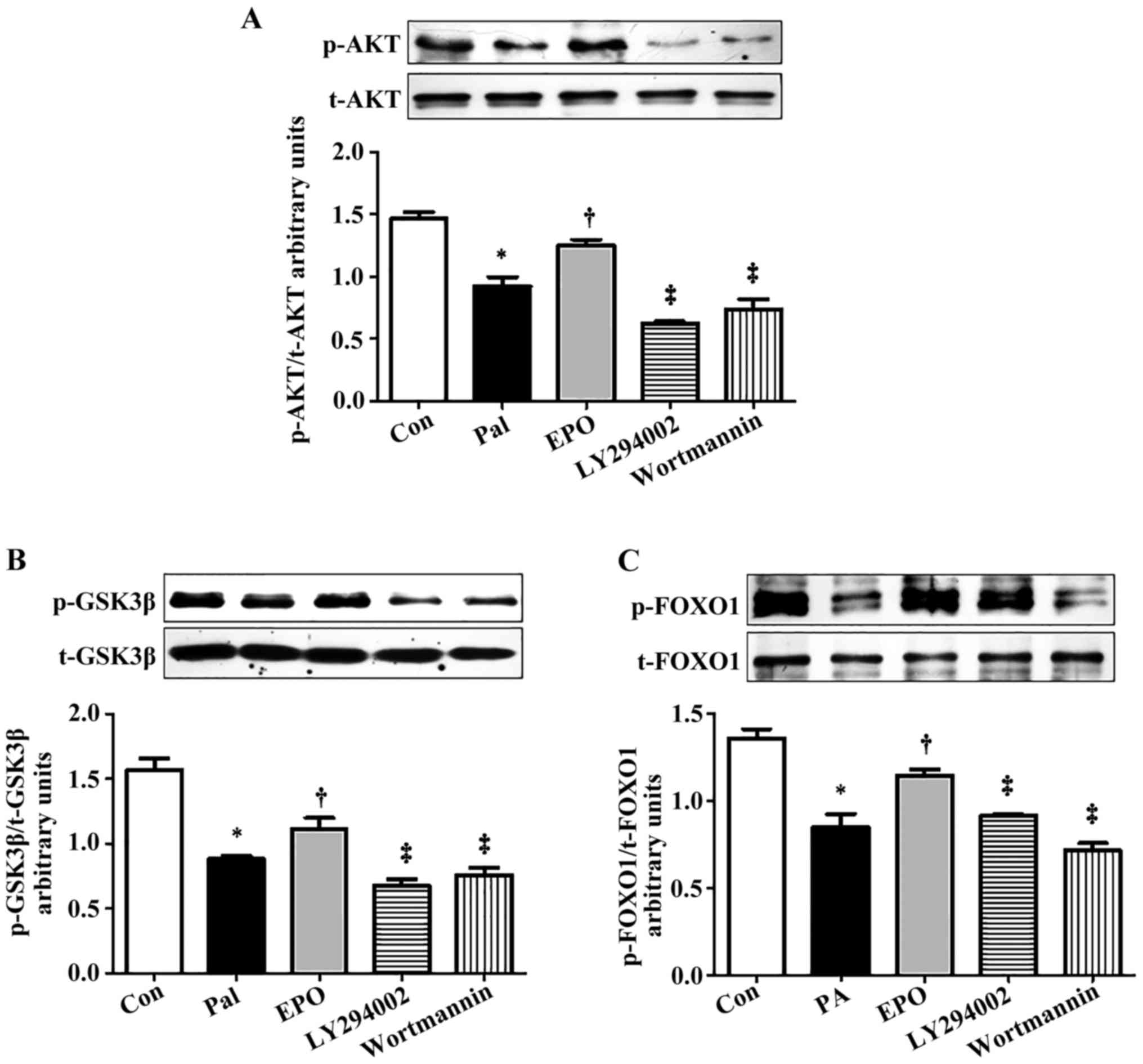 | Figure 3.Effects of PI3K inhibitors on the
phosphorylation of PI3K/AKT/FOXO1 and GSK-3β in PA-induced HepG2
cells treated with EPO. Western blot and quantitative analyses of
the activity of (A) AKT, (B) p-GSK-3β/GSK-3β and (C) p-FOXO1/FOXO1
following interference with PI3K inhibitors. Data shown are
representative images and expressed as the mean ± standard error of
the mean. *P<0.05, compared with the Con group;
†P<0.05, compared with the Pal group;
‡P<0.05, compared with the EPO group. Con, control;
Pal, palmitic acid; EPO, erythropoietin; EPO 10: 10 U/ml EPO
treatment; PI3K, phosphatidylinositol 3-kinase; LY294002, PI3K
inhibitor with LY294002 pretreatment; wortmannin, PI3K inhibitor
with wortmannin pretreatment; AKT, protein kinase B; FOXO1,
forkhead box O1; GSK-3, glycogen synthase kinase 3; p-,
phosphorylated; t-, total. |
EPO improves glucose metabolism in
insulin-resistant HepG2 cells
The results of the present study revealed that the
insulin-stimulated glycogen content in the insulin-resistant HepG2
cells was significantly decreased, whereas the protein expression
of phosphoenolpyruvate carboxykinase (PEPCK), a rate-limiting
enzyme in the process of gluconeogenesis, was significantly
increased. Following treatment with 5 or 10 U/ml of EPO, the
glycogen level (Fig. 4A) was
upregulated and the protein expression of PEPCK was downregulated
(Fig. 4B).
