Introduction
Heart failure is a progressive and complex clinical
syndrome that leads to impaired functional cardiac ability. It is
defined as a symptom resulting in ventricular dysfunction (1) and is characterized by high morbidity
and mortality. Contractile dysfunction is often linked to chronic
energy deficiency and ventricular remodeling. It has previously
been reported that ~50–60% of patients with heart failure have an
enlarged left ventricle (LV) chamber and reduced ejection fraction
(2,3). The majority of patients with heart
failure have a history of hypertension and LV hypertrophy (4). A pronounced decrease in free fatty
acid (FFA) β-oxidation in dilated cardiomyopathy has been
identified in patients with Class II and III heart failure
according to the New York Heart Association classifications when
compared with age-matched healthy individuals (5,6). The
same study additionally reported that during the progression of
congestive heart failure, the increased ventricular remodeling
elevated local oxygen consumption, and worsened the induced energy
deficiency and ejection function. As a result, the heart enters a
cycle of impaired cardiac functioning. However, the understanding
of differential protein expression in post-myocardial infarction
(post-MI) heart failure remains relatively limited.
Linear Trap Quadropole (LTQ) OrbiTrap mass
spectrometry is a protein quantification strategy that provides
relative and absolute measurements of proteins in complex mixtures
(7). The present study was
undertaken to investigate the differential protein expressions that
are associated with advanced heart failure irrespective of
treatment. LTQ OrbiTrap mass spectrometry was used to analyze the
expression profiles of energy metabolism-associated,
calcium-binding and cytoskeletal proteins in post-MI heart failure
and control groups. The aim of the present study was to identify
the target proteins that are associated with the disease in order
to provide novel diagnostic markers and alternative therapies to
reconstitute the energetic state and disrupt the damaging cycle of
the failing heart.
Materials and methods
Acute heart failure model
All animal procedures and experiments were performed
in accordance with the Guide for the Care and Use of Laboratory
Animals published by the US National Institutes of Health [NIH
publication 85(23), revised in
1996] (8). A total of 18 male
Wistar rats (age, 12–14 weeks; weight, 250–300 g) were provided by
the experimental animal center of Shandong University (Shandong,
China). The rats were housed in a climate-controlled environment at
a constant temperature of 22°C, relative humidity 50% and a 12-h
dark/light cycle. Rats were fed standard laboratory chow, and
allowed access to food and water ad libitum. The present study was
conducted with approval from the Ethics Committee of Shandong
University (Shandong, China).
The left anterior descending coronary artery of 12
rats was occluded as described previously (9). Rats were anaesthetized on
postoperative days 1 and 14 with an intraperitoneal injection of 40
mg/100 g chloral hydrate and the hearts were exposed. The left
anterior descending coronary artery was ligated at ~2–3 mm below
the left auricle. The heart was repositioned in the chest and the
chest was closed with a purse string suture. The animals were
randomly divided into the following 3 groups (6 rats/group):
Control group (untreated rats), the 1st day group (rats were
euthanized on the 1st postoperative day) and the 14th day group
(rats were euthanized on the 14th postoperative day). The left
ventricles were obtained immediately following animal sacrifice and
cut into two sections. The hearts were removed rapidly, the left
and right atrium and right ventricle were removed and the left
ventricle was divided into two parts along the long axis. One
section was immersed in 4% paraformaldehyde at room temperature
overnight, and the other section was immediately snap-frozen and
stored in liquid nitrogen for further analysis.
On the 14th postoperative day (prior to euthanasia),
acquired echocardiography was used to measure the left ventricular
end-diastolic diameter (LVEDD), left ventricular end-systolic
diameter (LVESD), left ventricular posterior wall end diastole and
end systole (LVPWs and LVPWd, respectively), left ventricular
ejection fraction (LVEF) and fractional shortening (FS).
Protein extraction
A 50 mg sample was taken from the LV of each rat
following animal sacrifice, which was immediately snap-frozen and
stored in liquid nitrogen. The samples were placed in liquid
nitrogen and ground into a fine powder, homogenized in a lysate
buffer (8 mol/l urea, 1 mol/l DTT, cocktail of protease inhibitors,
1 mg/ml leupeptin, 1 mg/ml aprotinin, 1 mg/ml pepstatin,
radioimmunoprecipitation buffer and 0.1% PSMF) as previously
described (10) and then incubated
on ice for 30 min. The samples were further lysed by ultrasound for
3 cycles of 10 sec. The whole lysate was centrifuged for 15 min at
4°C and 14,000 × g, and the supernatant was collected.
Sample processing
Protein concentrations of the lysates were
determined using a Bicinchoninic Acid Protein Assay kit (Beyotime
Institute of Biotechnology, Haimen, China). Protein extract (30 µl)
was mixed with 200 µl urea-acetate (UA) buffer (8 M urea in 0.1
mol/l Tris/HCl; pH 8.5) in a filter unit, centrifuged at 14,000 × g
at 4°C for 15 min and then washed three times with 100 µl UA
buffer. The flow-through was discarded from the collection tube.
The concentrate was mixed with 100 µl indole acetic acid buffer
(0.05 mol/l iodoacetamide in UA) and incubated in the dark at room
temperature for 20 min, which was followed by centrifugation for 10
min at 14,000 × g. The concentrate was then washed twice with 100
µl UA buffer followed by two washes with 100 µl ammonium
bicarbonate (ABC) buffer (0.05 M NH4HCO3 in
water). A total of 40 µl ABC buffer with trypsin (1:100) was added
to the filter and incubated overnight at 37°C to achieve complete
digestion. A further 40 µl ABC buffer was added and centrifuged at
14,000 × g for 10 min. The final solution was dried under a vacuum
and stored in a freezer at −80°C.
Sample purification with C18 Ziptip
column
Samples were diluted in 40 µl 0.1% trifluoroacetic
acid (TFA). A total of 200 µl 100% acetonitrile was added to a
Ziptip (Merck KGaA, Darmstadt, Germany) and centrifuged at 800 × g
for 2 min; this step was repeated twice. The concentrate was mixed
with 200 µl 0.1% TFA and centrifuged at 800 × g for 2 min. TFA was
added and centrifugation was performed eight times. The Ziptip was
washed twice with 0.1% TFA and centrifuged at 800 × g for 2 min.
The peptides were eluted with 40 µl formic acid, dried under a
vacuum and stored in a freezer at −80°C.
LTQ OrbiTrap mass spectrometry
A total of four injections were made into a Nano LC
1000 (Proxeon; Thermo Fisher Scientific, Inc.) interface of the LTQ
OrbiTrap elite mass spectrometer (Thermo Fisher Scientific, Inc.)
via a nano source. Samples of 2 µg were diluted in Solvent A (99.9%
water/0.1% formic acid) and loaded onto a 150 µm × 2 cm peptrap 300
Å C18 pre-column. A total of 2 µg peptide was eluted into a 75 µm ×
25 cm 100 Å C18 analytical column (self-packed) and separated with
a linear gradient of 5–30% Solvent B (99.9% acetonitrile/0.1%
formic acid) for 5 min and then 69% Solvent B for 115 min. The flow
rate was 250 nl/min. The survey scans were acquired in the OrbiTrap
analyzer with 60,000 resolution at 400 m/z and 275°C. The automated
gain control target was set at the level of 1×106. The 25 most
intense ions were fragmented using collisionally induced
dissociation in the linear ion trap. The precursor ions were
fragmented with helium gas for 30 msec with a normalized collision
energy of 35. The dynamic exclusion parameters were set to exclude
ions previously selected for fragmentation for 1 min. All data were
acquired in the reduced profile mode to accommodate further
downstream processing.
Protein identification and
quantification
Protein identification was accomplished using
Proteome Discoverer v1.4 (Thermo Fisher Scientific, Inc.) and
Mascot Server v2.4 software (www.matrixscience.com/server.html). The Mascot search
engine was used to identify consolidated data in the Uniprot rat
protein database (www.uniprot.org), with carbamidomethylation + 57,005
selected as the fixed modification and oxidation of methionine +
15,995 set as the variable modification. The mass tolerance was set
at 10 ppm and the MS/MS tolerance was set at 0.8 Da (10). The trypsin enzymolysis maximum
leakage cut-off value was set at 2, and the important threshold
value was set at 0.01 to ensure a false discovery rate of <1%.
