Introduction
The liver is a vital organ involved in regulating
many important functions, including metabolism, secretion and
storage. It serves a key role in regulating many physiological
processes (1). Furthermore, it is
involved in detoxification and biotransformation of drugs and
xenobiotics; therefore, has demonstrated increased susceptibility
to toxicity from these agents (2).
Liver diseases have become a worldwide problem as a result of
extremely poor prognoses and high mortality rates attributed to the
lack of effective prevention methods or therapeutic drugs (3). As such, attempts are perpetually
being made to discover novel treatments for liver diseases. The
drug discovery process has paid great attention to the
investigation of the efficacy of plant-based drugs used in
traditional medicine, as they are cheaper and have fewer side
effects. Natural products extracted from medicinal plants are
considered to be an effective and safe alternative to conventional
treatments for hepatotoxicity. Therefore, present study aimed to
investigate the hepatoprotective activity of Pien Tze Huang Gan Bao
(PZH-GB) against carbon tetrachloride (CCl4)-induced
hepatotoxicity in rats. PZH-GB, a classical traditional Chinese
medicine, has been used as a protective remedy for thousands of
years across China and Southeast Asian countries. PZH-GB has
demonstrated therapeutic effects against liver injury induced by
excessive alcohol and smoking. To date, however, there are no
studies available regarding the protective effect of PZH-GB in
response to chemical-induced liver injury.
The present study investigated the protective effect
of PZH-GB against CCl4-induced hepatotoxicity, and aimed
to determine the mechanisms by which it may confer its protection
on the rat liver against CCl4-induced chemical damage.
The effect of PZH-GB on liver injury was compared to that of
silymarin treatment, a substance used clinically in Europe and Asia
for the treatment of liver diseases. For evaluation of the
hepatoprotective mechanisms of PZH-GB, liver oxidative damage and
anti-oxidant defense potential, proinflammatory mediators including
inducible nitric oxide synthase (iNOS) and cyclooxygenase-2
(COX-2), and apoptosis-associated factors were determined.
Materials and methods
Reagents
PZH-GB was obtained from and authenticated by the
sole manufacturer Zhangzhou Pien Tze Huang Pharmaceutical Co., Ltd.
(Chinese FDA approval no. HPK-08411; Zhangzhou, China). A lactate
dehydrogenase assay kit (cat no. 2P56-21) was purchased from Abbott
Pharmaceutical Co., Ltd. (Lake Bluff, IL, USA). TRIzol reagents
were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). A PrimeScript™ RT reagent kit with gDNA Eraser was purchased
from Takara Bio, Inc. (Otsu, Japan). Caspase-3 (cat no. KHZ0022)
and −9 (cat no. KHZ0102) colorimetric protease assays were
purchased from Invitrogen; Thermo Fisher Scientific, Inc. Rabbit
monoclonal antibodies for COX-2 (cat no. 12282S; 1:1,000 dilution)
and GAPDH (cat no. 5174S; 1:1,000 dilution) were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA), and rabbit
monoclonal antibodies for iNOS (cat no. ab3523), cytochrome P450
family 2 subfamily E member 1 (CYP2E1; cat no. ab151544), p53 (cat
no. ab109396), B-cell lymphoma 2 (Bcl-2; cat no. ab59348) and Bcl-2
X-associated protein (Bax; cat no. ab32503) were obtained from
Abcam (1:1,000 dilution; Cambridge, MA, USA). Horseradish
peroxidase-conjugated secondary antibodies (cat no. ab205718) were
purchased from Abcam (1:2,000 dilution). CCl4 was
purchased from Shanghai Ling Feng Chemical Reagent Co., Ltd.
(Shanghai, China).
Animals
Male Sprague-Dawley rats (age, 8 weeks; weight,
180–200 g; n=60; Slike Co., Ltd., Shanghai, China), were housed
five per cage in an environmentally-controlled room at a
temperature of 22±1°C in a relative humidity 40–60%. Animals were
housed in an air ventilation setting of 12–18 times/h and a 12-h
light/dark cycle of 150–300 lux conditions, with feed and water
provided ad libitum for one week before the start of the
experiments. The animal studies were approved by the Fujian
Institute of Traditional Chinese Medicine Animal Ethics Committee
(Fuzhou, China). The experimental procedures were carried out in
accordance with the Guidelines for Animal Experimentation of Fujian
University of Traditional Chinese Medicine (Fuzhou, China).
