Introduction
Atherosclerotic arterial disease is one of the
leading causes of morbidity, mortality and disability worldwide,
and the prevalence of atherosclerosis (AS) is increasing in
developing countries (1,2). AS is initiated by circulating
monocytes infiltrating the subendothelial space in response to
multiple stimuli, such as hyperlipidemia, and then differentiating
into macrophages. These macrophages devour massive amounts of
cholesteryl esters to become foam cells, a hallmark of the early
stage of atherosclerotic lesions (3). Mechanistically, lipid metabolism
disorder and chronic immuno-inflammation are the two
well-recognized mechanism sunder lying foam cell formation.
Recently, mammalian target of rapamycin (mTOR) has
drawn much attention as it plays a role in a variety of human
diseases including AS, obesity, diabetes and cancer (4–7). The
relationship between mTOR and AS is an emerging topic of research
(8,9). For instance, rapamycin, an mTOR
inhibitor, has been shown to interfere with atherogenes is by
attenuating inflammation and enhancing atherosclerotic plaque
stability in various mammalian atherosclerotic models. Rapamycin,
an mTOR inhibitor, mediates its effects without altering serum
lipid levels, although it is currently used to treat hyperlipidemia
and hypercholesterolemia (10,11).
The discrepant findings regarding rapamycin's effects on
circulating lipid levels in animal models and human patients remain
to be addressed. In addition, mTOR inhibition prevents lipid
accumulation and upregulates cholesterol efflux (12,13).
For example, the mTOR signaling pathway was shown to be activated
during THP-1 (Human acute monocytic leukemia cells) foam cell
formation, and rapamycin or silencing mTOR suppressed this
activation and increased the expression of cellular lipid efflux
mediator adenosine triphosphate-binding cassette transporter A1
(ABCA1) (14). Collectively, these
findings point to a potential correlation between mTOR and
inflammation and cholesterol metabolism during foam cell formation
in AS.
Recent studies also implicated mTOR in regulating
diverse functions of professional antigen-presenting cells (APC),
including dendritic cells (DCs) and macrophages (15). Consistently, rapamycin selectively
promotes migration of mouse DCs to lymph nodes in vivo by
enhancing the expression of C-C chemokine receptor type 7 (CCR7)
(16), which is required for foam
cell formation. CCR7 expression is mediated in part by liver X
receptor (LXR) activation in atherosclerotic lesions, and both CCR7
and LXR are involved in plaque regression in ApoE−/−
mice (17,18). Thus, both mTOR and LXR signaling
mediate expression of CCR7 during plaque regression, suggesting a
potential functional link between mTOR and LXR signaling.
The transcription factor nuclear factor-κB (NF-κB)
regulates various cytokines and chemical factors and inflammatory
responses, which are predominant characteristics of AS development
(19). Regulation of NF-κB
activity has been well studied, and one mechanism involves Sirtuin
1 (SIRT1). SIRT1 suppresses autophagy through activating NF-κB
(20), but we revealed that SIRT1
can also prevent AS by activating LXR and inhibiting NF-κB
signaling (21). Thus, SIRT1
appears to function upstream of the LXR/NF-κB axis. Whether SIRT1
mediates NF-κB in a positive or a negative way appears to be
context-dependent.
Accumulating evidence indicates that there is a
functional interaction between SIRT1 and mTOR. For example,
rapamycin restored SIRT1-induced suppression of autophagy (20), and SIRT1 was required for the
rapamycin-mediated effects on high glucose-induced mesangial cell
senescence (22), suggesting a
functional link between mTOR and SIRT1. Indeed, it was reported
that SIRT1 negatively regulates mTOR and that mTOR inhibition
increases SIRT1 activity (23,24).
These findings point to the possibility that mTOR and SIRT1 may be
part of the same signaling pathway in AS pathogenesis, in which
foam cell formation and egression are two important processes.
Herein, we hypothesized that mTOR signaling promotes
monocyte-derived foam cell formation and inhibits foam cell egress
through downregulating SIRT1/LXR/CCR7 and upregulating NF-κB
signaling. To test our hypothesis, we investigated the expression
of key factors in mTOR and SIRT1/LXR/CCR7 signaling in U937-derived
foam cells in vitro and discussed the functional
relationship between the two signaling pathways.
Materials and methods
Reagents
Roswell Park Memorial Institute-1640 (RPMI-1640)
medium was obtained from Gibco (Grand Island, NY, USA). Oil red O
was purchased from Bio Basic Inc. (Markham, ON, Canada).