Effects of EPO on PA-induced
inflammatory signaling
The gene expression levels of TNF-α and MCP-1 were
increased in the PA-induced HepG2 cells, and were significantly
decreased following treatment with 5 or 10 U/ml EPO (Fig. 5A and B), whereas the IL-1β gene
showed a significant decline only following treatment with 10 U/ml
EPO (Fig. 5C). Furthermore, the
increased activity of JNK1 (the ratio of p-JNK1/JNK1) by PA was
markedly suppressed by 5 and 10 U/ml EPO treatment, but the
activity of JNK2 was not (Fig.
5D), as detected using western blot analysis.
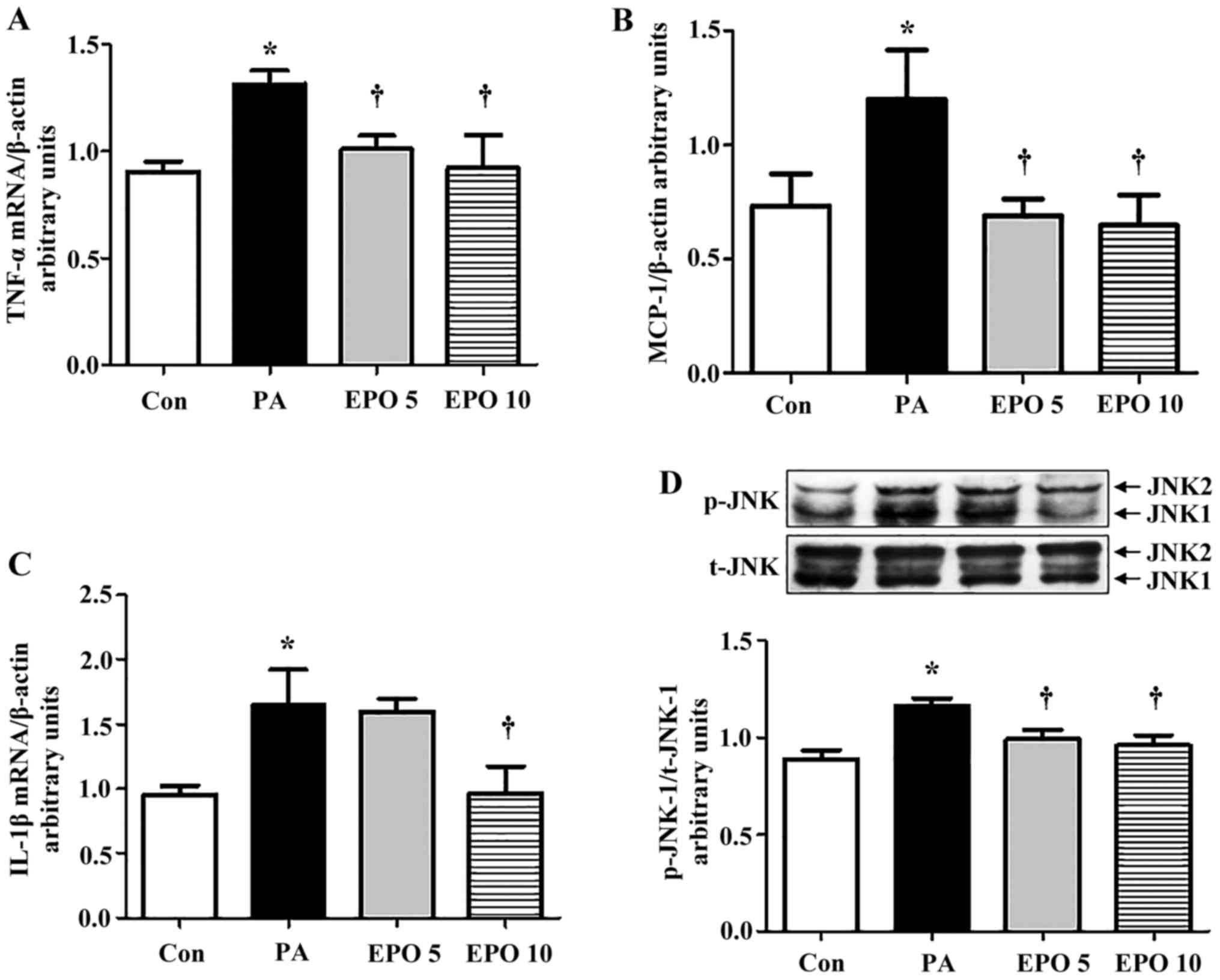 | Figure 5.Effect of EPO on expression levels of
inflammation-associated genes and proteins in HepG2 cells. The mRNA
expression levels of TNF-α, IL-1β, MCP-1 in PA and EPO-incubated
HepG2 cells were determined using reverse
transcription-quantitative polymerase chain reaction analysis. (A)
TNF-α; (B) MCP-1; (C) IL-1β. (D) Western blot and quantitative
analyses of the levels of JNK1 following EPO treatment. Data shown
are representative images and expressed as the mean ± standard
error of the mean. *P<0.05, compared with the Con group.
†P<0.05, compared with the PA group. Con, control;
PA, palmitic acid; EPO, erythropoietin; EPO 5: 5 U/ml EPO
treatment; EPO 10: 10 U/ml EPO treatment; TNF-α, tumor necrosis
factor-α; IL-1β, interleukin-1β; MCP-1; monocyte chemoattractant
protein-1; JNK, c-Jun N-terminal kinase; p-, phosphorylated; t-,
total. |
Discussion
Although initially identified in the hematopoietic
system, EPOR is expressed in various non-hematopoietic organs,
including the brain, heart, kidney, retina and vascular
endothelium. Ubiquitous EPOR has been associated with diverse
regulatory effects for EPO, including neurotrophic,
cardioprotective and renoprotective effects, and modulation of the
immunological, inflammatory response (16). In our previous study, it was found
that EPOR mRNA was expressed in the livers of mice (10). In the present study, the expression
of EPOR was detected in HepG2 cells, suggesting that EPO may
mediate its biological function on hepatic insulin resistance in
HepG2 cells. The results indicated that EPO treatment ameliorated
hepatic glucose metabolism and the insulin signaling pathway via
the IRS/AKT/FOXO1 and GSK-3β pathway, and through inhibition of the
inflammatory response.
Previous studies have suggested that EPO improves