CCl4-induced hepatotoxicity
and experimental design
The rats were divided into six groups according to
the following conditions: Group 1: control group (NO); group 2:
CCl4 model group (vehicle); group 3
(SM+CCl4): silymarin treated CCl4 group
(pre-treated with silymarin, 50 mg/kg), a positive control; group 4
(LoGB+CCl4): low dose PZH-GB treatment CCl4
group (pre-treated with PZH-GB, 150 mg/kg); group 5
(MeGB+CCl4): medium dose PZH-GB treatment
CCl4 group (pre-treated with PZH-GB, 300 mg/kg); and
group 6 (HiGB+CCl4): high dose PZH-GB treatment
CCl4 group (pre-treated with PZH-GB, 600 mg/kg). Each
group contained 10 rats. Rats from groups 3 to 6 were administered
an oral dose of either silymarin or PZH-GB while the rats from
control and CCl4 model groups were administered an oral
dose of PBS for 7 days before initial CCl4 exposure. On
the last day of pre-treatment, rats in groups 2 to 6 were
intraperitoneally (i.p.) injected with CCl4 at a dose of
2.0 ml/kg, provided as a 50% corn oil solution, while group 1
received 2.0 ml/kg of corn oil. A total of 24 h after
CCl4 administration, the animals were anesthetized (40
mg/kg pentobarbital i.p.; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), sacrificed by cervical dislocation, and subjected to
laparotomy, which involved opening the abdominal cavity and
exposing the abdominal aorta. Blood (15 ml) was collected by aorta
abdominalis into non-heparinized tubes and centrifuged at 980 × g
at 4°C for 10 min to obtain serum for biochemical tests. The livers
were quickly excised and washed, and part of each liver was cut and
fixed in formaldehyde saline (4%) solution for histological
analysis; the remaining liver was snap frozen in liquid nitrogen
and stored at −70°C until required for molecular analysis.
Measurement of LDH
The quantification of LDH5 was achieved by using the
pyruvate to lactate kinetic method, a two-point colometric method
for determination of LDH.
Pyruvate+NADH+H+⟷LDHL–Lactate+NAD+
Histopathology
Liver samples were fixed in 10% buffered formalin
for at least 48 h, processed and embedded in paraffin. Next, the
paraffin sections were prepared using an automatic tissue processor
and cut into 5-µm thick sections by a rotary microtome. The
sections were stained with hematoxylin and eosin (H&E) and
mounted on slides for examination of histopathological changes. The
results were recorded as photomicrographs using a Leica DMRB/E
light microscope (Leica Microsystems GmbH, Wetzlar, Germany). A
semi-quantitative scoring system was used to assess the severity of
the hepatic steatosis and inflammatory cell infiltration in 10
microscopic fields. In brief, the following criteria were used for
scoring hepatic steatosis: grade 0 (−), no inflammation or
ballooning degeneration; grade 1 (+), inflammation or ballooning
degeneration occupying 33% of the hepatic parenchyma; grade 2 (++),
inflammation or ballooning degeneration occupying 33–66% of the
hepatic parenchyma; grade 3 (+++), inflammation or ballooning
degeneration occupying >66% of the hepatic parenchyma.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from the remaining liver samples were
isolated using TRIzol reagent. The isolation was performed
according to the manufacturer's protocol, and RNA was subsequently
quantified by measuring the absorbance of the preparation at a
wavelength of 260 nm. cDNA synthesis was performed using the
PrimeScript™ RT reagent kit according to the manufacturer's
protocol. Total RNA (1.0 µg from each sample) was added to a
mixture of 4.0 µl 5X PrimeScript buffer, 1.0 µl PrimeScript RT
enzyme mix I, 1.0 µl Oligo dT primer (50 µM), 1.0 µl random 6 mers
(100 µM), and RNase-free water was added to a final volume of 20
µl. The final reaction mixture was kept at 25°C for 10 min, heated
to 37°C for 15 min, heated to 85°C for 5 sec, and then cooled to
4°C.
cDNA from the above preparation was subjected to PCR
amplification using SYBR Premix Ex Taq I in an ABI 7500 Fast
instrument. mRNA expression values were determined using the
2−∆∆Cq method (4).
GAPDH served as an internal reference control. All qPCR reactions
were conducted in triplicate. Primer sequences were as follows:
Forward, GCATCCTGTCCCCATCACCA and reverse, CCCAGCAACTACCAACCCATTC
for p52; forward, GAACTGTATCCCGCCCTGCTGGT and reverse,
CTTGCGTTGATGGTGGCTGTCTT for COX-2; forward, CCTCCTTGTTCAACTCACCTTCG
and reverse, ACCTCTGCCTGTGCGTCTCTTC for iNOS; forward,
CCTTTTGACCCCACATTTCTGAT and reverse, ATGGCTTCCAGGTAGGTATCGTAG for
CYP2E1; forward, GTGTATTTCACGGGACCTGGCT and reverse,
GATGCTCTTGAAGGTCTCGTAGGT for nuclear factor (NF)-κB; forward,
GCTGATGGCAACTTCAACTGGG and reverse, TTCTTCCAGATGGTGAGCGAGG for Bax;
forward, TACCGTCGTGACTTCGCAGAGAT and reverse,
AGGAGAAATCAAACAGAGGTCGC for Bcl-2; forward, CGGTCAGGTCATCACTATCGGC
and reverse, GTGTTGGCATAGAGGTCTTTACGG for GAPDH.