Protein quantification was obtained via unique peptides. P<0.05
was considered significant for protein quantification. To designate
significant alterations in protein expression, fold changes <2.0
were set as the cut-offs. This analysis was performed twice.
Bioinformatic analyses
Analyses of protein (and their genes) classification
were performed with tools available on the Protein Analysis Through
Evolutionary Relationships online classification system (PANTHER;
pantherdb.org).
Hematoxylin and eosin (H&E)
staining
The myocardial tissue was embedded in conventional
paraffin and sectioned into 5-µm-thick slides. Sections were
dewaxed in xylene at room temperature and rehydrated in graded
ethanol (dehydrated ethanol for 5 min, dehydrated ethanol for 5
min, 90% ethanol for 5 min, 90% ethanol for 5 min, 75% ethanol for
5 min and 75% ethanol for 5 min). Following the standard process of
H&E staining (10% hematoxylin for 3–5 min and 0.5% eosin for 1
min at room temperature) (11),
the specimens were observed under a light microscope (Leica
DM4000B; Leica Microsystems GmbH, Wetzlar, Germany; magnification,
×400) and the ratio of myocardial cells to capillaries, the
diameter of cardiomyocytes, cell density, capillary density,
intracellular substance and intercellular space were examined in 5
randomly selected fields in order to evaluate the extent of
myocardial hypertrophy.
Western blot analysis
The LTQ OrbiTrap protein expression results were
validated via western blot analysis. Total protein extracts used
for the western blot analysis were obtained using the
aforementioned procedure. The samples containing 100 µg of total
proteins were separated using 6% SDS-PAGE and transferred onto
polyvinylidene fluoride membranes (Merck KGaA) via
electro-blotting. The membranes were incubated in TBST containing
5% non-fat dried milk for 1 h at 25°C. The membranes were probed
overnight at 4°C with rabbit anti-myosin 7 polyclonal antibody
(cat. no. 22280-1-AP; ProteinTech Group, Inc., Chicago, IL, USA),
rabbit anti-Vitamin D binding protein (VDBP) polyclonal antibody
(cat. no. 16922-1-AP; ProteinTech), rabbit anti-gelsolin polyclonal
antibody (cat. no. PB0198; Wuhan Boster Biological Technology,
Ltd., Wuhan, China) and rabbit anti-Voltage-dependent L-type
calcium channel subunit α1D polyclonal antibody (cat. no. PB0286;
Wuhan Boster Biological Technology, Ltd.). GAPDH (cat. no.
10494-1-AP; ProteinTech) was used as an internal control. All
primary antibodies were used at a 1:1,000 dilution. Horseradish
peroxidase goat anti-rabbit IgG antibodies (cat. no. SA00001-2;
ProteinTech) were used as the secondary antibodies at a dilution of
1:2,000. The membranes were developed with enhanced
chemiluminescence plus reagent (Beyotime Institute of
Biotechnology) and bands were quantified by densitometry using
ImageJ2x software (version 2.14.7; National Institutes of Health,
Bethesda, MD, USA). Experiments were repeated independently 3
times.
Immunohistochemistry
The myocardial tissue was fixed in 4%
paraformaldehyde at room temperature overnight, embedded in
conventional paraffin and sectioned using an SP-9001 IHC staining
kit (ZSGD-BIO, Beijing, China) into 5-µm thickness in accordance
with the manufacturer's protocol. Sections were dewaxed in xylene
at room temperature, rehydrated in graded ethanol (dehydrated
ethanol for 5 min, dehydrated ethanol for 5 min, 90% ethanol for 5
min, 90% ethanol for 5 min, 75% ethanol for 5 min and 75% ethanol
for 5 min) and incubated with 0.3% hydrogen peroxide to inactivate
endogenous peroxidase activity. Antigen retrieval was achieved by
incubating the slides at high pressure for 2 min (~120°C) with
sodium citrate (pH 6.0). Following blocking with goat serum
(ZSGD-BIO) at 37°C for 30 min, the sections were incubated with
rabbit anti-myosin 7 polyclonal antibody (1:200; cat. no.
22280-1-AP; ProteinTech), rabbit anti-VDBP polyclonal antibody
(1:100; cat. no. 16922-1-AP; ProteinTech), rabbit anti-gelsolin
polyclonal antibody (1:50; cat. no. PB0198; Wuhan Boster Biological
Technology, Ltd.) or rabbit anti-Voltage-dependent L-type calcium
channel subunit α1D (Cav 1.3) polyclonal antibody (1:50; cat. no.
PB0286; Wuhan Boster Biological Technology, Ltd.) overnight at 4°C
and subsequently incubated with biotinylated secondary antibody
working fluid at 37°C for 30 min (cat. no. SP 9001; IHC staining
kit; ZSGB-BIO, Beijing, China). The specimens were stained with
0.05% 3,3′-diaminobenzidine for 1 min and re-dyed with 10%
hematoxylin for 3-5 min at room temperature. The specimens were
observed under a light microscope (Leica DM4000B; Leica
Microsystems GmbH; magnification, ×400) and photographed. Brown
reaction granules observed in the cells indicated a positive
result.
Statistical analysis
Data was analyzed using SPSS software (version 18.0;
SPSS, Inc., Chicago, IL, USA). One-way analysis of variance
followed by Tukey's multiple comparison test was used to determine
the statistical differences among the post-MI and control groups.
Data are presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
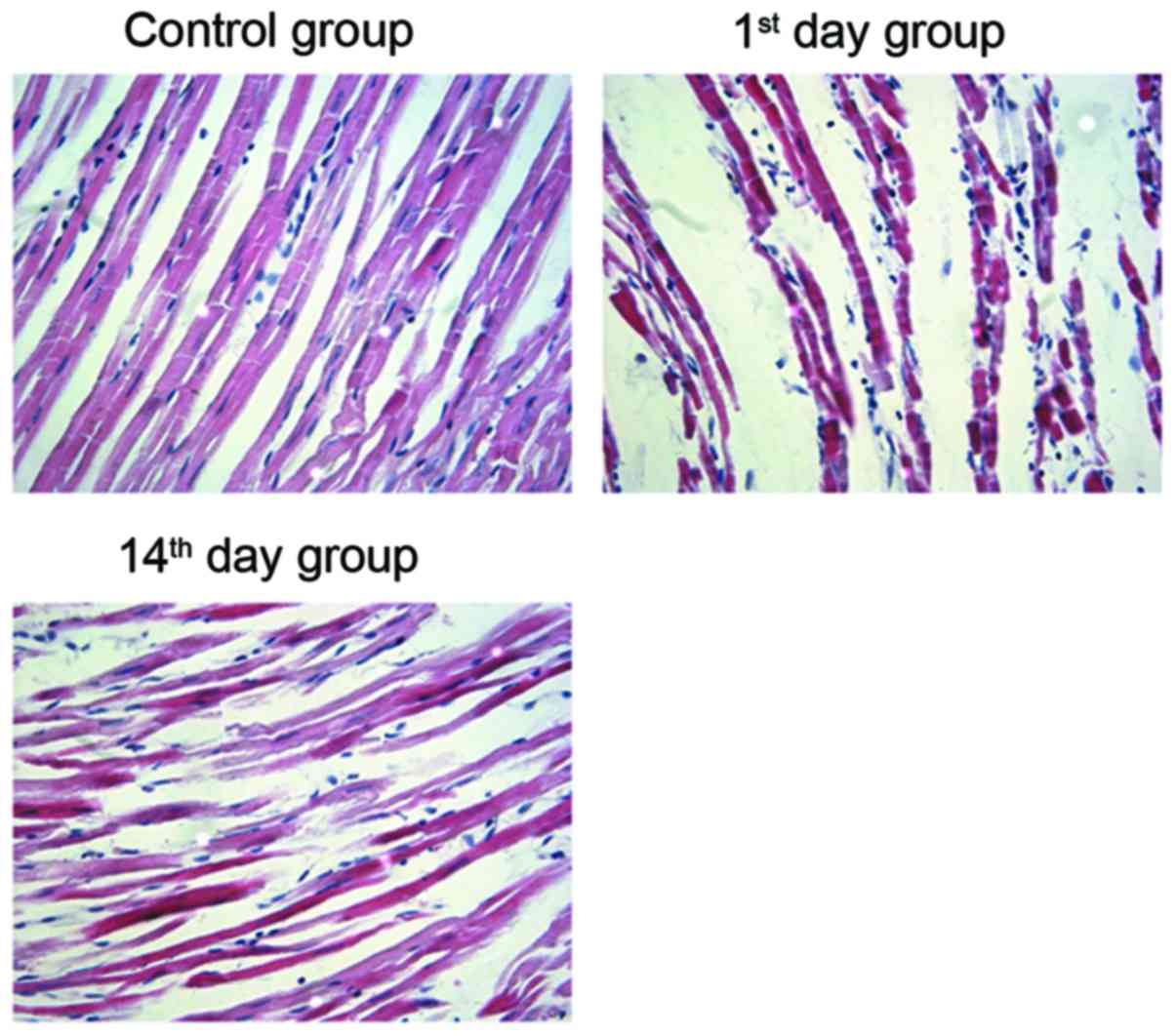
Morphological alterations post-MI
To verify the establishment of the myocardial
infarction model in rats, H&E staining of the cardiomyocytes
was performed. In the control group specimens, a neat arrangement
and clearly visible stripes were observed in cardiomyocytes.