several metabolic parameters when administered to patients with
chronic kidney disease (17–19).
The role and mechanism of EPO in regulating insulin resistance has
been reported previously. Mice lacking EPOR in non-hematopoietic
tissue became insulin resistant with abnormal glucose metabolism
(20). Similarly, mice
overexpressing the EPO gene in muscle tissue showed a significant
improvement in the levels of blood glucose through increases in
oxidative metabolism, fatty acid oxidation and key metabolic genes
in muscle (21). Evidence in
insulin-resistant 3T3L1 adipocytes suggested that EPO treatment
improves insulin resistance via the EPOR-associated phosphorylation
of signal transducer and activator of transcription 5 and AKT, and
inflammation (22). Consistent
with this data, a previous study involving EPOR-knockout mice and
adipocyte models demonstrated that the action of EPO in increasing
metabolic activity and browning of white adipocytes was mediated by
peroxisome proliferators-activated receptor α and sirtuin 1
(23). These data suggest that EPO
contributes to insulin resistance in the fat and muscle.
Previous studies have not examined the role and
mechanism of EPO in hepatic insulin resistance, in which the
insulin signaling cascade is different from that in fat and muscle
tissues. Our previous study in high-fat diet fed mice showed that
EPO regulated the phosphorylation of AKT and improved glucose
intolerance (10). In the present
study, it was found that EPO treatment significantly reduced the
protein expression of PEPCK and enhanced glycogen levels in
PA-induced HepG2 cells. The present study also observed for the
first time, to the best of our knowledge, that the serine
phosphorylation of IRS-1 and IRS-2 were inhibited by EPO treatment
in PA-exposed HepG2 cells, whereas the insulin-stimulated
phosphorylation of IR, an upstream molecule of IRS, was not altered
by EPO treatment. In addition, the levels of PI3K-p85, p-AKT,
p-FOXO1 and p-GSK-3β, downstream molecules of IRS, were increased
by EPO treatment. Through the inhibition of PI3K, it was
demonstrated that PI3K inhibitors, LY294002 and wortmannin,
significantly suppressed the phosphorylation of AKT, FOXO1 and
GSK-3β. Taken together, these results are consistent with the
hypothesis that EPO treatment improves hepatic insulin signaling in
PA-induced HepG2 cells via the IRS/PI3K/AKT/FOXO1 and GSK-3β
pathway.