Western blot analysis
Livers (n=4/group) were homogenized and total
protein lysate was prepared in Radioimmunoprecipitation Assay Lysis
and Extraction Buffer (Thermo Fisher Scientific, Inc.) containing
protease and phosphatase inhibitor cocktails, which was followed by
centrifugation at 12,000 × g for 15 min at 4°C. Protein
concentrations were determined with Bicinchoninic Acid protein
assay kit (Pierce; Thermo Fisher Scientific, Inc.). Protein samples
(50 µg) were separated by 10% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Membranes were blocked with TBS overnight at 4°C followed by
incubation with primary rabbit monoclonal antibodies against COX-2,
iNOS, CYP2E1, p53, Bax, Bcl-2 and GAPDH for 2 h at room
temperature. After three washes in TBS with 0.1% Tween-20 (TBST),
membranes were each probed with an anti-rabbit horseradish
peroxidase-conjugated secondary antibody (1:2,000) for 1 h at room
temperature, and the membranes were washed again in TBST followed
by enhanced chemiluminescence detection using SuperSignal™ West
Pico Chemiluminescent Substrate (Thermo Fisher Scientific,
Inc.).
Analysis of caspase-3 and −9
activation
The activities of caspase-3 and −9 were determined
by caspase-3 and −9 colorimetric activation kits following the
manufacturer's protocol. Briefly, six liver tissues from each group
were homogenized and lysed with lysis buffer for 30 min on ice. The
lysed tissues were centrifuged at 16,000 × g for 10 min at 4°C. The
protein concentration of the clarified supernatant was determined
and 100 µg protein was incubated with 50 µl colorimetric
tetrapeptides, Asp-Glue-Val-Asp (DEAD)-p-nitroaniline (pNA,
specific substrate of caspase-3) or Leu-Glue-His-Asp (LEHD)-pNA
(specific substrate of caspase-9) at 37°C in the dark for 2 h.
Samples were read at 405 nm on an ELISA plate reader (model ELX800;
BioTek, Winooski, VT, USA). The data were normalized to caspase
activity in control group liver tissue and are presented as a fold
of control.
High performance liquid chromatography
(HPLC)
Samples were analyzed on an Agilent 1200 HPLC system
(Agilent Technologies, Santa Clara, CA, USA) using a Welch Ultimate
XB-C18 (250×4.60 mm; 5 µm). The absorbance was measured at 274 nm.
The mobile phase consisted of methanol and 0.1% phosphoric acid
(45:55) at a flow rate of 0.9 ml/min with an injection volume of 5
µl. The column temperature was 20°C. Baicalin (Sigma-Aldrich; Merck
KGaA) served as a positive control.
Statistical analysis
All data are expressed as the means ± standard
deviation of three independent experiments. Data were analyzed
using SPSS version 11.5 (SPSS, Inc., Chicago, IL, USA). Statistical
analysis was performed by one-way analysis of variance followed by
a post hoc Fisher's least significant difference test, and
qualitative data was analyzed by Chi-squared test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of PZH-GB on serum LDH5
The effects of PZH-GB on CCl4-induced
hepatotoxicity in rats was evaluated by changes in serum LDH5. The
activities of LDH5 in control and all treated groups are presented
in Fig. 1. The data demonstrated
increased levels of serum LDH5 in the CCl4-treated
animals compared with the control group. This effect was reversed
in the animal groups that were given PZH-GB or silymarin
pretreatment. Treatment with 150, 300 and 600 mg/kg PZH-GB, and
silymarin at a dose of 50 mg/kg, exhibited a reduction of 19.29,
20.46, 27.97 and 56.80% on LDH5 levels, respectively.
 | Figure 1.Effects of pre-treatment of PZH-GB
and silymarin on serum LDH in CCl4-intoxicated rats.
Animals were pretreated with PZH-GB (150, 300 and 600 mg/kg),
silymarin (50 mg/kg) or vehicle. Data are expressed as the mean ±
standard deviation (n=10/group). #P<0.05 vs. control
group; *P<0.05 vs. CCl4-exposed group. PZH-GB, Pien
Tze Huang Gan Bao; LDH, lactate dehydrogenase; CCl4,
carbon tetrachloride; GB-L, GB-M, GB-H, PZH-GB low, medium and
high-dose treatment groups, respectively. |
Effect of GB on histopathological
changes
Histological analysis with H&E revealed advanced
centrilobular necrosis, broad infiltration of inflammatory cells,
ballooning degeneration and the loss of cellular boundaries in
liver sections of CCl4 model rats. Both PZH-GB and
silymarin groups exhibited an apparent reversal of these
histopathological effects, especially PZH-GB at the 600 mg/kg dose,
as presented in Fig. 2 and
Table I.
 | Table I.Quantification analysis for
histopathologic changes. |
Table I.
Quantification analysis for
histopathologic changes.
|
|
| Liver injury |
|---|
|
|
|
|
|---|
| Group | Rats (n) | − | + | ++ | +++ |
|---|
| Control | 10 | 10 | 0 | 0 | 0 |
| Model | 10 | 0 | 0 | 2 | 8a |
| Silymarin | 10 | 1 | 3 | 4 | 2b |
| GB-150 mg/kg | 10 | 0 | 4 | 3 | 3b |
| GB-300 mg/kg | 10 | 0 | 4 | 4 | 2b |
| GB-600 mg/kg | 10 | 2 | 4 | 2 | 2b |
Effect of PZH-GB on apoptosis
Cell apoptosis in liver tissues was determined via
immunohistochemical staining for terminal deoxynucleotidyl
transferase dUTP nick end labeling (TUNEL). As presented in
Fig. 3, the percentage of
TUNEL-positive cells in the CCl4 model group was
86.33±15.62%, which was increased significantly compared with the
control group (11.83±2.32%). Alternatively, the percentage of
TUNEL-positive cells in the silymarin and low-, medium- and
high-PZH-GB-pretreated groups was 66.67±18.19, 37.17±11.50,
41.00±3.03 and 36.33±14.46%, respectively. This reflected a
significant decrease compared with the CCl4 untreated
model group.