Conversely, the cardiomyocytes from the 1st and 14th day groups
exhibited varying sizes and uneven staining. In addition, an
increase in cell diameter and intercellular space, and an irregular
arrangement of cardiac muscle fibers was observed following MI
(Fig. 1). This suggests that the
arrangement of cardiomyocytes in post-MI groups is irregular when
compared with the control group.
LTQ OrbiTrap analysis
The present study then identified the differentially
expressed proteins in MI model rats. Data from the LTQ OrbiTrap
experiments contained 1,709 unique proteins from the cardiac
tissues of the 14th day group, which included well-known markers
associated with the cytoskeleton, energy metabolism and actin. In
addition, 1,722 unique proteins were identified in the cardiac
tissues of the 1st day group, and 2,148 unique proteins were
observed in the cardiac tissues of the control group (Table I). Furthermore, 1,080 and 437
proteins were differentially expressed in the cardiac tissues of
the 14th day group when compared with the 1st day group and the
control group, respectively. When compared with the control group,
1,574 proteins were differentially expressed in the cardiac tissues
of the 1st day group.
 | Table I.Proteins with statistically
significant differential expression in cardiac tissues among the
control, 1st and 14th day groups. |
Table I.
Proteins with statistically
significant differential expression in cardiac tissues among the
control, 1st and 14th day groups.
|
|
|
| Differentially
expressed proteins |
|---|
|
|
|
|
|
|---|
|
|
|
| Control vs. 1st day
(fold >2) | Control vs. 14th
day (fold >2) | 1st day vs. 14th
day (fold >2) |
|---|
|
|
|
|
|
|
|
|---|
| Group | Protein count | Peptide count | Upregulated | Downregulated | Upregulated | Downregulated | Upregulated | Downregulated |
|---|
| Control | 2148 | 9243 | 45 | 55 | 50 | 28 | 48 | 51 |
| 1st day | 1722 | 7473 |
|
|
|
|
|
|
| 14th day | 1709 | 7541 |
|
|
|
|
|
|
To identify proteins involved in post-MI heart
failure, <2.0-fold expression was used as the cut-off. The
results revealed that 99 proteins that were differentially
expressed in the 14th and 1st day groups may be associated with the
progression of heart failure. A further 100 proteins were
differentially expressed between the 1st day and control groups.
Comparisons between the control and 14th day groups revealed 78
differentially expressed proteins, including 50 upregulated
proteins and 28 downregulated proteins in the control group
(Table I).
The differentially expressed proteins in the control
and 14th day groups are closely associated with energy metabolism
(including glycolysis, mitochondrial tricarboxylic acid cycle and
fatty acid β-oxidation), contractile function [β-myosin heavy chain
isoforms (myosin-7)], calcium handling (Gelsolin, Cav 1.3,
Galectin-3 and VDBP), pathologic hypertrophy (Gelsolin and
Myosin-7) and cardiac remodeling (Fibrinogen β chain; Table II). Furthermore, dynamic
alterations in differential protein expression at different time
points (the 1st and 14th postoperative days) post-MI were observed
(Table III). For example,
Myosin-7 expression was almost unaltered on the 1st day post-MI
however, it was significantly upregulated on the 14th day post-MI
with the progression of heart failure, which was in accordance with
a previous study (12). In the
acute phase of heart failure, namely in the 1st day post-MI group,
expression of Gelsolin, Cav1.3 and VDBP increased significantly,
with a fold increase of 3.92, 7.09 and 11.77, respectively, when
compared with the control group.
| Table II.Proteins with statistically
significant differential expression between the control and 14th
day groups. |
Table II.
Proteins with statistically
significant differential expression between the control and 14th
day groups.
| Accession | Peptides |
P-valuea | Fold
changesa | Description | Function |
|---|
| RS7_RAT | 1 |
7.86×10−3 | 757.53 | 40S ribosomal
protein S7 |
|
| SUCA_RAT | 3 (2) |
3.10×10−5 | 64.97 | Succinyl-CoA ligase
[ADP/GDP-forming] subunit α | Oxidoreductase |
| STIM1_RAT | 6 (1) |
8.62×10−6 | 7.94 | Stromal interaction
molecule 1 |
|
| PER1_RAT | 6 (1) |
3.17×10−3 | 7.51 | Period circadian
protein homolog 1 | Transcription
cofactor |
| ERAP1_RAT | 6 (1) |
1.25×10−3 | 6.7 | Endoplasmic
reticulum aminopeptidase 1 |
Metalloprotease |
| P97573 | 9 (3) | 0.01 | 6.47 |
Phosphatidylinositol-3,4,5-trisphosphate
5-phosphatase 1 | Phosphatase |
| PALM_RAT | 4 (1) | 0.03 | 5.93 | Paralemmin-1 |
|
| MDHM_RAT | 7 (2) | 0.03 | 5.43 | Malate
dehydrogenase | Dehydrogenase |
| ES1_RAT | 3 (2) |
4.24×10−3 | 3.71 | ES1 protein
homolog |
|
| ACON_RAT | 8 (3) |
2.04×10−3 | 3.48 | Aconitate
hydratase | Dehydrogenase |
| ODPA_RAT | 5 (3) |
9.40×10−4 | 3.02 | Pyruvate
dehydrogenase E1 component subunit α, somatic form | Dehydrogenase |
| 3HIDH_RAT | 3 (2) | 0.05 | 2.91 |
3-hydroxyisobutyrate dehydrogenase | Dehydrogenase |
| H31_RAT | 5 (2) |
1.29×10−3 | 2.85 | Histone H3.1 |
|
| DESM_RAT | 7 (3) | 0.01 | 2.83 | Desmin | Structural
protein |
| KCRS_RAT | 9 (3) |
5.03×10−4 | 2.81 | Creatine kinase
S-type | Amino acid
kinase |
| DHSD_RAT | 1 |
1.09×10−3 | 2.74 | Succinate
dehydrogenase [ubiquinone] cytochrome b small subunit |