The fatty acid-associated activation of JNK1 and
production of inflammatory cytokines are important in hepatic
insulin resistance (11,24,25).
The activation of JNK1 directly interferes with insulin signaling
in hepatocytes. This interference is based on the direct
phosphorylation of IRS-1 and IRS-2 at inhibitory sites, which
inhibits recruitment to activated IR. The JNK-mediated
phosphorylation of IRS disrupts downstream events, including
activation of the PI3K/AKT cascade (26–28).
Therefore, further investigations are required to determine whether
improvements in the activities of IRS-1 and IRS-2 by EPO are
associated with the suppressive role of EPO on the inflammatory
pathway in PA-induced HepG2 cells. In the present study, it was
observed that the gene expression levels of TNF-α, IL-1β and MCP-1
were significantly decreased by EPO treatment in the PA-exposed
HepG2 cells. In agreement with alterations in pro-inflammatory
markers, the activity of JNK1 was suppressed by EPO treatment.
These findings indicated that the improvement of hepatic insulin
resistance by EPO may be, at least in part, mediated by inhibiting
inflammation-associated signaling.
In conclusion, the present study demonstrated that
EPO treatment significantly improved glucose metabolism in
PA-induced HepG2 cells. Furthermore, the data revealed a key
mechanism of EPO in regulating hepatic insulin resistance, which
ameliorated the phosphorylation of IRS-1 and IRS-2, and the
downstream activation of AKT/FOXO1 and GSK-3β. These effects may be
associated with the inhibitory role of EPO on the inflammatory
response. These findings offer a novel mechanistic understanding of
the beneficial effects of EPO on hepatic insulin resistance, to
assist in the treatment of insulin resistance and diabetes in the
future.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China Grant Award (grant no.
81200595/81400807), the Project of Six High-peak Talents of Jiangsu
Province (grant no. WSN-101), the Natural Science Foundation of
Jiangsu Province (grant no. KA037), the Research Project of Jiangsu
Province 333 Engineering (grant no. BRA2016232), and the Research
Project of Jiangsu Provincial Commission of Health and Family
Planning (grant nos. H201667 and F201549). Some of the data were
presented as an abstract at the American Diabetes Association 73th
Scientific Sessions (ADA) in 2013 (number 3664).
References
|
1
|
Weickert MO and Pfeiffer AF: Signalling
mechanisms linking hepatic glucose and lipid metabolism.
Diabetologia. 49:1732–1741. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Samuel VT and Shulman GI: Mechanisms for
insulin resistance: Common threads and missing links. Cell.
148:852–871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Solinas G, Naugler W, Galimi F, Lee MS and
Karin M: Saturated fatty acids inhibit induction of insulin gene
transcription by JNK-mediated phosphorylation of insulin-receptor
substrates. Proc Natl Acad Sci USA. 103:16454–16459. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hirosumi J, Tuncman G, Chang L, Görgün CZ,
Uysal KT, Maeda K, Karin M and Hotamisligil GS: A central role for
JNK in obesity and insulin resistance. Nature. 420:333–336. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galbo T, Olsen GS, Quistorff B and
Nishimura E: Free fatty acid-induced PP2A hyperactivity selectively
impairs hepatic insulin action on glucose metabolism. PLoS One.
6:e274242011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bahlmann FH and Fliser D: Erythropoietin
and renoprotection. Curr Opin Nephrol Hypertens. 18:15–20. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mitsuma W, Ito M, Kodama M, Fuse K,
Okamura K, Minagawa S, Kato K, Hanawa H, Toba K, Nakazawa M and
Aizawa Y: Cardioprotective effects of recombinant human
erythropoietin in rats with experimental autoimmune myocarditis.
Biochem Biophys Res Commun. 344:987–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villa P, Bigini P, Mennini T, Agnello D,
Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman
TR, et al: Erythropoietin selectively attenuates cytokine
production and inflammation in cerebral ischemia by targeting
neuronal apoptosis. J Exp Med. 198:971–975. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rasic-Milutinovic Z, Perunicic-Pekovic G,
Cavala A, Gluvic Z, Bokan L and Stankovic S: The effect of
recombinant human erythropoietin treatment on insulin resistance
and inflammatory markers in non-diabetic patients on maintenance
hemodialysis. Hippokratia. 12:157–161. 2008.PubMed/NCBI
|
|
10
|
Meng R, Zhu D, Bi Y, Yang D and Wang Y:
Erythropoietin inhibits gluconeogenesis and inflammation in the
liver and improves glucose intolerance in high-fat diet-fed mice.