4′6-diamidino-2-phenylindole dihydrochloride (DAPI) was purchased
from Santa Cruz Biotechnology (Dallas, TX, USA). Oxidized low
density lipoprotein (ox-LDL) was obtained from Yi Yuan
Biotechnologies (Guangzhou, China). The following reagents were
purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA): sodium
palmitate (PA), phorbol 12-myristate 13-acetate (PMA), rapamycin,
lysophosphatidic acid (LPA), polyvinyl alcohol mounting medium with
DABCO (PVA-DABCO; Sigma-Aldrich).
Antibodies
Monoclonal antibodies against mTOR and
phosphorylated (p)-mTOR were purchased from Cell Signaling
Technology (Danvers, MA, USA). p-ribosomal protein S6 kinase
(p70S6K), SIRT1, LXRα and NF-κB antibodies were purchased from
Santa Cruz Biotechnology. Antibodies against CCR7 and tumor
necrosis factor-α (TNF-α) and donkey anti-rabbit IgG H&L (Alexa
Fluor® 594) were purchased from Abcam (Cambridge, UK).
β-actin antibody was obtained from Zhon Shan Golden Brid (Beijing,
China).
U937 cell differentiation and foam
cell formation
The human monocytic cell line U937 was purchased
from the Type Culture Collection (Chinese Academy of Sciences,
Shanghai, China) and cultured in a humidified atmosphere at 37°C
containing 5% CO2 in RPMI-1640 supplemented with 10%
fetal bovine serum (FBS; Biowest, Logan, UT, USA), penicillin (100
U/ml) and streptomycin (100 mg/ml). U937 cells were cultured in
6-well plates (2×106 cells/well) and incubated with 160
nM PMA for 24 h, the differentiated macrophages were then
harvested. Foam cell formation was induced by incubation of
U937-derived macrophages with PA (0.2 mM) and ox-LDL (80 µg/ml) for
another 24 h.
Oil red O staining
The U937-derived foam cells were induced
successfully in 6-well cell culture clusters. Media was aspirated
and the cells were fixed in 4% paraformaldehyde for half an hour.
Subsequently, cells were stained with freshly diluted 0.3% Oil red
O solution for 30 min at room temperature (RT). Thereafter, Oil red
O solution was removed and cells were re-stained with hematoxylin
for 10 sec before mounting with glycerin gelatin. Cells were
observed under a light microscope (Leica DM2500; Leica
Microsystems, Mannheim, Germany) at ×400 magnification and then
photographed using a BX-51 camera (Olympus, Tokyo, Japan).
Cell viability and proliferation
assay
U937 cell viability was measured by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, cells were cultured in 96-well plates
(1×104cells/well) with 100 µl medium. The medium was
refreshed with different concentrations of rapamycin (0.01–100 nM)
and cultured for 24 h. Thereafter, MTT (5 mg/ml) was added to each
well including control (one set of wells with MTT but no cells),
and the cells were cultured for another 4 h at 37°C Triple liquid
[10% sodium dodecyl sulfate (SDS), 5% isobutanol, 0.012 mol/l
hydrochloric acid, dissolved in distilled water] was added to each
well. After covering with tinfoil and agitating cells on an orbital
shaker for 15 min, the absorbance of each sample was measured at
570 nm by a microtiter plate reader (Multiskan MK3; Thermo
Scientific, Helsinki, Finland). All experiments were repeated in
triplicate.
Immunofluorescence staining
For immunofluorescence analysis, U937-derived foam
cells were fixed in 4% paraformaldehyde in 1X phosphate-buffered
saline (PBS) for 15 min and permeabilized with 0.1% Triton X-100 in
1X PBS for 10 min. After being blocked with 3% bovine serum albumin
(BSA) in 1X PBS for 30 min, cells were incubated with NF-κB p65
antibody at 4°C overnight and washed twice with 0.1% Tween-20 in 1X
PBS every 5 min, followed by incubation for 1 h with Alexa
594-labeled anti-rabbit IgG secondary antibody at RT. Cell nuclei
were stained with 200 ng/ml DAPI for 10 min at RT and the cells
were mounted in PVA-DABCO. Images were captured using a FV1000
confocal laser scanning microscope (Olympus).
Real-time reverse transcription
quantitative PCR (real-time RT-PCR) analysis
Total RNA was extracted from cells using TRIzol
reagent RNAiso Plus (Takara, Dalian, China) according to the
manufacturer's instructions. The concentration and purity of the
RNA were measured and determined by NanoDrop 2000c (Thermo
Scientific, Waltham, MA, USA) and the OD260/280 value was 1.9–2.1.