|
| FUMH_RAT | 11 (4) | 0.01 | 2.71 | Fumarate
hydratase | Lyase |
| CH60_RAT | 3 (1) | 0.01 | 2.64 | 60 kDa heat shock
protein | Chaperonin |
| ITB1_RAT | 7 (4) | 0.01 | 2.52 | Integrin β-1 |
|
| SMC3_RAT | 25 (4) |
2.99×10−4 | 2.43 | Structural
maintenance of chromosomes protein 3 |
|
| THIOM_RAT | 2 (1) | 0.04 | 2.4 | Thioredoxin |
|
| ATPO_RAT | 9 (5) |
3.86×10−3 | 2.39 | ATP synthase
subunit Ol | ATP synthase |
| GRP75_RAT | 16 (3) | 0.03 | 2.34 | Stress-70
protein |
|
| ACADL_RAT | 8 (4) |
7.38×10−3 | 2.3 | Long-chain specific
acyl-CoA dehydrogenase | Transferase |
| ALDH2_RAT | 7 (3) | 0.03 | 2.28 | Aldehyde
dehydrogenase |
|
| ATP5E_RAT | 2 (1) |
5.64×10−3 | 2.19 | ATP synthase
subunit epsilon | ATP synthase |
| ATPB_RAT | 4 (3) |
7.99×10−4 | 2.14 | ATP synthase
subunit β | ATP synthase |
| ECHM_RAT | 4 (1) |
2.97×10−3 | 2.1 | Enoyl-CoA
hydratase |
Acetyltransferase |
| MAVS_RAT | 2 | 0.03 | 2.02 | Mitochondrial
antiviral-signaling protein |
|
| ANXA6_RAT | 10 (7) |
1.92×10−5 | −2.07 | Annexin A6 |
|
| SPA3N_RAT | 2 (1) | 0.02 | −2.08 | Serine protease
inhibitor A3N | Serine protease
inhibitor |
| PSB1_RAT | 3 (2) |
1.83×10−3 | −2.1 | Proteasome subunit
β type-1 | Protease |
| PGK1_RAT | 9 (3) |
2.47×10−4 | −2.16 | Phosphoglycerate
kinase 1 | Carbohydrate
kinase |
| RL26_RAT | 4 (1) |
9.18×10−4 | −2.24 | 60S ribosomal
protein L26 | Ribosomal
protein |
| CDK7_RAT | 5 (2) | 0.01 | −2.25 | Cyclin-dependent
kinase 7 (Fragment) | Non-receptor
serine/threonine protein kinase |
| FINC_RAT | 9 (6) |
3.50×10−3 | −2.28 | Fibronectin | Signaling
molecule |
| CHD8_RAT | 8 (2) |
1.55×10−3 | −2.31 |
Chromodomain-helicase-DNA-binding protein
8 | DNA helicase |
| CDS2_RAT | 1 | 0.03 | −2.46 | Phosphatidate
cytidylyltransferase 2 |
Nucleotidyltransferase |
| MYH7_RAT | 101 (17) |
9.06×10−6 | −2.48 | Myosin-7 | G-protein
modulator |
| APOH_RAT | 1 |
1.44×10−3 | −2.53 | β-2-glycoprotein
1 | Apolipoprotein |
| IGG2B_RAT | 2 |
6.88×10−3 | −2.56 | Ig γ-2B chain C
region |
|
| PABP1_RAT | 7 (1) |
9.54×10−5 | −2.77 |
Polyadenylate-binding protein 1 | Transcription
factor |
| SELS_RAT | 3 (1) |
5.32×10−3 | −2.78 | Selenoprotein
S |
|
| KACA_RAT | 1 |
3.62×10−5 | −2.87 | Ig κ chain C
region, A allele |
|
| PPIB_RAT | 3 (1) | 0.02 | −2.88 | Peptidyl-prolyl
cis-trans isomerase B | Isomerase |
| P06399 | 36 (10) |
3.00×10−6 | −2.88 | Fibrinogen α
chain |
|
| HSDL2_RAT | 6 (1) |
7.17×10−3 | −2.9 | Hydroxysteroid
dehydrogenase-like protein 2 | Dehydrogenase |
| DJC14_RAT | 5 (2) |
5.49×10−4 | −2.98 | Dnaj homolog | Chaperone |
| C4BPA_RAT | 6 (3) |
8.39×10−3 | −3.02 | C4b-binding protein
α chain | Apolipoprotein |
| EXOC8_RAT | 6 (1) |
2.70×10−3 | −3.09 | Exocyst complex
component 8 |
|
| GDIB_RAT | 3 (1) |
1.76×10−4 | −3.12 | Rab GDP
dissociation inhibitor β |
Acyltransferase |
| NDUA9_RAT | 3 (2) |
6.17×10−4 | −3.2 | NADH dehydrogenase
[ubiquinone] 1 α subcomplex subunit 9 | Dehydrogenase |
| MVP_RAT | 4 (2) |
1.37×10−3 | −3.26 | Major vault
protein |
Ribonucleoprotein |
| S10A3_RAT | 1 |
1.41×10−3 | −3.28 | Protein
S100-A3 | Calmodulin |
| HBB2_RAT | 5 (1) |
7.25×10−3 | −3.58 | Hemoglobin subunit
β-2 |
|
| FIBB_RAT | 10 (6) |
1.68×10−4 | −3.75 | Fibrinogen β
chain | Signaling
molecule |
| CEP41_RAT | 4 (1) |
4.12×10−4 | −3.9 | Centrosomal protein
of 41 kDa |
|
| AT5F1_RAT | 3 (1) | 0.01 | −3.93 | ATP synthase
subunit b, mitochondrial |
|
| GRM4_RAT | 5 (2) |
6.43×10−5 | −4.01 | Metabotropic
glutamate receptor 4 | G-protein coupled
receptor |
| PRELP_RAT | 4 (1) |
1.31×10−3 | −4.03 | Prolargin | Extracellular
matrix protein |
| FIBG_RAT | 8 (4) |
8.81×10−5 | −4.18 | Fibrinogen γ chain
OS | Signaling
molecule |
| IGG2A_RAT | 4 (1) |
2.06×10−3 | −4.76 | Ig γ-2A chain C
region |
|
| HEMO_RAT | 6 (2) |
1.10×10−3 | −4.84 | Hemopexin | Transfer/carrier
protein |
| LSG1_RAT | 4 (1) |
5.76×10−6 | −4.84 | Large subunit
GTPase 1 homolog | Signaling
molecule |
| ZBT38_RAT | 11 (2) |
1.86×10−3 | −5.56 | Zinc finger and BTB
domain-containing protein 38 | KRAB box
transcription factor |
| RL10A_RAT | 5 (1) |
3.82×10−4 | −5.64 | 60S ribosomal
protein L10a | Ribosomal
protein |
| PGS2_RAT | 4 (1) |
6.25×10−6 | −5.7 | Decorin | Extracellular
matrix protein |
| KNT1_RAT | 3 (1) |
1.71×10−3 | −5.96 | T-kininogen 1 |
|
| IGHG1_RAT | 3 (1) |
5.69×10−5 | −6.37 | Ig γ-1 chain C
region |
|
| PTGIS_RAT | 9 (3) |
2.05×10−5 | −6.76 | Prostacyclin
synthase | Oxidoreductase |
| CAC1D_RAT | 7 (1) |
3.56×10−3 | −7.46 | Voltage-dependent
L-type calcium channel subunit α-1D | Voltage-gated
calcium channel |
| ECI1_RAT | 4 (1) | 0.04 | 9.73 | Enoyl-CoA ∆
isomerase 1 |
Acetyltransferase |
| KNT2_RAT | 3 (1) |
1.41×10−4 | −16.47 | T-kininogen 2 |
|
| RS13_RAT | 2 (1) |
1.04×10−3 | −23.58 | 40S ribosomal
protein S13 | Ribosomal
protein |
| SPRY4_RAT | 3 (1) | 0.04 | −28.28 |
Sprydomain-containing protein 4 |
|
| LEG3_RAT | 2 (1) |
6.19×10−6 | −36.14 | Galectin-3 | Signaling
molecule |
| PRP19_RAT | 3 (1) | 0.02 | −68.84 | Pre-mRNA-processing
factor 19 | mRNA splicing
factor |
| HD_RAT | 17 (1) |
5.69×10−3 | −366.94 | Huntingtin,
subfamily C member 14 |
|
| Table III.Dynamic changes in differential
protein expression in cardiac tissues among the control, 1st and
14th day groups. |
Table III.