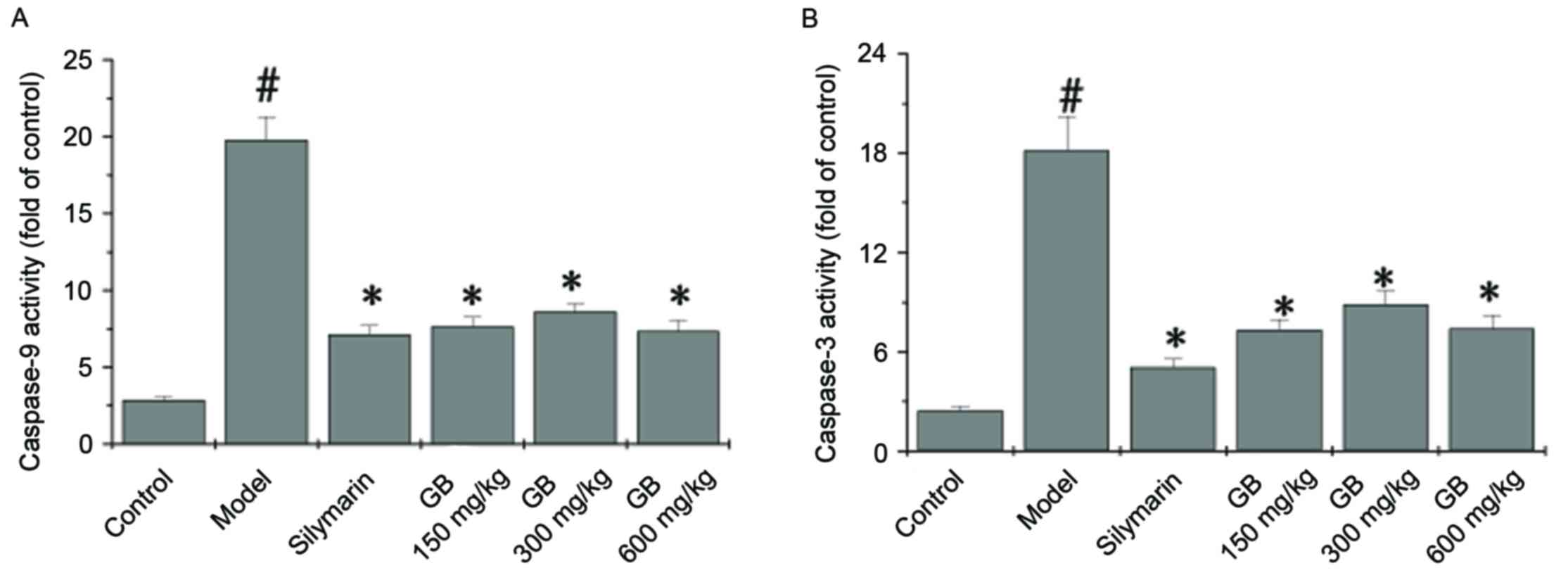
To identify the downstream effectors involved in the
CCl4 apoptotic signaling pathway, the activation of
caspase-9 and caspase-3 was examined by a colorimetric assay using
specific chromophores. Activation of caspase-9 (Fig. 4A) and caspase-3 (Fig. 4B) in the liver tissue was
significantly induced by CCl4. Both PZH-GB and silymarin
treatment inhibited the activation of caspase-9 and caspase-3
(P<0.05 vs. model group).
To further investigate the mechanism by which PZH-GB
confers its anti-apoptotic activity, RT-qPCR and western blot
analyses were performed to determine the mRNA and protein
expression levels of p53, Bcl-2 and Bax in liver tissue. As
presented in Fig. 5A, the mRNA
expression levels of p53 and Bax were upregulated in
CCl4-induced liver, whereas both PZH-GB and silymarin
treatment reversed the expression of p53, Bcl-2 and Bax, albeit in
slightly different patterns. The western blotting results were
consistent with this (Fig. 5B).
Collectively, these results suggested that CCl4 induced
apoptosis of liver cells, whereas PZH-GB and silymarin inhibited
apoptosis by decreasing the expression of the Bax/Bcl-2 ratio and
p53.
 | Figure 5.Effect of Pien Tze Huang Gan Bao on
the expression of p53, Bax and Bcl-2 in carbon
tetrachloride-exposed rats. (A) mRNA and (B) protein expression
levels of p53, Bcl-2 and Bax, as determined by reverse
transcription-quantitative polymerase chain reaction and western
blotting, respectively. GAPDH served as the internal control. Data
are presented as the mean ± standard deviation (n=3/group).
