Introduction
Cleft palate is one of the most common congenital
craniofacial disorders (1,2). The palate consists of two parts, the
primary and the secondary palate. Secondary palatal development
involves several steps: First, the two palatal shelves grow
vertically, and then reorient to a horizontal position above the
tongue. Subsequently, the opposite palatal shelves come into
contact and fuse (3). Vertical
growth and elevation are important during palatal development. When
palatal shelf growth and elevation fail to occur, a cleft palate
forms.
Retinoic acid (RA), which is the active metabolite
of vitamin A, is essential for numerous biological processes and
serves a critical role in vertebrate morphogenesis (4). Conversely, exogenous RA is a potent
teratogen during embryonic development. According to the stage,
i.e. early or late, of palatogenesis, excess RA administration can
induce cleft palate of varying degrees. Exposure of pregnant mice
to RA on embryonic day (E)10 has been reported to result in small
palatal shelves that fail to elevate, whereas RA exposure on E12
has been demonstrated to lead to normal-sized palatal shelves that
can form contacts but are incapable of fusion (5,6).
Notch signaling is an evolutionarily conserved
signaling pathway in mammals that serves important roles in the
regulation of cellular survival and apoptosis (7,8).
Notch signaling is activated when the Notch receptor (Notch1-4) is
bound by a ligand, such as Jagged1 and 2 or Delta-like-1, −3 and
−4, which is expressed on the surface of an adjacent cell. The
functions of Notch2 and Notch3 have been studied extensively in
vascular smooth muscle cells; however, the complex roles of Notch2
in vascular smooth muscle cell proliferation have yet to be
elucidated. A previous study identified the differential regulation
of the unique functions of Notch2 and Notch3 during vascular smooth
muscle cell proliferation (9). In
addition, it has previously been reported that Notch2 may inhibit
vascular smooth muscle cell proliferation through the regulation of
the cyclin-dependent kinase inhibitor p27kip1 (10). The relationship between Notch and
RA has also been investigated. RA treatment was demonstrated to
induce cell cycle arrest at G1 phase in glioblastoma
stem cells, via downregulating cyclin D1 and upregulating p27
expression (11). Jagged2-Notch1
signaling has been implicated in physiological palatogenesis during
embryonic development (12).
However, the role of Notch2 following RA exposure during palatal
development has yet to be elucidated. The aim of the present study
was therefore to investigate the role of Notch signaling in
RA-induced mouse embryonic palate mesenchymal (MEPM) cell
proliferation inhibition.
Materials and methods
Antibodies
Anti-Notch1 (ab52627), anti-p21 (ab109199),
anti-cyclin D1 (ab134175) and anti-Ki-67 (ab15580) antibodies were
purchased from Abcam (Cambridge, UK). Anti-Notch2 (5732p),
anti-extracellular signal regulated kinase (ERK) (9102s)
anti-phosphorylated (p)-ERK (9101s) and anti-GAPDH (2118s)
antibodies were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Anti-Notch3 (AF1308-SP) antibody was purchased
from R&D Systems, Inc. (Minneapolis, MN, USA).
Mice
All animals used were purchased from Sun Yat-sen
University (Guangzhou, China). Female C57BL/6 mice (age, 10–12
weeks; weight, 25–30 g) were mated with mature males (age, 7–8
weeks; weight, 18–25 g) between 8 and 10 pm, and the detection of
the vaginal plug was designated E0. A total of 30 mice were used in
each control and treatment group. Female mice on E10 were
administered all-trans (at)RA (100 mg/kg; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) dissolved in corn oil by oral gavage.
Female mice in the control group were administered the same volume
of corn oil. atRA-treated and untreated pregnant mice were
sacrificed via cervical dislocation on E12.5, 13.5, 14.5 and 16.5.
All animal studies were approved by the Ethics Committee for Animal
Experiments of Sun Yat-sen University (approval no IACUC:
DB-15-0302; Guangzhou, China).