Extracted RNA was reverse-transcribed with PrimeScript™ RT reagent
kit with gDNA eraser (Takara). Real-time PCR was performed using
ABI Prism 7500 systems (Applied Biosystems, Foster City, CA, USA)
with GoTaq® qPCR Master Mix (Promega Corporation,
Madison, WI, USA). Amplification was carried out in a total volume
of 20 µl and 40 cycles after pre-denaturation (95°C for 10 min)
followed by: 95°C for 15 sec and 60°C for 1 min. The primer
sequences used in real-time-PCR are shown in Table I. Experiments were performed in
triplicate for each sample. The relative amount of mRNA was
calculated using the comparative CT method. Glyceraldehyde
3-phosphate dehydrogenase (GAPDH) served as the reference
housekeeping gene. A melting curve analysis was performed after
amplification to verify the accuracy of the amplicon.
 | Table I.Primer sequences used for
quantitative real-time PCR. |
Table I.
Primer sequences used for
quantitative real-time PCR.
| Gene | Forward sequences
(5′ to 3′) | Reverse sequences
(5′ to 3′) |
|---|
| mTOR |
CATCATGTTGCGGATGGCTC |
AGCATCAGGTTGGATGGGTG |
| p70S6K |
ACTAGTGTGAACAGAGGGCC |
CATTGCCTTTCCATAGCCCC |
| SIRT1 |
GCTCGCCTTGCTGTAGACTTCC |
GCAACCTGTTCCAGCGTGTCT |
| NF-kB |
TGTCCAGCTTCGGAGGAAAT |
TCTGACGTTTCCTCTGCACT |
| TNF-α |
TCTTCTGCCTGCTGCACTTT |
TCAGCTTGAGGGTTTGCTACA |
| LXR-α |
ATGCCGAGTTTGCCTTGC |
CATCCGTGGGAACATCAGT |
| CCR7 |
CATCAGCATTGACCGCTACG |
GTATCCAGATGCCCACACAGG |
| ABCA1 |
TCCTCCTGGTGAGTGCTTTG |
GGGACTCCTCTCAAAAGGGC |
| GAPDH |
TGAACGGGAAGCTCACTGG |
TCCACCACCCTGTTGCTGTA |
Western blot analysis
Cells were lysed using RIPA lysis buffer containing
150 mM NaCl, 1.0% NP-40, 50 mM Tris·HCl (pH 7.4), 0.5% sodium
deoxycholate, and 0.1% SDS. Proteins were separated by 10%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred
onto nitrocellulose membranes (Merck Millipore, Darmstadt,
Germany). After being washed once with Tris-buffered saline (TBS),
the membranes were blocked in 5% skimmed milk in TBS-Tween (TBST)
for 1 h and incubated with primary antibodies at 4°C overnight,
followed by another incubation with an appropriate secondary
antibody at RT for 1 h. The immunoblots were visualized by enhanced
chemiluminescence (Advansta, Menlo Park, CA, USA). Signal
intensities were measured by Quantity One software (Bio-Rad,
Hercules, CA, USA).
Statistics
Data are presented as means ± standard deviation
(SD) from at least three independent experiments. Results were
statistically analyzed with unpaired Student's t-test or one-way
analysis of variance (ANOVA) using the statistical software
GraphPad Prism 5.01 (La Jolla, CA, USA). In all tests, P<0.05
was considered statistically significant.
Results
PA and ox-LDL treatment induces lipid
droplet accumulation and upregulates p-mTOR and p-p70S6K during
foam cell formation
Foam cell formation was induced in U937-derived
macrophages by PA and ox-LDL treatment. Following macrophage
differentiation induced by PMA for 24 h, the U937-derived
macrophages were divided into three groups: the control group, the
PA group, and the PA+ ox-LDL group. After incubation for another 24
h, a larger number of lipid droplets accumulated in the cells in
the PA+ ox-LDL group compared to the control and PA groups, as
revealed by Oil red O staining (Fig.
1A-C). We also evaluated changes in p-mTOR and p-p70S6K protein
levels in cells from these three groups by Western blot analysis.
As demonstrated in Fig. 1D and E,
p-mTOR and p-p70S6K expression were upregulated in the PA+ ox-LDL
group compared to the other two groups. Thus, foam cell formation
upregulates expression of p-mTOR and p-p70S6K.
LPA upregulates p-mTOR and p-p70S6K
but decreases SIRT1 expression
LPA has been reported to promote atherosclerotic
lesion formation by promoting monocyte migration from the
bloodstream into the vascular wall and decreasing monocyte-derived
cell emigration from the vessel wall (25). To better understand the effects of
atherogenetic conditions on mTOR signaling, U937-derived
macrophages were incubated with different LPA concentrations (0,
10, 20 and 30 µM) to induce foam cell formation. We used Oil red O
staining to observe lipid accumulation and found that 30 µM LPS
increased the number of lipid droplets in the cultured cells
(Fig. 2A and B). Next, the effects
of LPA on the expression of p-mTOR, p-p70S6K and SIRT1 were
assessed by western blot analysis. As shown in Fig. 2E and F, p-mTOR and its downstream
factor p-p70S6K were upregulated by 30 µM LPA, while SIRT1
expression was downregulated. Therefore, 30 µM LPA was chosen for
the subsequent foam cell formation experiments.