PLoS One. 8:e535572013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao D, Nong S, Huang X, Lu Y, Zhao H, Lin
Y, Man Y, Wang S, Yang J and Li J: The effects of palmitate on
hepatic insulin resistance are mediated by NADPH Oxidase 3-derived
reactive oxygen species through JNK and p38MAPK pathways. J Biol
Chem. 285:29965–29973. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Copps KD and White MF: Regulation of
insulin sensitivity by serine/threonine phosphorylation of insulin
receptor substrate proteins IRS1 and IRS2. Diabetologia.
55:2565–2582. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real time quantitative PCR and
the 2(−Delta Delta C(T) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Taniguchi CM, Ueki K and Kahn R:
Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of
metabolism. J Clin Invest. 115:718–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piro S, Maniscalchi ET, Monello A, Pandini
G, Mascali LG, Rabuazzo AM and Purrello F: Palmitate affects
insulin receptor phosphorylation and intracellular insulin signal
in a pancreatic alpha-cell line. Endocrinology. 151:4197–4206.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Arcasoy MO: The non-haematopoietic
biological effects of erythropoietin. Br J Haemato. 141:14–31.
2008. View Article : Google Scholar
|
|
17
|
Allegra V, Mengozzi G, Martimbianco L and
Vasile A: Early and late effects of erythropoietin on glucose
metabolism in maintenance hemodialysis patients. Am J Nephrol.
16:304–308. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khedr E, El-Sharkawy M, Abdulwahab S,
Eldin EN, Ali M, Youssif A and Ahmed B: Effect of recombinant human
erythropoietin on insulin resistance in hemodialysis patients.
Hemodial Int. 13:340–346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tuzcu A, Bahceci M, Yilmaz E, Bahceci S
and Tuzcu S: The comparison of insulin sensitivity in non-diabetic
hemodialysis patients treated with and without recombinant human
erythropoietin. Horm Metab Res. 36:716–720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Teng R, Gavrilova O, Suzuki N, Chanturiya
T, Schimel D, Hugendubler L, Mammen S, Yver DR, Cushman SW, Mueller
E, et al: Disrupted erythropoietin signalling promotes obesity and
alters hypothalamus proopiomelanocortin production. Nat Commun.
2:5202011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hojman P, Brolin C, Gissel H, Brandt C,
Zerahn B, Pedersen BK and Gehl J: Erythropoietin over-expression
protects against diet-induced obesity in mice through increased fat
oxidation in muscles. PLoS One. 4:e58942009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan Y, Shu JL, Gu HF, Zhou DC, Liu XL,
Qiao QY, Fu SK, Gao FH and Jin HM: Erythropoietin improves insulin
resistance via the regulation of its receptor-mediated signaling
pathway in 3T3L1 adipocytes. Mol Cell Endocrinol. 367:116–123.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Teng R, Di L, Rogers H, Wu H, Kopp
JB and Noguchi CT: PPARα and Sirt1 mediate erythropoietin action in
increasing metabolic activity and browning of white adipocytes to
protect against obesity and metabolic disorders. Diabetes.
62:4122–4131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haus JM, Solomon TP, Marchetti CM, Edmison
JM, González F and Kirwan JP: Free fatty acid-induced hepatic
insulin resistance is attenuated following lifestyle intervention
in obese individuals with impaired glucose tolerance. J Clin
Endocrinol Metab. 95:323–327. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Solinas G and Karin M: JNK1 and IKKbeta:
Molecular links between obesity and metabolic dysfunction. FASEB J.
24:2596–2611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanti JF and Jager J: Cellular mechanisms
of insulin resistance: Role of stress-regulated serine kinases and
insulin receptor substrates (IRS) serine phosphorylation. Curr Opin
Pharmacol. 9:753–762. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Taniguchi CM, Kondo T, Sajan M, Luo J,
Bronson R, Asano T, Farese R, Cantley LC and Kahn CR: Divergent
regulation of hepatic glucose and lipid metabolism by
phosphoinositide 3-kinase via Akt and PKClambda/zeta. Cell Metab.
3:343–353. 2006. View Article : Google Scholar : PubMed/NCBI
|