Histological staining
Embryos dissected from atRA-treated and control mice
on E12.5, 13.5, 14.5 and 16.5 were fixed in 4% paraformaldehyde for
24 h at room temperature and embedded in paraffin. For
immunohistochemistry and hematoxylin-eosin (H&E) staining,
deparaffinized sections (4 µm) were processed using standard
procedures. Briefly, sections were deparaffinized and hydrated
stepwise, heated in a microwave for 20 min for antigen retrieval,
and blocked with goat serum (Wuhan Boster Biological Technology,
Ltd., Wuhan, China) following nonspecific peroxidase blocking in 3%
hydrogen peroxide solution. Sections were incubated with primary
antibodies (Notch1, 1:500; Notch2, 1:250; Notch3, 1:250; Ki67,
1:500) diluted in 5% bovine serum albumin (BSA; Wuhan Boster
Biological Technology, Ltd.) at 4°C overnight, and then with the
secondary antibody (1:200; cat. no. 7074s; Cell Signaling
Technology, Inc.) diluted in 1% BSA at 37°C for 30 min. Sections
were subsequently stained with 3,3′-diaminobenzidine (DAB) and
hematoxylin. Following gradual dehydration, sections were sealed
with neutral gum. For H&E staining, sections were
deparaffinized and hydrated stepwise, immersed in haematoxylin,
washed in differentiation liquid, and then immersed in eosin.
Following gradual dehydration, sections were sealed with neutral
gum. IHC-positive mesenchymal cells were counted in a fixed area
near the tip of the palatal shelf. The ratio of IHC-positive cells
to the total cells was calculated and processed. Images were
captured using an Axio Imager 2 light microscope (Carl Zeiss,
Oberkochen, Germany). A fixed area near the tip of the palatal
shelf (20×20 µm) was selected for quantification using Image-Pro
Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Briefly, in the fixed areas, positive immunostaining for Notch2 was
selected, and the integrated optical density (OD) was measured. The
% of relative OD units compared with control was calculated as
described previously (13).
Collection of palatal shelves and
isolation of MEPM cells
Embryonic palatal shelves were isolated from
atRA-treated or untreated pregnant female mice between E12.5 and
E14.5. The epithelium was removed using tungsten needles following
incubation in 1 U/ml dispase II (Roche Diagnostics, Indianapolis,
IN, USA) at 37°C for 20 min. The palatal shelves were digested in
0.25% trypsin for 3 min and agitated to obtain a cell suspension.
Suspended cells were collected for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) or
western blot analysis.
Cell culture and treatments
Primary MEPM cells were isolated from the palatal
shelves of control untreated mice on E13.5 and cultured in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal
bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.).
Cells were maintained at 37°C in a 5% CO2 atmosphere.
The culture medium was replaced every 24 h. BMS493, an RA receptor
(RAR) antagonist, was purchased from Sigma-Aldrich (Merck KGaA).
MEPM cells were treated with 3 µM BMS493 or with 0, 1, 5 and 10 µM
atRA, for 48 h at 37°C.
RNA interference
Notch2 small interfering (si)RNA and control siRNA
were transfected into cultured MEPM cells using
Lipofectamine® RNAiMAX Reagent in Opti-MEM reduced serum
medium (Invitrogen; Thermo Fisher Scientific, Inc.) at 50 nM,
according to the manufacturer's protocol. The control siRNA
(Lot:0,0806) was purchased from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China). The Notch2 siRNA was chemically synthesized by
Guangzhou RiboBio Co., Ltd., with the following sequence: CCA GTA
CAG TGA AAT GTT T. A total of 24 h post-transfection, cells were
treated with atRA (5 µM) in DMEM without FBS for 48 h at 37°C and
collected for analysis.
Western blot analysis
Proteins of interest isolated from atRA-treated or
control embryos and cultured MEPM cells were analyzed using western
blot analysis, as previously described (14). Total protein was extracted from
palatal shelves and MEPM cells using radioimmunoprecipitation assay
lysis buffer (Thermo Fisher Scientific, Inc.) supplemented with
protease and phosphatase inhibitors at 4°C for 30 min, and
centrifuged at 15,000 × g at 4°C for 30 min. Protein content was
determined by bicinchoninic acid assay. Equal amounts (30 µg) of
extracted protein samples were separated by 10% SDS-PAGE and
transferred onto nitrocellulose membranes (EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% evaporated
skimmed milk at 37°C for 1 h and incubated with primary antibodies
(Notch1, Notch2, Notch3, P21, cyclinD1, p-ERK, ERK, ki67, GAPDH;
1:1,000 dilution) in 5% evaporated skimmed milk at 4°C for 12 h,
and horseradish peroxidase-conjugated secondary antibody (1:2,000
dilution; cat. no. 7074s; Cell Signaling Technology, Inc.) in 5%
evaporated skimmed milk at 37°C for 1 h. Protein bands were
visualized using an enhanced chemiluminescence kit (Cell Signaling
Technology, Inc.) and the ImageQuant Las4000mini system (GE
Healthcare Life Sciences, Chalfont, UK). GAPDH was used as an
internal control. The signal intensities were quantified using
ImageJ version 1.48u software (National Institutes of Health,
Bethesda, MD, USA) (15).