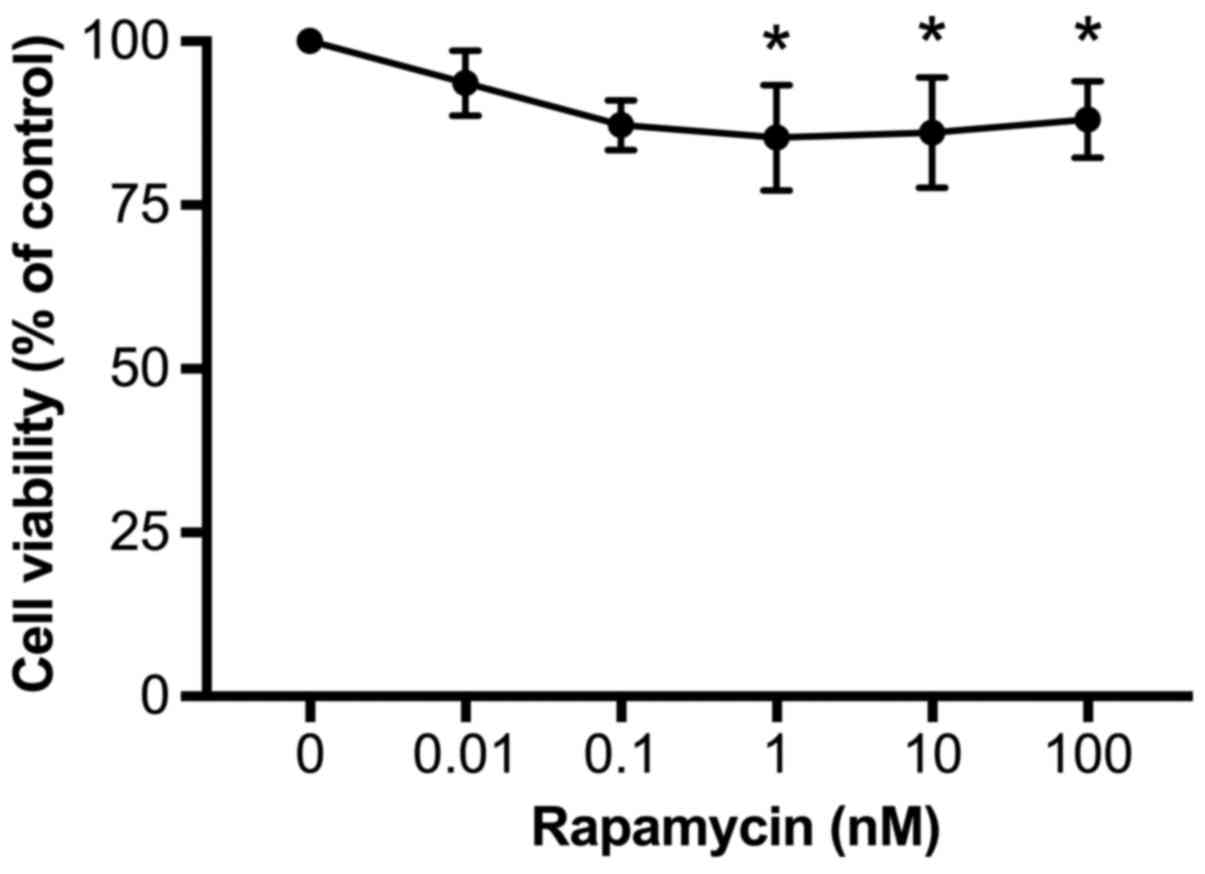
Rapamycin reduces cell viability
To determine whether rapamycin affects cell
viability, U937 cells were incubated with rapamycin at different
concentrations (0, 0.01, 0.1, 1, 10, and 100 nM) in RPMI-1640 for
24 h and then cell viability was evaluated by the MTT method. As
demonstrated in Fig. 3, rapamycin
treatment dose-dependently decreased cell viability, from 94±2% at
0.01 nM to 85±4% at 1 nM (P<0.05, vs. 0). Increases in rapamycin
concentration to 10 or 100 nM did not further reduce cell viability
(86±4 and 88±2%, respectively). Therefore, in order to achieve the
maximal inhibitory effects, 100 nM rapamycin was used for the
subsequent experiments.