#P<0.05 vs. control group; *P<0.05 vs. model
group. Bax, B cell lymphoma-2 X-associated protein; Bcl-2, B-cell
lymphoma 2; GB, Pien Tze Huang Gan Bao; L, low-dose; M,
medium-dose; H, high-dose. |
Effect of PZH-GB on oxidative stress
and inflammation
The effect of PZH-GB on iNOS, COX-2 and CYP2E1
expression was examined by RT-qPCR and western blotting.
CCl4 upregulated the mRNA (Fig. 6A) and protein (Fig. 6B) expression levels of iNOS, COX-2
and CYP2E1; however, pre-treatment with PZH-GB and silymarin
ameliorated this effect for CYP2E1 and iNOS. Silymarin had no
effect on COX-2 expression levels; however, 600 mg/kg PZH-GB
significantly reduced them.
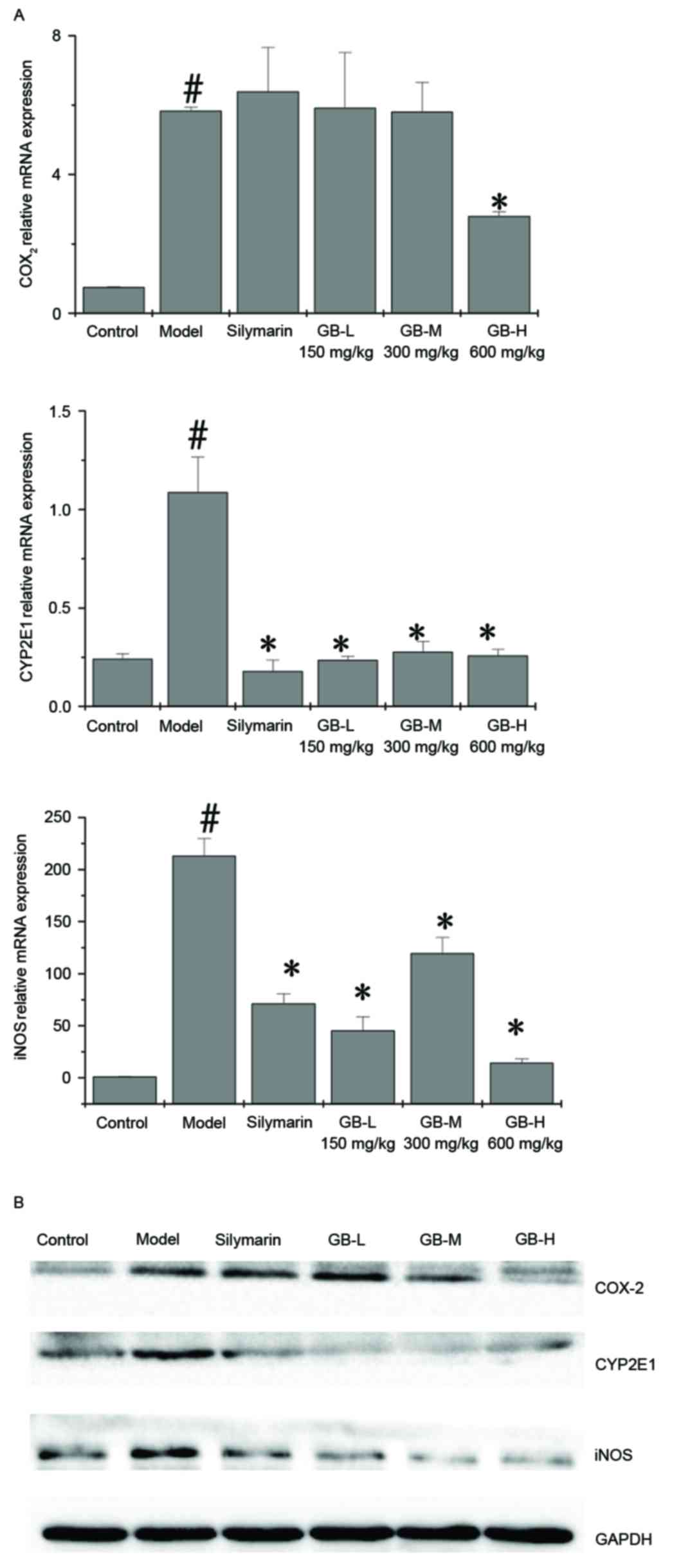 | Figure 6.Effect of Pien Tze Huang Gan Bao on
the expression of COX-2, CYP2E1 and iNOS in carbon
tetrachloride-exposed rats. (A) mRNA and (B) protein expression
levels of COX-2, CYP2E1 and iNOS, as determined by reverse
transcription-quantitative polymerase chain reaction and western
blotting, respectively. GAPDH served as the internal control. Data
are presented as the mean ± standard deviation (n=3/group).
#P<0.05 vs. control group; *P<0.05 vs. model
group. COX-2, cyclooxygenase 2; iNOS, inducible nitric oxide;
CYP2E1, cytochrome P450 family 2 subfamily E member 1; GB, Pien Tze
Huang Gan Bao; L, low-dose; M, medium-dose; H, high-dose. |
HPLC
Based on the fingerprint as presented in Fig. 7, an optimum and easily controlled
procedure for preparing PZH-GB was established, as mentioned above.