RT-qPCR
Total RNA was extracted from MEPM cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. cDNA was reverse
transcribed from 1 µg of total RNA using a Transcriptor
First-Strand cDNA Synthesis kit (Roche Diagnostics, Burgess Hill,
UK). qPCR was performed using the SYBR-Green I reaction mix (Roche
Diagnostics), as per the manufacturer's instructions. The
thermocycling conditions were as follows: 95°C for 5 min, then 40
cycles of 95°C for 10 sec, 60°C for 20 sec, and 72°C for 20 sec.
qPCR reactions were performed in a Roche Light Cycler 480 system
(Roche Diagnostics) The following primers were used: β-actin,
forward 5′-TCAC-CCACACTGTGCCCATCTACGA-3′ and reverse
5′-GGATGCCACAGGATTCCATACCC-A-3′; RARα, forward
5′-TCAGTGCCATCTGCCTCAT-3′ and reverse
5′-CGTAGACTTT-CAGTGCTTCCAG-3′; RARβ, forward
5′-GTGCCATCTGTTTAATCTGTGG-3′ and reverse 5′-TGCTGGGTCGTCGTTTTC-3′;
RARγ, forward 5′-ATACCCCAGAGCAG-GACACTA-3′ and reverse
5′-GGCAAAGGCAAAGACGAG-3′; Notch1, forward
5′-AACATGGGCCGTACTCCGTTA-3′ and reverse
5′-AGCCAGGATCAGTGGAGTTGTG-3′; Notch2, forward
5′-ACAAGTGAAGTGCAGGAGAGGGG-3′ and reverse
5′-CAGCGGC-AGGAATAGTGAGGAG-3′; and Notch3, forward
5′-CTCTGTGGTGATGCTGGAGATTGA-3′ and reverse
5′-TGCTGACAAGGCTCCCAGGTAG-3′. Target genes were normalized to
β-actin, and relative fold changes in mRNA expression were
calculated using the formula 2−ΔΔCq (16).
Proliferation assay
A Cell Counting Kit-8 (Wylton Chemistry Co., Ltd.,
Tongren, China) assay was used to assess cellular proliferation.
Primary MEPM cells were seeded into a 96-well collagen-coated plate
(1×104 cells/well) and treated with control siRNA,
control siRNA+atRA (5 µM), Notch2 siRNA and Notch2 siRNA+atRA (5
µM), prior to measuring proliferation as per the kit's
instructions.
Bead implantation in palatal
shelves
Palatal shelves of E13.5 mouse embryos were gently
removed from the maxilla in cold PBS (Invitrogen; Thermo Fisher
Scientific, Inc.). Dissected palatal shelves were placed on
MF-Millipore™ membrane filters. Biggers, Gwatkin, and
Judah (Fitton-Jackson modification) medium (BGJb; Invitrogen;
Thermo Fisher Scientific, Inc.) was used for culture. Anion
exchange resin beads (Dowex® 1×4, 100–200 mesh;
Sigma-Aldrich; Merck KGaA) served as atRA carriers. Following
incubation in 20–50 µM atRA at room temperature for 45 min, the
beads were washed with BGJb medium for 15 min. Control beads were
treated with vector alone. Beads were implanted into palate shelves
using fiber probe under a stereomicroscope (Carl Zeiss AG). Samples
were cultured for 48 h and then collected for immunohistochemical
analysis.
Statistical analyses
The statistical significance of the difference
between groups was assessed by Student's t-test and one-way
analysis of variance, using SPSS 19.0 software (IBM SPSS, Armonk,
NY, USA). Data was expressed as mean ± standard deviation. All
experiments were repeated at least 3 times. P<0.05 was
considered to indicate a statistically significant difference.