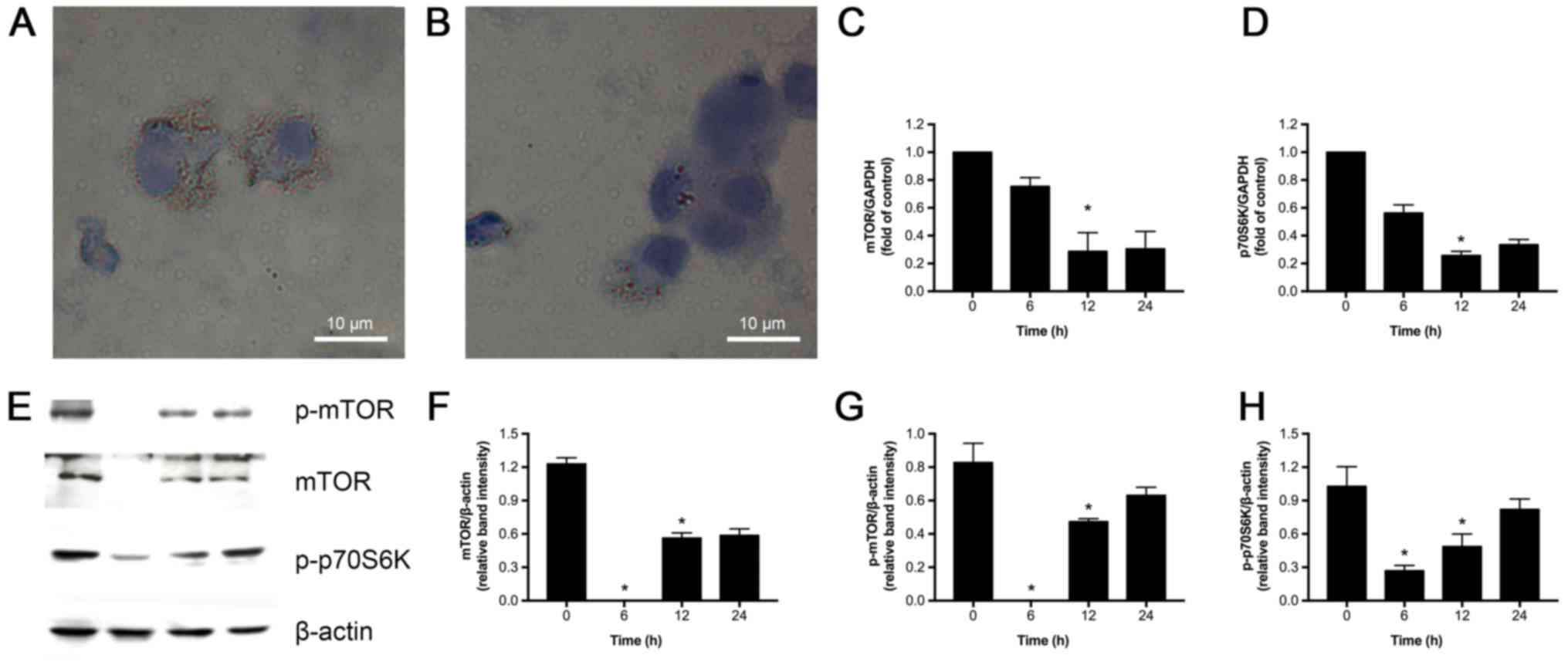
Rapamycin reduces lipid droplet
accumulation and inhibits mTOR signaling in ox-LDL, PA and LPA
induced foam cells
We next investigated if rapamycin could inhibit mTOR
in U937-derived foam cells induced by PA, ox-LDL and LPA.As
expected, Oil red O staining revealed accumulation of lipid
droplets in the foam cells treated with PA, ox-LDL and LPA, which
was significantly decreased by 100 nM rapamycin (Fig. 4A and B). The effects of rapamycin
on mRNA expression of mTOR and p70S6K were also assessed. As shown
in Fig. 4C and D, exposure to 100
nM rapamycin at varying time-points (6, 12, and 24 h) decreased
mTOR and p70S6K expression compared to control (0 h), with the most
significant inhibitory effect observed at 12 h. Consistent with the
above observations, western blot analysis revealed that 12 h
rapamycin treatment substantially reduced mTOR, p-mTOR, and
p-p70S6K (P<0.05) protein expression (Fig. 4E-H). Based on these observations,
we conclude that rapamycin suppresses mTOR signaling, and we used a
12 h treatment in the subsequent experiments.