The control sample was composed of baicalin.
Discussion
CCl4, a well-known compound for producing
chemical hepatic injury, is metabolized by the hepatic microsomal
cytochrome P450 2E1 system (5,6).
During metabolism, an unstable trichloromethyl (CCl3) free radical
is formed and rapidly converted to trichloromethyl peroxide
(Cl3COO−) (3,7). This results in a large amount of
reactive oxygen species (ROS) generation, which in turn may
increase oxidative stress (8).
Previous studies have revealed that the ROS accumulation can
overwhelm the intrinsic antioxidant defense system and trigger
oxidative stress representative of significant cytotoxic effects in
liver and other tissues (9).
Mitochondria are the main organelles involved in
intracellular oxidation reactions and are also the main location
for ROS production (10). During
the CCl4 metabolism process, nicotinamide adenine
dinucleotide (NAD) serves as a co-enzyme in the presence of large
free radical consumption, resulting in a reduction in the ratio of
intracellular NAD/NADH. In turn, this event may promote
mitochondrial respiratory chain circulation and generate excess ROS
(11). When mitochondrial ROS
generation occurs beyond the antioxidant capacity of scavengers,
ROS can damage the mitochondria and overflow into the cytoplasm,
leading to cellular damage (12).
Mitochondrial damage can then trigger the release of caspase-3 into
the cytoplasm, which in turn signals the onset of apoptosis. A
growing number of studies suggest that apoptosis serves an
important role in liver disease etiology (13,14).
The Bcl-2 family of proteins (including the anti-apoptotic protein
Bcl-2 and the pro-apoptotic protein Bax) serve a key regulatory
role in the apoptotic process. The ratio of Bax/Bcl-2 represents
the degree of apoptosis to a certain extent (15).
In the current study, evidence that pre-treatment
with PZH-GB had a protective effect against CCl4-induced
hepatic apoptosis in rats was provided. It was demonstrated that
PZH-GB attenuates chemical-induced liver injury, and that
histopathological changes caused by CCl4, such as
cellular ballooning, are clearly ameliorated by PZH-GB. This
finding was consistent with observed changes of liver enzyme
levels, such as LDH5 (a LDH isoenzyme which is synthesized by the
liver). LDH is a key enzyme marker associated with liver injury.
Following liver injury, LDH levels were elevated. In the present
study, increased LDH was induced by CCl4. PZH-GB
treatment could decrease the LDH level, but the LDH level of GB-H
group was lower than that of the control group. This result may
indicate that GB-H treatment can excessively reduce the stress
response induced by CCl4. The anti-apoptotic effect of
PZH-GB was further evidenced by the demonstration that PZH-GB
decreased the number of TUNEL-positive hepatic cells, and reduced
caspase-3 activation. Steatosis and ballooning of hepatocytes are
the earliest, most frequent and most striking pathological changes
observed in CCl4-induced liver injury (16,17).
The present study confirmed this pathological change by H&E
staining, and determined that PZH-GB reversed this change.
Apoptosis of hepatocytes is a critical mechanism of
liver injury (18). To date, the
TUNEL assay had been used as a marker of apoptosis. In the present
study, TUNEL-positive cells in the CCl4 group were
significantly increased compared with the control group. More
importantly, PZH-GB reduced the number of TUNEL-labeled cells.
However, the TUNEL assay is not a specific marker of
apoptosis-multiple factors activate apoptotic cascade proteins.
Previous evidence supports the role of caspase-3 as an executioner
of apoptosis, and it is understood that caspase-3 serves a dominant
role after hepatic injury (19).
In addition, the Bcl-2 family serves an important role in
hepatocyte intrinsic apoptosis, particularly Bcl-2 and Bax. The
Bcl-2/Bax ratio is considered to determine cell fate after an
apoptotic stimulus (20,21). The present study demonstrated that
the caspase-9 and −3 activity in the group treated with PZH-GB was
remarkably decreased compared with the untreated
CCl4-induced group. Furthermore, the PZH-GB treated
group exemplified a restored Bcl-2/Bax ratio. These data
collectively suggested that PZH-GB alleviated
CCl4-induced hepatic injury by suppressing apoptosis and
the associated members of the mitochondrial signaling pathway which
confer this apoptotic stimulus.
It has been demonstrated that hepatocyte apoptosis
can induce liver injury, and p53 is accumulated in hepatocytes in
several liver diseases (22–25).
It has been reported that CCl4 upregulates the p53
expression (26). Therefore,
transcriptional mechanisms are indeed involved in p53 activation by
CCl4. RT-qPCR and western blot analysis further
demonstrated that p53 was markedly upregulated in the
CCl4 group compared with the control group. This
indicated that p53 was activated after CCl4
administration, and that PZH-GB was able to normalize that
activation. However, the mRNA expression levels of p53, Bax and
Bcl-2 were increased in the GB-M group compared with the GB-L and
GB-H groups. These data indicated that the effect of PZH-GB was
unstable at the transcription level. These findings suggested that
intervention of PZH-GB along the mitochondrial apoptotic pathway
was p53-mediated in a rat model of CCl4-induced liver
disease.