Results
atRA exposure on E10.0 induces cleft
palate in mouse embryos
atRA exposure on E10.0 was demonstrated to induce
cleft palate in 99% of the embryos (99/100), whereas no embryos
exhibited cleft palate in the control group (0/100). H&E
staining revealed the morphological changes of palatal shelves on
E12.5, 13.4, 14.5 and 16.5 in atRA-treated embryos and controls
(Fig. 1A). On E12.5, atRA-exposed
embryos exhibited palatal shelves of smaller volume compared with
controls, as determined by visual observation. On E13.5, palatal
shelves grew vertically and no differences were apparent between
atRA-treated and control embryos. On E14.5, palatal shelves
elevated and contacted with the opposing palates in control
embryos, whereas atRA-exposed embryos exhibited unelevated shelves.
On E16.5, a firm palate formed and the medial edge epithelium
disappeared in untreated embryos; however, in atRA-exposed embryos,
palatal shelves failed to elevate and a cleft palate developed.
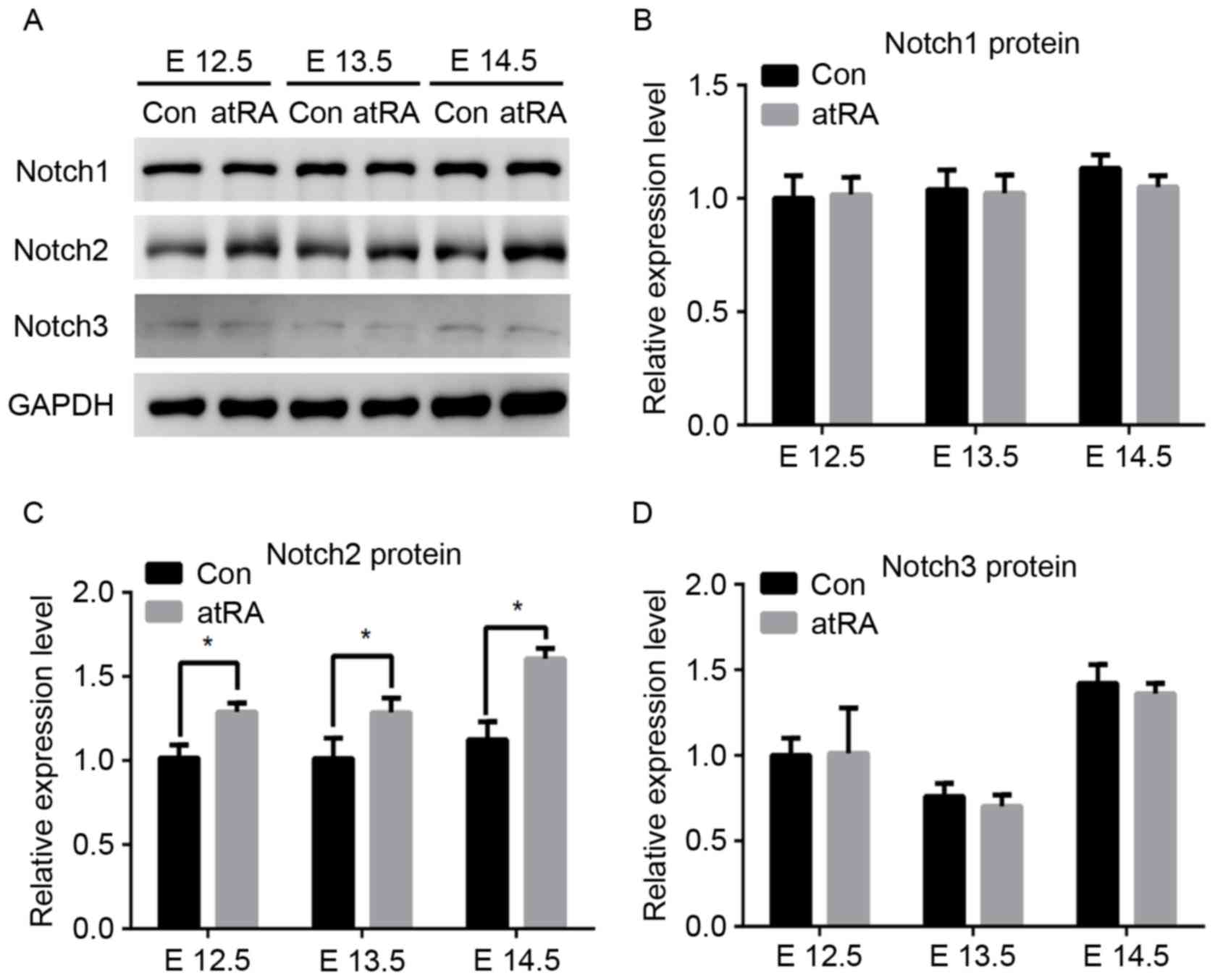
Notch2 expression is upregulated in
MEPM cells from atRA-treated embryos via RAR activation
To identify atRA-induced Notch signaling events
impairing palatogenesis, protein expression levels of Notch1,
Notch2 and Notch3 were assessed in MEPM cells isolated from
atRA-treated and untreated embryos between E12.5 and E14.5.