Rapamycin increases SIRT1 signaling
but decreases NF-κB activity in foam cells
Next, we investigated the functional relationship
between mTOR and SIRT1 signaling. To do so we measured the
expression of SIRT1 signaling factors, including SIRT1, LXRα, CCR7,
ABCA1 and NF-κB, in the foam cells in the absence or presence of
rapamycin using real-time RT-PCR and Western blot analyses. SIRT1,
LXRα, CCR7 and ABCA1 mRNA levels were significantly decreased but
NF-κB and its downstream factor TNF-α was upregulated in foam
cells. However, rapamycin (100 nM) treatment for 12 h significantly
increased SIRT1, LXRα, CCR7 and ABCA1 mRNA levels but decreased the
NF-κB and TNF-α (Fig. 5A-F). In
line with the above observations, SIRT1, LXRα and CCR7 protein
expression were decreased but NF-κB and TNF-α were increased in
foam cells, and these changes were reversed by 12 h rapamycin
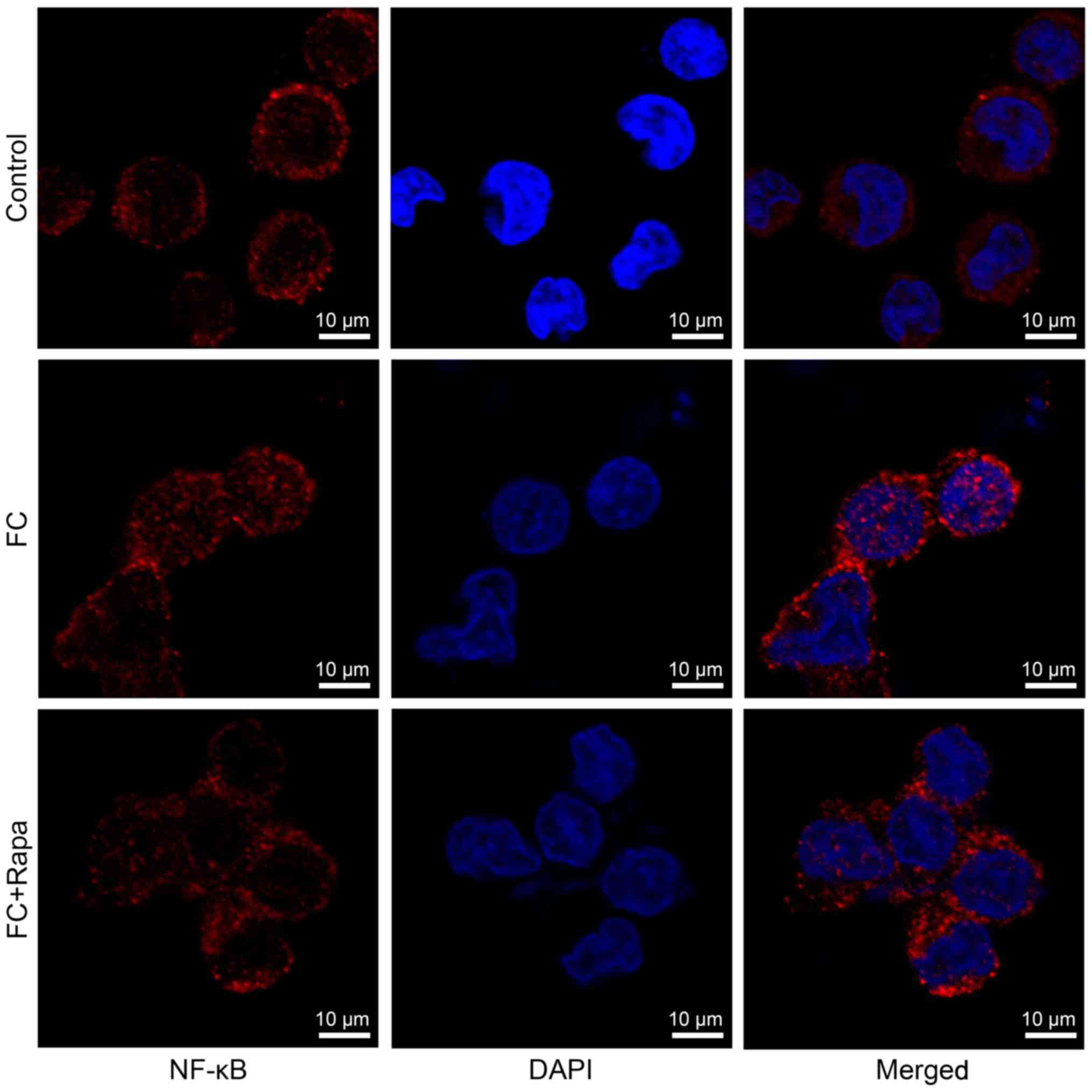
treatment (Fig. 5G-L).
Immunofluorescence analysis showed that foam cells exhibited more
NF-κB nuclear staining compared to the control cells, suggesting
that NF-κB signaling is activated in foam cells. However, this
increased nuclear expression of NF-κB was decreased by rapamycin
treatment (Fig. 6). Thus, our data
indicate that rapamycin increases SIRT1 signaling but decreases
NF-κB activity in foam cells.