Dynamic changes in differential
protein expression in cardiac tissues among the control, 1st and
14th day groups.
| Accession | Description | Fold
changea |
P-valuea | Fold
changeb |
P-valueb | Fold
changec |
P-valuec | Function |
|---|
| MYH7_RAT | Myosin-7 | −1.01 |
2.15×10−5 | −2.48 |
9.06×10−6 | 2.45 |
1.05×10−5 | G-protein
modulator |
| GELS_RAT | Gelsolin | −3.92 |
1.70×10−4 | −1.81 |
1.50×10−5 | −2.16 |
4.40×10−6 | Non-motor actin
binding protein, calcium-binding protein |
| CAC1D_RAT | Cav1.3 | −7.09 |
3.94×10−3 | −7.46 |
3.56×10−3 | 1.05 |
1.25×10−3 | Voltage-gated
calcium channel |
| VTDB_RAT | VDBP | −11.77 | 0.01 | 1.23 | 0.01 | −13.65 |
8.10×10−4 |
|
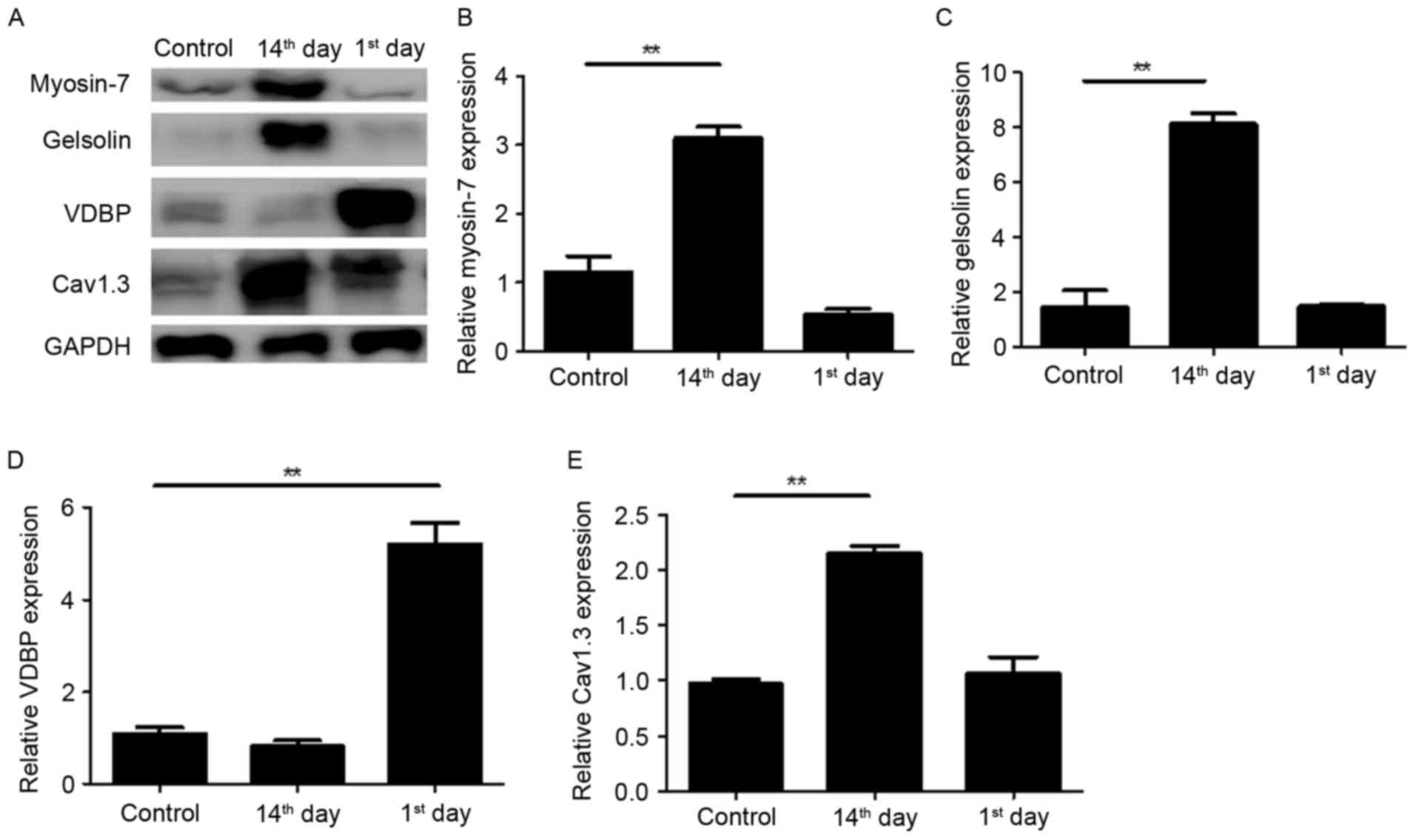
Verification of protein expression by
western blotting
The four dynamically altered proteins during the
development of advanced heart failure were associated with a number
of biological processes (Table
III), thus their expression in myocardial tissues was verified
by western blotting (Fig. 2).
Consistent with the results of LTQ OrbiTrap, the expression of
Myosin-7 was significantly upregulated on the 14th day post-MI with
an unnoticeable alteration on the 1st day (Fig. 2A and B). The expression of VDBP was
markedly upregulated on the 1st day post-MI (Fig. 2A and D) and the expression of
Cav1.3 was significantly upregulated on the 14th day post-MI
(Fig. 2A and E). The upregulation
of Gelsolin on the 1st day post-MI was not observed by western
blotting however, a marked upregulation was observed on the 14th
day (Fig. 2A and C). These results
suggest that the present proteomics results are relatively
reliable.
Verification of protein expression by
immunohistochemistry
The present study further verified the
aforementioned results via immunohistochemistry in the myocardial
tissue at different time points post-MI. As presented in Fig. 3, Gelsolin, Myosin-7 and Cav1.3 were
observed in the cytoplasm and nucleus of myocardial cells, and VDBP
was highly expressed in the cell membrane. A similar expression
profile for the four proteins during the progression of advanced
heart failure was additionally observed.
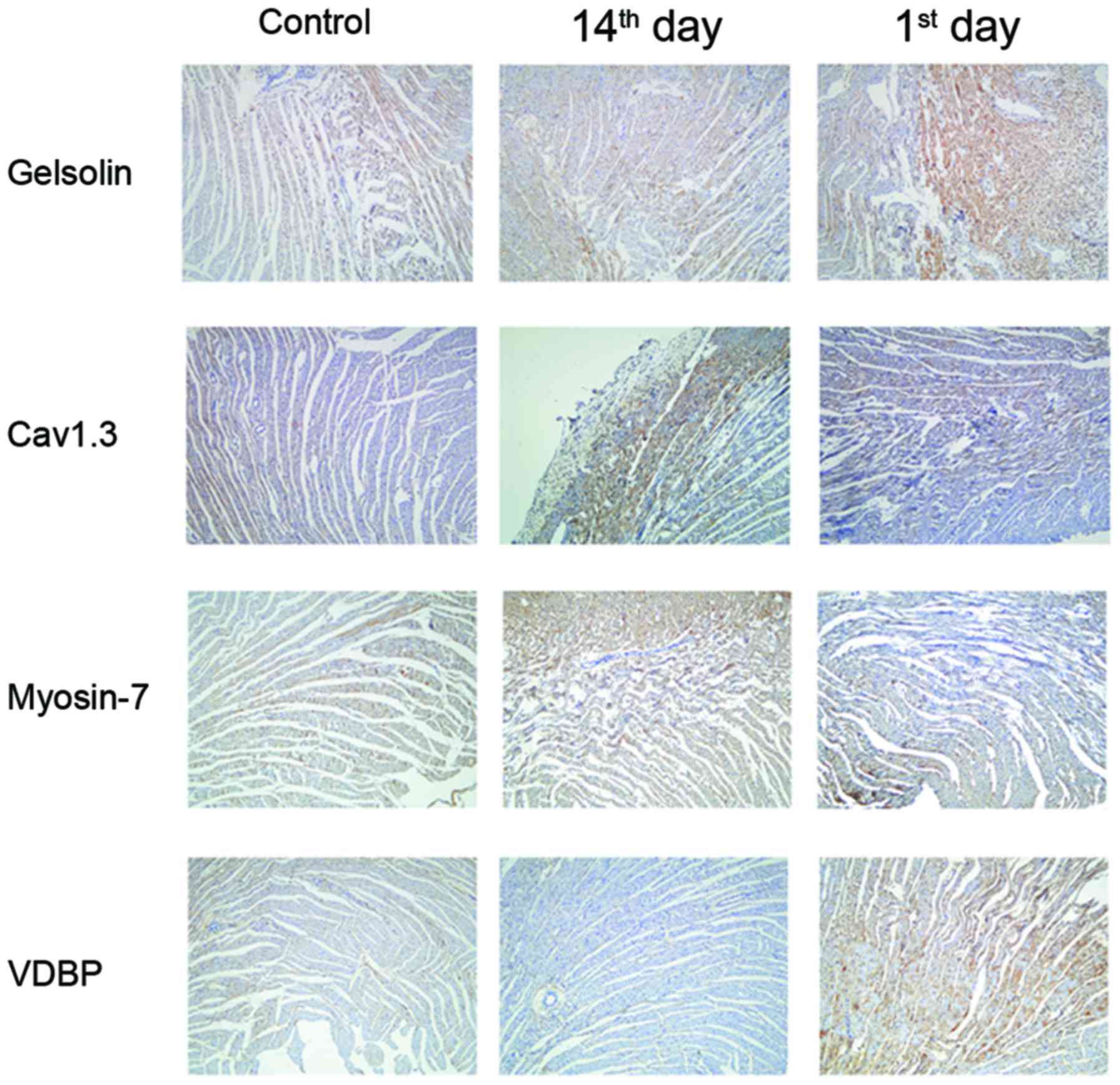 | Figure 3.Immunohistochemistry validations of
the alterations in the selected proteins identified by proteomic
analysis. Myocardial tissues from untreated (control), 1st day
post-MI and 14th day post-MI rats were embedded in conventional
paraffin and the expression of Myosin-7, VDBP, Gelsolin and Cav1.3
were detected by immunohistochemistry staining using the
appropriate antibodies (magnification, ×400). VDBP, vitamin D
binding protein; Cav1.3, voltage-dependent L-type calcium channel
subunit α1D; MI, myocardial infarction; control group, operation
with no occlusion (untreated); 1st day group, tissues were taken
from rats 1 day following surgical procedures; 14th day group,
tissues were taken from rats 14 days following surgical
procedures. |
Discussion
In the present study, quantitative proteomics based
on LTQ OrbiTrap technology was used to evaluate differential
protein expression during the development of heart failure post-MI
in myocardial tissues. According to the PANTHER classification, a
number of key enzymes in energy metabolism, including succinyl-CoA
ligase (ADP/GDP-forming) subunit α, isocitrate dehydrogenase
(nicotinamide adenine dinucleotide) subunit α, fumarate hydratase,
aconitate hydratase, enoyl-CoA hydratase, enoyl-CoA ∆ isomerase 1,
malate dehydrogenase, pyruvate dehydrogenase E1 component subunit
α, long-chain specific acyl-CoA dehydrogenase, very long-chain
specific acyl-CoA dehydrogenase, carnitine O-acetyltransferase,
carnitine O-palmitoyl transferase 2, creatine kinase S-type and
rhosphoglycerate kinase 1, were downregulated on the 1st and 14th
day following MI when compared with the control group, which
indicated that in the early and end stages of heart failure, the
process of glycolysis and fatty acid β-oxidation are significantly
decreased. Carnitine-palmitoyl transferase I (CPT1) is a
rate-limiting enzyme mediating the mitochondrial uptake of fatty
acid. It locates to the mitochondrial outer membrane to form fatty
acylcarnitine, which catalyzes the conversion of long-chain acyl
CoA to long-chain acylcarnitine. CPT2, located on the mitochondrial
inner membrane, converts acylcarnitine back to long-chain acyl CoA,
releasing carnitine (13).
Enoyl-CoA hydratase and acyl CoA dehydrogenase catalyze the
rate-limiting step in mitochondrial fatty acid β-oxidation
(14).
The majority of the previous, relevant studies
demonstrated a pronounced decrease in the protein levels of various
FFA β-oxidation enzymes in heart failure models, including human
dilated cardiomyopathy (6), canine
tachycardia induced heart failure (15,16),
rat aortic banding model (17) and
rat chronic coronary ligation model (18). The decrease of FFA β-oxidation
enzymes at the mRNA level was additionally reported in explanted