ROS formed during the biotransformation of
CCl4 are more reactive and toxic than the parental
compound. Biotransformation of CCl4 occurs in the
endoplasmic reticulum and the isoenzyme implicated in this process
is CYP2E1 (27,28). Our preliminary studies have
demonstrated that the rats exposed to CCl4 generated
more oxidative products, including malondialdehyde (MDA) and TBARs
thiobarbituric acid reactive substances, and PZH-GB treatment could
decrease the level of MDA and TBARs. In this study, the results
revealed that the active free radical/intermediate CCl4
caused a reduction in CYP2E1, which was markedly restored by PZH-GB
pre-treatment. Taken together, this suggested that ROS was
generated upon exposure to CCl4.
The liver is a major inflammatory organ, and
inflammatory processes contribute to a number of pathological
events following exposure to various hepatotoxins (29). NO is a highly reactive oxidant that
is produced via the action of iNOS, and serves important roles as a
vasodilator and neurotransmitter, and in the immunological system
as a defense against tumor cells, parasites and bacteria (30). However, there is evidence that
excessive NO production by iNOS may also lead to hepatic damage
(31,32). COX-2 is the enzyme responsible for
the catalysis of prostaglandin E2 from arachidonic acid (33), and the induction of COX-2 is
closely associated with NO production (34). The current study confirmed that
significant increases in hepatic iNOS and COX-2 expression are
exhibited after CCl4 administration. These alterations
were attenuated by PZH-GB pre-treatment, which suggests that PZH-GB
suppressed iNOS and COX-2 protein secretion and/or enhanced the
degradation of these proteins. Accordingly, at least one other
potential mechanism of PZH-GB protection against
CCl4-induced hepatotoxicity appears to be, at least in
part, due to the suppression of inflammatory responses.
In conclusion, the results of the present study
demonstrated that PZH-GB exerted anti-apoptotic effects via a
p53-dependent mitochondrial pathway. It also protected against
CCl4-induced hepatocyte apoptosis by regulating the
Bcl-2 family of proteins and caspase activity. These anti-apoptotic
effects were associated with decreases in the expression of
pro-apoptotic proteins in the cytoplasm and the inhibition of
proteins associated with apoptosis in the mitochondria.
Additionally, PZH-GB exhibited a protective effect against
CCl4-induced acute histological liver injury, likely via
the suppression of inflammatory responses. Coupled with an
anti-apoptotic capacity and its ability as a free radical
scavenger, PZH-GB has exhibited an effective therapeutic quality in
a rat model of chemical-induced liver disease that may be
clinically useful as a safer alternative to present treatment
options.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81303125), and the
Developmental Fund of Chen Keji Integrative Medicine (CKJ2013015).
The authors would like to thank Clarity Manuscript Consultant LLC
(Indianapolis, IN, USA) for language editing.
References
|
1
|
Kumar CH, Ramesh A, Kumar JN Suresh and
Ishaq BM: A review on hepatoprotective activity of medicinal
plants. Int J Pharmaceut Sci Res. 23:501–515. 2011.
|
|
2
|
Ahsan R, Islam KM, Bulbul IJ, Musaddik A
and Haque E: Hepatoprotective activity of methanol extract of some
medicinal plants against carbon tetrachloride-induced
hepatotoxicity in rats. Eur J Sci Res. 37:302–310. 2009.
|
|
3
|
Chattopadhyay RR: Possible mechanism of
hepatoprotective activity of Azadirachta indica leaf extract: part
II. J Ethnopharmacol. 89:217–219. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Recknagel RO, Glende EA Jr, Dolak JA and
Waller RL: Mechanisms of carbon tetrachloride toxicity. Pharmacol
Ther. 43:139–154. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Williams AT and Burk RF: Carbon
tetrachloride hepatotoxicity: An example of free radical-mediated
injury. Semin Liver Dis. 10:279–284. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Slater TF: Free radicals as reactive
intermediates in tissue injury. Adv Exp Med Biol. 136:575–589.
1981.PubMed/NCBI
|
|
8
|
Brattin WJ, Glende EA Jr and Recknagel RO:
Pathological mechanisms in carbon tetrachloride hepatotoxicity. J
Free Radic Biol Med. 1:27–38. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cederbaum AI: Role of CYP2E1 in
ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig
Dis. 28:802–811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartosz G: Reactive oxygen species:
Destroyers or messengers? Biochem Pharmacol. 77:1303–1315. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cadenas E and Davies KJ: Mitochondrial
free radical generation, oxidative stress, and aging. Free Radic
Biol Med. 29:222–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi J, Aisaki K, Ikawa Y and Wake K:
Evidence of hepatocyte apoptosis in rat liver after the
administration of carbon tetrachloride. Am J Pathol. 153:515–525.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoek JB, Cahill A and Pastorino JG:
Alcohol and mitochondria: A dysfunctional relationship.
Gastroenterology. 122:2049–2063. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Volkmann N, Marassi FM, Newmeyer DD and
Hanein D: The rheostat in the membrane: BCL-2 family proteins and
apoptosis. Cell Death Differ. 21:206–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takebe H, Sato I, Tajima S, Ikeda Y, Ito K
and Nose T: Effects of cianidanol (KB-53) on liver cirrhosis
induced by CCl4 in rats: A pathological investigation.