Discussion
In the present study, we established a cell model of
foam cell formation, which is the main pathological process in
early AS. When U937 cells were cultured in the presence of high fat
and high cholesterol, mTOR and p70S6K expression, as well as their
phosphorylated forms, were upregulated indicating that mTOR
signaling was activated during foam cell formation. These findings
are consistent with accumulating evidence showing that mTOR
signaling plays an imperative role in lipid metabolism (26) and AS (27). In our in vitro model, we
further explored the functional link between mTOR and SIRT1
signaling during the formation of foam cells and concluded that
mTOR promotes foam cell formation through suppressing SIRT1
signaling.
Recent studies supported the notion that mTOR
signaling contributes to AS pathogenesis and that mTOR inhibitors,
such as rapamycin, have pleiotropic anti-atherosclerotic effects
that prevent or delay AS progression (8). In the present study, we used U937
cells to establish a cell model of foam cell formation induced by
PA + ox-LDL, and we also measured the expression levels of mTOR and
SIRT1in foam cells treated with LPA, which mimics atherosclerotic
lesions in vivo. LPA, as a lipid mediator, has been reported
to play important roles in inflammation and AS (28,29).
Consistent with the above reports, we observed increased levels of
mTOR and its downstream p70S6K in foam cells, suggesting that mTOR
signaling is activated during foam cell formation. As expected,
these increases were attenuated by 100 nM rapamycin, with a maximum
inhibition achieved at 12 h in PA + ox-LDL-induced foam cells.
Intriguingly, the increase in mTOR activity was coincidental with
the decrease in SIRT1 expression, and rapamycin also suppressed the
decrease in SIRT1. Similarly, in the LPA induced atherosclerotic
lesion model, mTOR signaling was significantly increased but SIRT1
was decreased. Previous findings revealed that SIRT1 negatively
mediated mTOR under stress conditions (30), and that SIRT1 negatively regulated
mTOR and vice versa (23,24). Our findings further support the
potential link between elevated mTOR signaling and decreased SIRT1
activity in foam cells, although the evidence for a direct
functional correlation between these two remains unknown.
Normal mTOR activity in endothelial cells requires
proper cholesterol trafficking (31). LXRα and its downstream factors,
CCR7 and ABCA1, contribute to cholesterol trafficking, and thus are
involved in AS regression. For instance, increased expression of
CCR7, as a requisite factor for DC migration, was shown to play a
crucial role in AS regression in ApoE−/−mice
(17). Also, LXR-CCR7 signaling
participated in monocyte-derived cell egression during AS
regression in mice (18).
Therefore, activation of the LXRα-CCR7-ABCA1axis is believed to
mitigate AS. Indeed, in the present study, LXRα expression, and its
downstream factors CCR7 and ABCA1, were reduced during foam cell
formation. However, rapamycin treatment rescued this reduction.
Combined with our previous findings suggesting that SIRT1 prevents
AS through LXRα signaling (21),
it is possible that mTOR inhibition may retard AS pathogenesis and
promote foam cell egression by upregulating SIRT1, further
enhancing the LXRα-CCR7 signaling pathway. Therefore, we believe
that the SIRT1-LXRα-CCR7 signaling pathway mediates
rapamycin-induced beneficial effects on AS.
AS is also regarded as a low level chronic
immumo-inflammatory disease. NF-κB, a core transcription factor
involved in the pro-inflammatory response, controls expression of
several important genes (including TNF-α) directing the initiation
and progression of AS (19).
Activated endothelial NF-κB signaling promotes macrophage
recruitment to atherosclerotic plaques (32). More specifically,
macrophage-derived foam cells can secrete TNF-α and other cytokines
to recruit immune cells, contributing to the formation of
atherosclerotic plaques, further increasing the risk of
cardiovascular events (19). Also,
inflammation downregulated the expression of ABCA1 and LXRα, which
may reduce cholesterol efflux (33). However, SIRT1 inhibits the
transcriptional activity of NF-κB via deacetylation (34), and regulates the efficiency of
NF-κB signaling via altering pro-inflammatory responses (35). Consistent with the above
observations, our study showed that NF-κB expression and its target
gene, TNF-α, were enhanced during foam cell formation, but this
effect was reversed by rapamycin, indicating that mTOR inhibition
may suppress foam cell formation by enhancing SIRT1-NF-κB
signaling.