hearts (19), dogs with end-stage
tachycardia-induced heart failure (20) and dogs with
microembolization-induced heart failure (21). In addition, mRNA of the key enzymes
involved in fatty acid uptake and FFA β-oxidation were decreased to
a greater extent compared with their proteins and enzymatic
activities, in the end stages of heart failure (22). It has been reported that peroxisome
proliferator-activated receptor γ coactivator-1α (23), estrogen-related receptor α
(20), peroxisome
proliferator-activated receptor-α (21,24)
and retinoid X receptor α (25)
may regulate the mRNA expression of genes involved in the
mitochondrial fatty acid metabolism pathway in human, mouse, rat
and dog heart failure models (20,23).
Notably, in addition to carbohydrates and lipids, other
metabolites, including certain amino acids and aldehydes, may
influence energy status. Therefore, the defects in energy
metabolism and decrease in cardiac muscle contractions are
important factors in heart failure post-MI.
During systole, the opening of the L-type
Ca2+ channel (LTCC) triggers sarcoplasmic reticulum (SR)
Ca2+ release via the ryanodine-2 (RyR2) channels, and
the SR Ca2+ reuptake is conducted by the SR
Ca2+ ATPase (SERCA). Conversely, the sodium-calcium
exchanger (NCX) extrudes Ca2+ from the cardiomyocyte to
maintain a steady-state condition. The [Ca2+]i transient
is conducted by sarcolemmal Ca2+ channels, which results
in Ca2+ flux released from the SR via RyR2 channels.
This Ca2+-induced Ca2+ release (CICR) is
regulated by LTCC, which is localized to T-tubuli organization. The
amplitude of the [Ca2+]i transient is dependent on the
SR Ca2+ content. The diastolic Ca2+
concentration is regulated by the [Ca2+]i transient
decline, which is primarily due to SR Ca2+ reuptake
through SERCA and extrusion of Ca2+ via the NCX. Calcium
ion transport is abnormal in heart failure due to the increased
diastolic Ca2+ levels, reduced Ca2+
sensitivity of myofilaments and decreased Ca2+ reuptake,
which results in a diastolic Ca2+ overload (26–28).
In the present study, two proteins involved in
calcium ion transport, Gelsolin and Cav1.3, were upregulated in the
rat model of heart failure. Gelsolin is a widely-distributed
calcium-regulated actin-binding protein which mediates cell
motility, ion channel regulation, signal transduction (29) and multiple cytoskeletal remodeling
(30). In addition, gelsolin has
anti-apoptotic and pro-apoptotic functions (29). A previous study demonstrated with a
post-MI model, that it is highly expressed in animal and human
hearts and that it is associated with the progression of heart
failure following MI, suggesting that gelsolin may serve an
important role in cardiac remodeling post-MI (30). Li et al (31) used gelsolin-null mice and
wild-type littermates to clarify the role of gelsolin in heart
failure and the mechanism for gelsolin-stimulated apoptosis. The
group revealed that a deficiency in gelsolin protects the heart
post-MI. This protection is due in part, to the absence of
gelsolin-mediated apoptosis following MI. Gelsolin is cleaved by
caspase-3 between residues Asp352 and Gly353, and the N-terminal
gelsolin fragment may induce apoptosis. Therefore, gelsolin acts as
actin in a Ca2+-independent manner and it may promote
morphological alterations during apoptosis, indicating that
gelsolin facilitates MI-induced cardiomyocyte apoptosis. The
results of the present study are in agreement with the findings of
Li et al (31) as the
gelsolin protein was upregulated in the 1st and 14th day groups
post-MI.
Cav1.3 (α1D) subunit (D-LTCC) is a component of
LTCCs, which are vital for Ca2+ influx and are
responsible for Ca2+ entry into cardiomyocytes during
action potentials (32). Cav1.3
Ca2+ channels are highly expressed in cardiac pacemaking
tissues [sinoatrial (SA) and atrioventricular nodes], and serve an
important role in the spontaneous diastolic depolarization and pace
making activities within SA node cells (33). Zhang et al (33) used a Cav1.3-null mutant
mouse to illustrate that Cav1.3 Ca2+ channels were
expressed in mouse atrial, however not ventricular tissues. The
present study demonstrated that during the process of heart failure
post-MI, the expression of Cav1.3 was upregulated in the left
ventricular muscle. Therefore, it is possible that in the acute
phase of heart failure, the β-adrenaline receptor is activated to
trigger the opening of the LTCC and thus induces the influx of
Ca2+, which in turn activates Ca2+ released
from the SR.
Petrone et al (34) randomly selected 464 cases of heart
failure and 464 controls to examine the expression of VDBP and
revealed that there was no significant association between plasma
levels of VDBP and risk of heart failure. In the present study,
VDBP was significantly upregulated on the 1st day post-MI, then
expression gradually declined with the progression of the disease.