Immunohistochemistry revealed that Notch2 expression was elevated
in the atRA-treated embryos compared with in the controls
(P<0.05; Fig. 1B). Notably, no
significant differences were detected in the expression of Notch1
and Notch3 between atRA-exposed and control embryos (Fig. 2). Furthermore, Notch mRNA and
protein expression levels were assessed using western blot analysis
(Fig. 3) and RT-qPCR (Fig. 4). In accordance with the results of
immunohistochemical analysis, Notch2 mRNA and protein expression
levels were significantly upregulated in atRA-treated embryos
compared with in the control group. Conversely, protein (Fig. 3B and D) and mRNA (Fig. 4A and C) expression levels of Notch1
and Notch3 were not significantly different between atRA-exposed
embryos and controls.
To investigate whether RAR activation was implicated
in the atRA-induced Notch2 upregulation in MEPM cells, RAR mRNA
expression levels were assessed in the presence of various atRA
concentrations using RT-qPCR (Fig.
5). mRNA expression levels of RARβ and RARγ increased
significantly following atRA treatment compared with untreated
cells (P<0.05; Fig. 5), but no
significant differences were observed in the mRNA expression levels
of RARα. To further investigate whether the upregulation of Notch2
resulted from the direct action of atRA on RARs, the RAR antagonist
BMS493 was applied to MEPM cells prior to atRA treatment. Notably,
Notch2 expression was downregulated following pretreatment with
BMS493 (P<0.05; Fig. 6).
MEPM cellular proliferation is
suppressed in atRA-treated embryos
To examine the proliferation rate of MEPM cells in
control and atRA-exposed embryos, the expression of Ki-67 was
assessed in MEPM cells isolated from E12.5–14.5 embryos, using
immunohistochemistry. There were fewer Ki-67-positive MEPM cells in
atRA-treated embryonic tissue compared with in control tissue on
E12.5 and 13.5 (P<0.05); however, no significant difference was
observed on E14.5 (Fig. 7A).
Western blot analysis demonstrated a downregulation in Ki-67
protein expression levels in MEPM cells in E12.5 and 13.5
atRA-exposed embryos compared with in age-matched controls
(P<0.05; Fig. 7B).
 | Figure 7.MEPM cellular proliferation rate was
decreased in atRA-treated embryos. (A) Ki-67 expression in palatal
shelves isolated from control and atRA-treated embryos between
E12.5 and E14.5. Scale bars, 20 µm. (B) Protein expression levels
of p-ERK, total ERK1/2, p21, cyclin D1 and Ki-67 in MEPM cells
isolated from control and atRA-treated embryos (n=3/group).
*P<0.05 vs. Con group. MEPM, mouse embryonic palate mesenchymal;
atRA, all-trans retinoic acid; E, embryonic day; p, phosphorylated;
ERK, extracellular signal-regulated kinase; Con, control. |
To further investigate downstream Notch signaling
events, total and p-ERK protein expression levels were assessed.
p-ERK was significantly downregulated in E12.5 and 13.5
atRA-treated embryos compared with in the control group
(P<0.05). Cell cycle-related proteins p21 and cyclin D1 were
also analyzed. p21 protein expression levels were increased,
whereas cyclin D1 levels were decreased in the atRA-treated group
compared with in controls between E12.5 and E14.5 (P<0.05;
Fig. 7B).
Notch2 silencing attenuates
atRA-induced inhibition of MEPM cellular proliferation
To investigate whether Notch2 may be involved in
atRA-induced inhibition of MEPM cellular proliferation, Notch2
siRNA was used. atRA was demonstrated to decrease p-ERK protein
expression levels; however, Notch2 knockdown attenuated the
atRA-induced p-ERK downregulation (P<0.05; Fig. 8A). As a potential signaling pathway
downstream of ERK, p21 expression levels were decreased in the
Notch2 knockdown group compared with in the control group
(P<0.05; Fig. 8A). Conversely,
cyclin D1 protein levels increased in MEPM cells not expressing
Notch2 compared with in control cells (P<0.05; Fig. 8A).