Nihon Yakurigaku Zasshi. 81:585–591. 1983.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Merino N, González R, González A and
Remirez D: Histopathological evaluation on the effect of red
propolis on liver damage induced by CCl4 in rats. Arch
Med Res. 27:285–289. 1996.PubMed/NCBI
|
|
18
|
Yadav SS, Sindram D, Perry DK and Clavien
PA: Ischemic preconditioning protects the mouse liver by inhibition
of apoptosis through a caspase-dependent pathway. Hepatology.
30:1223–1231. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perry DK, Smyth MJ, Stennicke HR, Salvesen
GS, Duriez P, Poirier GG and Hannun YA: Zinc is a potent inhibitor
of the apoptotic protease, caspase-3. A novel target for zinc in
the inhibition of apoptosis. J Biol Chem. 272:18530–18533. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao Y, Huang C, Li ZF, Wang AY, Liu LY,
Zhao XG, Luo Y, Ni L, Zhang WG and Song TS: Exogenous
phosphatidylethanolamine induces apoptosis of human hepatoma HepG2
cells via the bcl-2/Bax pathway. World J Gastroenterol.
15:1751–1758. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin S and Dai CL: Attenuation of
reperfusion-induced hepatocyte apoptosis is associated with
reversed bcl-2/bax ratio in hemi-hepatic artery-preserved portal
occlusion. J Surg Res. 174:298–304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu B, Shi Y, Peng W, Zhang Q, Liu J, Chen
N and Zhu R: Diosmetin induces apoptosis by upregulating p53 via
the TGF-β signal pathway in HepG2 hepatoma cells. Mol Med Rep.
14:159–164. 2016.PubMed/NCBI
|
|
23
|
Ortega JF, de Conti A, Tryndyak V, Furtado
KS, Heidor R, Horst MA, Fernandes LH, Tavares PE, Pogribna M,
Shpyleva S, et al: Suppressing activity of tributyrin on
hepatocarcinogenesis is associated with inhibiting the p53-CRM1
interaction and changing the cellular compartmentalization of p53
protein. Oncotarget. 7:24339–24347. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Allam A, Gabr S, Ajarem J and
Abdel-Maksoud M: Bcl-2 and p53 expression in hepatic tissues of
Egyptian patients with Chronic Hepatitis C. J Pak Med Assoc.
65:1186–1192. 2015.PubMed/NCBI
|
|
25
|
Li X, Yu J, Brock MV, Tao Q, Herman JG,
Liang P and Guo M: Epigenetic silencing of BCL6B inactivates p53
signaling and causes human hepatocellular carcinoma cell resist to
5-FU. Oncotarget. 6:11547–11560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo XL, Liang B, Wang XW, Fan FG, Jin J,
Lan R, Yang JH, Wang XC, Jin L and Cao Q: Glycyrrhizic acid
attenuates CCl4-induced hepatocyte apoptosis in rats via
a p53-mediated pathway. World J Gastroenterol. 19:3781–3791. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rechnagel RO and Glende EA Jr: Carbon
tetrachloride hepatotoxicity: An example of lethal cleavage. CRC
Crit Rev Toxicol. 2:263–297. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al-Shabanah OA, Alam K, Nagi MN, Al-Rikabi
AC and Al-Bekairi AM: Protective effect of aminoguanidine, a nitric
oxide synthase inhibitor, against carbon tetrachloride induced
hepatotoxicity in mice. Life Sci. 66:265–270. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Badger DA, Sauer JM, Hoglen NC, Jolley CS
and Sipes IG: The role of inflammatory cells and cytochrome P450 in
the potentiation of CCl4-induced liver injury by a
single dose of retinol. Toxicol Appl Pharmacol. 141:507–519. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lowenstein CJ and Snyder SH: Nitric oxide,
a novel biologic messenger. Cell. 70:705–707. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Inoue T, Kwon AH, Oda M, Kaibori M,
Kamiyama Y, Nishizawa M, Ito S and Okumura T: Hypoxia and heat
inhibit inducible nitric oxide synthase gene expression by
different mechanisms in rat hepatocytes. Hepatology. 32:1037–1044.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nadler EP, Dickinson EC, Beer-Stolz D,
Alber SM, Watkins SC, Pratt DW and Ford HR: Scavenging nitric oxide
reduces hepatocellular injury after endotoxin challenge. Am J
Physiol Gastrointest Liver Physiol. 281:G173–G181. 2001.PubMed/NCBI
|
|
33
|
Chun KS and Surh YJ: Signal transduction
pathways regulating cyclooxygenase-2 expression: Potential
molecular targets for chemoprevention. Biochem Pharmacol.
68:1089–1100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang YC, Li PC, Chen BC, Chang MS, Wang
JL, Chiu WT and Lin CH: Lipoteichoic acid-induced nitric oxide
synthase expression in RAW 264.7 macrophages is mediated by
cyclooxygenase-2, prostaglandin E2, protein kinase A, p38 MAPK, and
nuclear factor-κB pathways. Cell Signal. 18:1235–1243. 2006.
View Article : Google Scholar : PubMed/NCBI
|