VDBP is an acute phase reactant (35) and its expression levels are
upregulated in the acute phase of inflammation (36). VDBP expression may be increased by
the pro-inflammatory cytokine interleukin-6 (37). In addition, this protein may
associate with inflammatory cell surfaces (37). VDBP is a multifunctional transport
protein for vitamin D metabolites (38), as vitamin D metabolism serves an
important role in the maintenance of calcium homeostasis (39). VDBP binds to fatty acids and actin,
preventing their polymerization, which may be detrimental in the
circulatory system. VDBP may exert immune functions by inhibiting
the production of 1,2,5(OH)2D3 in T-cells
(40). It has been reported that
~85–90% of 2,5(OH)D3 and
1,2,5(OH)2D3 in the circulation is bound to
VDBP (41,42). Haddad et al (43) reported that serum VDBP does not
decrease during vitamin D deficiency. Therefore, VDBP may protect
against vitamin D deficiency, and it is fundamental for vitamin D
dynamic homeostasis, as demonstrated in VDBP-null mice (39).
Vitamin D serves a major role in cardiac function by
suppressing the parathyroid hormone, inhibiting renin, upregulating
vascular endothelial growth factor and modulating calcium influx.
Previous studies in animals (44,45)
have revealed the association between vitamin D and the
cardiovascular system. Rats with experimentally induced vitamin D
deficiency have been observed to develop heart failure with
hypertension and cardiomegaly (46). Pilz et al (47) demonstrated the association between
vitamin D deficiency and left ventricular hypertrophy. The group
identified a negative correlation between 25-hydroxy vitamin D
levels and the impairment of left ventricular function in a
cross-sectional study of patients with coronary angiography
(47).
Myosin-7, also known as cardiac β-myosin heavy
chain, is a myocardial growth fetal gene that is associated with
ventricular systolic function and remodeling ventricular
pathological hypertrophy (48). In
ventricular pathological hypertrophy and heart failure, contractile
proteins and sarcomeres increase via the activation of myocardial
growth fetal genes, including Myosin-7 (49). Abraham et al (50) studied the genes associated with
phenotypic modulation in idiopathic dilated cardiomyopathy, and
observed an increase in the mRNA expression of Myosin-7. In other
models of ventricular pathological hypertrophy and heart failure, a
coordinated decrease in α-MyHC mRNA and increase in
Myosin-7 mRNA were associated with a reduction in shortening
velocity (12). In addition,
Myosin-7 has a lower myofibrillar Ca2+-stimulated ATPase
activity than the α-isoform, resulting in a reduction in shortening
velocity and myocardial systolic function (12). Therefore, it has become apparent
that intracellular Ca2+ homeostasis is vital for
myocardial contractility (7,9,10),
and the capacity of the cardiac muscle to produce contractile force
is dependent upon myofibrillar Ca2+-stimulated ATPase
activity (51). Therefore, the
relative amount of α- and β-myosin heavy chain isoforms determines
myosin ATPase activity (52).
Machackova et al (53)
investigated the association between the alterations in gene
expression and the heart failure phenotype. It was demonstrated
that the β-myosin heavy chain proportion increased from 6.3 to
77.7% of total myosin heavy chain, whereas the α-myosin heavy chain
proportion decreased from 93.7 to 22.3% in post-MI heart
failure.
In conclusion, the present proteomics study
demonstrated that the profile of proteins associated with metabolic
remodeling, calcium regulation and contractile function was altered
in the presence of post-MI heart failure. At different time points
(the 1st and 14th day post-MI), there are dynamic alterations in
differential protein expression. Myosin-7, Gelsolin, VDBP and
Cav1.3 were upregulated with the development of heart failure and,
to the best of our knowledge, this is the first proteomic analysis
of Myosin-7, Gelsolin, Cav1.3 and VDBP in a post-MI rat model using
LTQ OrbiTrap. Therefore, these results may provide a comprehensive
insight into the underlying mechanisms of the development of heart
failure.
Acknowledgements
The authors would like to express their appreciation
for the support provided by the cardiovascular internal medical
staff at Shandong Provincial Hospital Affiliated with Shandong
University (Jinan, China) during the present study. They would also
like to thank the staff at the Cancer Center of the Medical College
of Shandong University. The present study was supported by the
Medical Science and Technology Development Program of Shandong
Province (grant no. 2014WSA01015).
References
|
1
|
Hunt SA, Baker DW, Chin MH, Cinquegrani
MP, Feldman AM, Francis GS, Ganiats TG, Goldstein S, Gregoratos G,
Jessup ML, et al: ACC/AHA Guidelines for the evaluation and
management of chronic heart failure in the adult: Executive summary
a report of the American college of cardiology/american heart
association task force on practice guidelines (Committee to Revise
the 1995 Guidelines for the Evaluation and Management of Heart
Failure): Developed in collaboration with the international society
for heart and lung transplantation; endorsed by the heart failure
society of America. Circulation. 104:2996–3007. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Owan TE, Hodge DO, Herges RM, Jacobsen SJ,
Roger VL and Redfield MM: Trends in prevalence and outcome of heart
failure with preserved ejection fraction. N Engl J Med.
355:251–259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhatia RS, Tu JV, Lee DS, Austin PC, Fang
J, Haouzi A, Gong Y and Liu PP: Outcome of heart failure with
preserved ejection fraction in a population-based study. N Engl J
Med. 355:260–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gradman AH and Alfayoumi F: From left
ventricular hypertrophy to congestive heart failure: Management of
hypertensive heart disease. Prog Cardiovasc Dis. 48:326–341. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal
JS and Stanley WC: Myocardial fatty acid metabolism in health and
disease. Physiol Rev. 90:207–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paolisso G, Gambardella A, Galzerano D,
D'Amore A, Rubino P, Verza M, Teasuro P, Varricchio M and D'Onofrio
F: Total-body and myocardial substrate oxidation in congestive
heart failure. Metabolism. 43:174–179. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ross PL, Huang YN, Marchese JN, Williamson
B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et
al: Multiplexed protein quantitation in Saccharomyces cerevisiae
using amine-reactive isobaric tagging reagents. Mol Cell
Proteomics. 3:1154–1169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th.
National Academies Press; Washington, DC: 2011
|
|
9
|
Dixon IM, Lee SL and Dhalla NS:
Nitrendipine binding in congestive heart failure due to myocardial
infarction. Circ Res. 66:782–788. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang B, Li Y, Zhao L, Yuan S, Wang Z, Li B
and Chen Q: Stable isotope dimethyl labeling combined with LTQ mass
spectrometric detection, a quantitative proteomics technology used
in liver cancer research. Biomed Rep. 1:549–554. 2013.PubMed/NCBI
|
|
11
|
de Carvalho HF and Taboga SR: Fluorescence
and confocal laser scanning microscopy imaging of elastic fibers in
hematoxylin-eosin stained sections. Histochem Cell Biol.
106:587–592. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
VanBuren P, Harris DE, Alpert NR and
Warshaw DM: Cardiac V1 and V3 myosins differ in their hydrolytic
and mechanical activities in vitro. Circ Res. 77:439–444. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shibayama J, Yuzyuk TN, Cox J, Makaju A,
Miller M, Lichter J, Li H, Leavy JD, Franklin S and Zaitsev AV:
Metabolic remodeling in moderate synchronous versus dyssynchronous
pacing-induced heart failure: Integrated metabolomics and
proteomics study. PLoS One. 10:e01189742015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Andresen BS, Olpin S, Poorthuis BJ,
Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A,
Pourfarzam M, Bartlett K, et al: Clear correlation of genotype with
disease phenotype in very-long-chain acyl-CoA dehydrogenase
deficiency. Am J Hum Genet. 64:479–494. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Osorio JC, Stanley WC, Linke A, Castellari
M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD and Recchia FA:
Impaired myocardial fatty acid oxidation and reduced protein
expression of retinoid X receptor-alpha in pacing-induced heart
failure. Circulation. 106:606–612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nikolaidis LA, Sturzu A, Stolarski C,
Elahi D, Shen YT and Shannon RP: The development of myocardial
insulin resistance in conscious dogs with advanced dilated
cardiomyopathy. Cardiovasc Res. 61:297–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Allard MF, Schonekess BO, Henning SL,
English DR and Lopaschuk GD: Contribution of oxidative metabolism
and glycolysis to ATP production in hypertrophied hearts. Am J
Physiol. 267:H742–H750. 1994.PubMed/NCBI
|
|
18
|
Heather LC, Cole MA, Lygate CA, Evans RD,
Stuckey DJ, Murray AJ, Neubauer S and Clarke K: Fatty acid
transporter levels and palmitate oxidation rate correlate with
ejection fraction in the infarcted rat heart. Cardiovasc Res.