In the absence of Notch2, MEPM cells treated with
atRA exhibited increased proliferation compared with control cells
(P<0.05; Fig. 8B). Notch2
knockdown was demonstrated to partially attenuate the atRA-induced
inhibition of MEPM cellular proliferation. Western blot analysis of
Ki-67 protein expression levels also supported these results
(P<0.05; Fig. 8A).
Stimulation of Notch2 expression in
palatal shelves by atRA
Palatal shelves isolated from E13.5 embryos were
cultured for 48 h with implanted beads soaked in atRA or control
vector (Fig. 9A). Notch2 protein
expression was assessed using immunohistochemistry. Notch2
expression appeared to be upregulated in MEPM cells surrounding the
atRA-releasing bead compared with in control cells (P<0.05;
Fig. 9B). Conversely, Ki-67
expression appeared to be decreased in atRA-exposed palatal shelves
compared with in controls (P<0.05; Fig. 9C).
Discussion
In the present study, the effects of atRA on MEPM
cells were investigated during palatogenesis. The present results
indicated that atRA inhibited MEPM cellular proliferation in
vitro and in vivo. Furthermore, Notch2 appeared to be
involved in the atRA-mediated inhibition of cellular
proliferation.
It has previously been reported that Notch signaling
is inhibited during RA-induced inhibition of glioblastoma stem cell
growth (11). Notably, the present
study revealed that Notch2 expression was upregulated in
atRA-treated E12.5–14.5 embryos, whereas Notch1 and Notch3
expression remained unaltered. In accordance with the results of a
previous study (17), p21
expression was upregulated and cyclin D1 expression was
downregulated in atRA-treated E12.5–14.5 embryos. Since ERK
signaling is implicated in the regulation of cellular
proliferation, total and p-ERK levels were assessed. p-ERK protein
expression levels were revealed to be decreased in atRA-treated
E12.5–14.5 embryos.
It is well established that the effects of atRA are
mediated by the activation of RARs (18). Therefore, the present study
investigated whether Notch2 upregulation may result from the direct
action of atRA on RARs. The present results demonstrated that RAR
inhibition prevented the atRA-induced Notch2 upregulation in MEPM
cells. The upregulation of Notch2 observed during the cellular
response to atRA suggested that Notch2 signaling may participate in
the mechanisms underlying atRA-induced cleft palate. Other pathways
may also participate in this process. Previous studies had
indicated that Smad2/3 and Wnt/β-catenin signaling were involved in
atRA-induced MEPM growth inhibition (14,18).
ERK1/2 signaling is implicated in the regulation of
cellular proliferation (19,20).
The ERK pathway has been reported to serve a critical role in
cellular proliferation, via inducing the transcription of cyclin D1
(21). In addition, ERK2 has been
demonstrated to enhance cellular proliferation via promoting p21
degradation (22). A previous
study revealed that the regulation of MEK/ERK signaling by Notch2
is implicated in vascular smooth muscle cell proliferation
(9). In the present study,
knockdown of Notch2 increased p-ERK protein expression levels and
attenuated the atRA-induced p-ERK downregulation. In addition, p21
expression was downregulated, whereas cyclin D1 expression was
upregulated following Notch2 silencing. These results suggested
that atRA, via the upregulation of Notch2 protein expression, may
inhibit the proliferation of MEPM cells, possibly through the
implication of ERK signaling pathways.
In summary, administration of atRA inhibited the
proliferation of mouse embryo mesenchymal cells and blocked the
elevation of palatal shelves, thus leading to a cleft palate. The
present study identified that Notch signaling was involved in
atRA-induced inhibition of MEPM cell proliferation. These findings
highlighted the molecular and teratogenic actions of atRA, and
might contribute to the development of potential novel treatments
for cleft palate in the future.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (grant nos. 81300862 and 81100739), the
Project of Health and Family Planning Commission of Shenzhen
Municipality (grant no. 201302202) and the Natural Science
Foundation of Guangdong Province (grant no. S2011040004190).