72:430–437. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sack MN, Rader TA, Park S, Bastin J,
McCune SA and Kelly DP: Fatty acid oxidation enzyme gene expression
is downregulated in the failing heart. Circulation. 94:2837–2842.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lei B, Lionetti V, Young ME, Chandler MP,
d'Agostino C, Kang E, Altarejos M, Matsuo K, Hintze TH, Stanley WC
and Recchia FA: Paradoxical downregulation of the glucose oxidation
pathway despite enhanced flux in severe heart failure. J Mol Cell
Cardiol. 36:567–576. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosca MG, Vazquez EJ, Kerner J, Parland W,
Chandler MP, Stanley W, Sabbah HN and Hoppel CL: Cardiac
mitochondria in heart failure: Decrease in respirasomes and
oxidative phosphorylation. Cardiovasc Res. 80:30–39. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sihag S, Cresci S, Li AY, Sucharov CC and
Lehman JJ: PGC-1alpha and ERRalpha target gene downregulation is a
signature of the failing human heart. J Mol Cell Cardiol.
46:201–212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barger PM, Brandt JM, Leone TC, Weinheimer
CJ and Kelly DP: Deactivation of peroxisome proliferator-activated
receptor-alpha during cardiac hypertrophic growth. J Clin Invest.
105:1723–1730. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morgan EE, Chandler MP, Young ME,
McElfresh TA, Kung TA, Rennison JH, Tserng KY, Hoit BD and Stanley
WC: Dissociation between gene and protein expression of metabolic
enzymes in a rodent model of heart failure. Eur J Heart Fail.
8:687–693. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bers DM: Altered cardiac myocyte Ca
regulation in heart failure. Physiology (Bethesda). 21:380–387.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hobai IA and O'Rourke B: Decreased
sarcoplasmic reticulum calcium content is responsible for defective
excitation-contraction coupling in canine heart failure.
Circulation. 103:1577–1584. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Piacentino V III, Weber CR, Chen X,
Weisser-Thomas J, Margulies KB, Bers DM and Houser SR: Cellular
basis of abnormal calcium transients of failing human ventricular
myocytes. Circ Res. 92:651–658. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanaka M, Müllauer L, Ogiso Y, Fujita H,
Moriya S, Furuuchi K, Harabayashi T, Shinohara N, Koyanagi T and
Kuzumaki N: Gelsolin: A candidate for suppressor of human bladder
cancer. Cancer Res. 55:3228–3232. 1995.PubMed/NCBI
|
|
29
|
McGough AM, Staiger CJ, Min JK and
Simonetti KD: The gelsolin family of actin regulatory proteins:
Modular structures, versatile functions. FEBS Lett. 552:75–81.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nishio R and Matsumori A: Gelsolin and
cardiac myocyte apoptosis: A new target in the treatment of
postinfarction remodeling. Circ Res. 104:829–831. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li GH, Shi Y, Chen Y, Sun M, Sader S,
Maekawa Y, Arab S, Dawood F, Chen M, de Couto G, et al: Gelsolin
regulates cardiac remodeling after myocardial infarction through
DNase I-mediated apoptosis. Circ Res. 104:896–904. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja
D, Namkung Y, Shin HS and Chiamvimonvat N: Functional roles of
Ca(v)1.3 (alpha(1D)) calcium channel in sinoatrial nodes: Insight
gained using gene-targeted null mutant mice. Circ Res. 90:981–987.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Z, He Y, Tuteja D, Xu D, Timofeyev
V, Zhang Q, Glatter KA, Xu Y, Shin HS, Low R and Chiamvimonvat N:
Functional roles of Cav1.3(alpha1D) calcium channels in atria:
Insights gained from gene-targeted null mutant mice. Circulation.
112:1936–1944. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Petrone AB, Weir NL, Steffen BT, Tsai MY,
Gaziano JM and Djousse L: Plasma vitamin D-binding protein and risk
of heart failure in male physicians. Am J Cardiol. 112:827–830.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Guha C, Osawa M, Werner PA, Galbraith RM
and Paddock GV: Regulation of human Gc (vitamin D-binding) protein
levels: Hormonal and cytokine control of gene expression in vitro.
Hepatology. 21:1675–1681. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Petrini M, Galbraith RM, Werner PA,
Emerson DL and Arnaud P: Gc (vitamin D binding protein) binds to
cytoplasm of all human lymphocytes and is expressed on B-cell
membranes. Clin Immunol Immunopathol. 31:282–295. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gomme PT and Bertolini J: Therapeutic
potential of vitamin D-binding protein. Trends Biotechnol.
22:340–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chun RF: New perspectives on the vitamin D
binding protein. Cell Biochem Funct. 30:445–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Christakos S, Ajibade DV, Dhawan P,
Fechner AJ and Mady LJ: Vitamin D: Metabolism. Endocrinol Metab
Clin North Am. 39:243–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kongsbak M, von Essen MR, Levring TB,
Schjerling P, Woetmann A, Odum N, Bonefeld CM and Geisler C:
Vitamin D-binding protein controls T cell responses to vitamin D.
BMC Immunol. 15:352014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Safadi FF, Thornton P, Magiera H, Hollis
BW, Gentile M, Haddad JG, Liebhaber SA and Cooke NE: Osteopathy and
resistance to vitamin D toxicity in mice null for vitamin D binding
protein. J Clin Invest. 103:239–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bikle DD, Gee E, Halloran B, Kowalski MA,
Ryzen E and Haddad JG: Assessment of the free fraction of
25-hydroxyvitamin D in serum and its regulation by albumin and the
vitamin D-binding protein. J Clin Endocrinol Metab. 63:954–959.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haddad JG, Fraser DR and Lawson DE:
Vitamin D plasma binding protein. Turnover and fate in the rabbit.
J Clin Invest. 67:1550–1560. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Weishaar RE and Simpson RU: Vitamin D3 and
cardiovascular function in rats. J Clin Invest. 79:1706–1712. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Weishaar RE, Kim SN, Saunders DE and
Simpson RU: Involvement of vitamin D3 with cardiovascular function.
III. Effects on physical and morphological properties. Am J
Physiol. 258:E134–E142. 1990.PubMed/NCBI
|
|
46
|
Dorsch MP, Nemerovski CW, Ellingrod VL,
Cowger JA, Dyke DB, Koelling TM, Wu AH, Aaronson KD, Simpson RU and
Bleske BE: Vitamin D receptor genetics on extracellular matrix
biomarkers and hemodynamics in systolic heart failure. J Cardiovasc
Pharmacol Ther. 19:439–445. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pilz S, Mürz W, Wellnitz B, Seelhorst U,
Fahrleitner-Pammer A, Dimai HP, Boehm BO and Dobnig H: Association
of vitamin D deficiency with heart failure and sudden cardiac death
in a large cross-sectional study of patients referred for coronary
angiography. J Clin Endocrinol Metab. 93:3927–3935. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nadal-Ginard B and Mahdavi V: Molecular
basis of cardiac performance. Plasticity of the myocardium
generated through protein isoform switches. J Clin Invest.
84:1693–1700. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nakao K, Minobe W, Roden R, Bristow MR and
Leinwand LA: Myosin heavy chain gene expression in human heart
failure. J Clin Invest. 100:2362–2370. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Abraham WT, Gilbert EM, Lowes BD, Minobe
WA, Larrabee P, Roden RL, Dutcher D, Sederberg J, Lindenfeld JA,
Wolfel EE, et al: Coordinate changes in Myosin heavy chain isoform
gene expression are selectively associated with alterations in
dilated cardiomyopathy phenotype. Mol Med. 8:750–760.
2002.PubMed/NCBI
|
|
51
|
Machackova J, Barta J and Dhalla NS:
Myofibrillar remodeling in cardiac hypertrophy, heart failure and
cardiomyopathies. Can J Cardiol. 22:953–968. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dhalla NS, Saini-Chohan HK,
Rodriguez-Leyva D, Elimban V, Dent MR and Tappia PS: Subcellular
remodelling may induce cardiac dysfunction in congestive heart
failure. Cardiovasc Res. 81:429–438. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Machackova J, Sanganalmath SK, Elimban V
and Dhalla NS: β-adrenergic blockade attenuates cardiac dysfunction
and myofibrillar remodelling in congestive heart failure. J Cell
Mol Med. 15:545–554. 2011. View Article : Google Scholar : PubMed/NCBI
|