References
|
1
|
Chai Y and Maxson RE Jr: Recent advances
in craniofacial morphogenesis. Dev Dyn. 235:2353–2375. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wyszynski DF and Beaty TH: Phenotypic
discordance in a family with monozygotic twins and nonsyndromic
cleft lip and palate: Follow-up. Am J Med Genet. 110:182–183. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hilliard SA, Yu L, Gu S, Zhang Z and Chen
YP: Regional regulation of palatal growth and patterning along the
anterior-posterior axis in mice. J ANAT. 207:655–667. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ross SA, McCaffery PJ, Drager UC and De
Luca LM: Retinoids in embryonal development. Physiol Rev.
80:1021–1054. 2000.PubMed/NCBI
|
|
5
|
Birnbaum LS, Harris MW, Stocking LM, Clark
AM and Morrissey RE: Retinoic acid and
2,3,7,8-tetrachlorodibenzo-p-dioxin selectively enhance
teratogenesis in C57BL/6N mice. Toxicol Appl Pharmacol. 98:487–500.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abbott BD, Harris MW and Birnbaum LS:
Etiology of retinoic acid-induced cleft palate varies with the
embryonic stage. Teratology. 40:533–553. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cao H, Hu Y, Wang P, Zhou J, Deng Z and
Wen J: Down-regulation of Notch receptor signaling pathway induces
caspase-dependent and caspase-independent apoptosis in lung
squamous cell carcinoma cells. APMIS. 120:441–450. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang T, Holt CM, Xu C, Ridley C, Jones
POR, Baron M and Trump D: Notch3 activation modulates cell growth
behaviour and cross-talk to Wnt/TCF signalling pathway. Cell
Signal. 19:2458–2467. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baeten JT and Lilly B: Differential
regulation of NOTCH2 and NOTCH3 contribute to their unique
functions in vascular smooth muscle cells. J Biol Chem.
290:16226–16237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boucher JM, Harrington A, Rostama B,
Lindner V and Liaw L: A receptor-specific function for Notch2 in
mediating vascular smooth muscle cell growth arrest through
cyclin-dependent kinase inhibitor 1B. Circ Res. 113:975–985. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ying M, Wang S, Sang Y, Sun P, Lal B,
Goodwin CR, Guerrero-Cazares H, Quinones-Hinojosa A, Laterra J and
Xia S: Regulation of glioblastoma stem cells by retinoic acid: Role
for Notch pathway inhibition. Oncogene. 30:3454–3467. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Casey LM, Lan Y, Cho ES, Maltby KM,
Gridley T and Jiang R: Jag2-Notch1 signaling regulates oral
epithelial differentiation and palate development. Dev Dyn.
235:1830–1844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He J, Chen Q, Wang L, Wu F and Du Z:
Corticotropin-releasing hormone receptor 1 coexists with
endothelin-1 and modulates its mRNA expression and release in rat
paraventricular nucleus during hypoxia. Neuroscience.
152:1006–1014. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang M, Huang H and Chen Y: Smad2/3 is
involved in growth inhibition of mouse embryonic palate mesenchymal
cells induced by all-trans retinoic acid. Birth Defects Res A Clin
Mol Teratol. 85:780–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okano J, Suzuki S and Shiota K:
Involvement of apoptotic cell death and cell cycle perturbation in
retinoic acid-induced cleft palate in mice. Toxicol Appl Pharmacol.
221:42–56. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu X, Gao J, Liao Y, Tang S and Lu F:
Retinoic acid alters the proliferation and survival of the
epithelium and mesenchyme and suppresses Wnt/β-catenin signaling in
developing cleft palate. Cell Death Dis. 4:e8982013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meloche S and Pouysségur J: The ERK1/2
mitogen-activated protein kinase pathway as a master regulator of
the G1- to S-phase transition. Oncogene. 26:3227–3239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kolch W: Coordinating ERK/MAPK signalling
through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 6:827–837.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weber JD, Raben DM, Phillips PJ and
Baldassare JJ: Sustained activation of
extracellular-signal-regulated kinase 1 (ERK1) is required for the
continued expression of cyclin D1 in G1 phase. Biochem J.
326:61–68. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hwang CY, Lee C and Kwon KS: Extracellular
signal-regulated kinase 2-dependent phosphorylation induces
cytoplasmic localization and degradation of p21Cip1. Mol Cell Biol.
29:3379–3389. 2009. View Article : Google Scholar : PubMed/NCBI
|