Calorie restriction (CR), which limits calorie
intake without compromising essential nutrients, could extend
lifespan and delay age-related diseases, such as AS, by increasing
SIRT1 activity (36). Our previous
study showed that SIRT1 expression was increased under CR (21). In addition, it was shown that CR
slowed aging and delayed age-associated diseases by deactivating
the mTOR pathway (37,38), indicating that both SIRT1 and mTOR
signaling are involved in CR-linked responses. Similarly, in the
present study we found that SIRT1 was activated by mTOR inhibition
in foam cells, followed by enhanced expression of LXR and CCR7 and
suppressed NF-κB signaling. Thus, it appears that the functional
interaction between mTOR and SIRT1 represents a general paradigm
that is involved in different pathophysiological settings. We
speculate that mTOR inhibition may selectively increase
SIRT1-LXR-CCR7 and decrease NF-κB signaling, thus repressing foam
cell formation and promoting foam cell egression (Fig. 7).
In conclusion, in the present study we demonstrated
that mTOR signaling is upregulated during foam cell formation
induced by PA and ox-LDL, as well as LPA-mediated AS that mimics a
high fat and high cholesterol environment. Furthermore, we showed
that potentiated mTOR signaling is coincident with enhanced NF-κB
and decreased SIRT1-LXRα-CCR7 signaling. The changes in the
above-mentioned signaling pathways were reversed by the mTOR
inhibitor, rapamycin. Hence, mTOR signaling may promote foam cell
formation and inhibit foam cell egression through suppressing
SIRT1-LXR-CCR7 and enhancing NF-κB signaling, while mTOR inhibition
or upregulation of SIRT1 signaling may promote foam cell egress and
reduce atherosclerotic plaques clinically.
Acknowledgements
The work was supported by grants from the Natural
Science Foundation of China (no. 81270382), the Medical Scientific
Research Foundation of Guangdong Province (no. A2014445) and the
Research Fund for the Doctoral Program of Higher Education (no.
20134402110004).
Glossary
Abbreviations
Abbreviations:
|
AS
|
atherosclerosis
|
|
SIRT1
|
sirtuin 1
|
|
LXR
|
liver X receptor
|
|
CCR7
|
C-C chemokine receptor type 7
|
|
NF-κB
|
nuclear factor-κB
|
|
mTOR
|
mammalian target of rapamycin
|
|
PMA
|
phorbol 12-myristate 13-acetate
|
|
TNF-α
|
tumor necrosis factor-α
|
|
LPA
|
lysophosphatidic acid
|
|
ABCA1
|
adenosine triphosphate-binding
cassette transporter A1
|
|
APC
|
antigen-presenting cells
|
|
DCs
|
dendritic cells
|
|
ox-LDL
|
oxidized low density lipoprotein
|
|
DAPI
|
4′6-diamidino-2-phenylindole
dihydrochloride
|
|
p70S6K
|
p-ribosomal protein S6 kinase
|
|
RT
|
room temperature
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
|
|
SDS
|
sodium dodecyl sulfate
|
|
BSA
|
bovine serum albumin
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
TBS
|
Tris-buffered saline
|
References
|
1
|
Yusuf S, Reddy S, Ounpuu S and Anand S:
Global burden of cardiovascular diseases: Part I: General
considerations, the epidemiologic transition, risk factors, and
impact of urbanization. Circulation. 104:2746–2753. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al:
Heart disease and stroke statistics-2014 update: A report from the
American Heart Association. Circulation. 129:e28–e292. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Falk E: Pathogenesis of atherosclerosis. J
Am Coll Cardiol. 47:(8 Suppl). C7–C12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cruzado JM: Nonimmunosuppressive effects
of mammalian target of rapamycin inhibitors. Transplant Rev
(Orlando). 22:73–81. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang Z and Ming XF: mTOR signalling: The
molecular interface connecting metabolic stress, aging and
cardiovascular diseases. Obes Rev. 13:(Suppl 2). S58–S68. 2012.
View Article : Google Scholar
|
|
6
|
Yang Q and Guan KL: Expanding mTOR
signaling. Cell Res. 17:666–681. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sciarretta S, Volpe M and Sadoshima J:
Mammalian target of rapamycin signaling in cardiac physiology and
disease. Circ Res. 114:549–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martinet W, De Loof H and De Meyer GR:
mTOR inhibition: A promising strategy for stabilization of
atherosclerotic plaques. Atherosclerosis. 233:601–607. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Johnson SC, Rabinovitch PS and Kaeberlein
M: mTOR is a key modulator of ageing and age-related disease.
Nature. 493:338–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mueller MA, Beutner F, Teupser D, Ceglarek
U and Thiery J: Prevention of atherosclerosis by the mTOR inhibitor
everolimus in LDLR−/− mice despite severe
hypercholesterolemia. Atherosclerosis. 198:39–48. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen WQ, Zhong L, Zhang L, Ji XP, Zhang M,
Zhao YX, Zhang C and Zhang Y: Oral rapamycin attenuates
inflammation and enhances stability of atherosclerotic plaques in
rabbits independent of serum lipid levels. Br J Pharmacol.
156:941–951. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma KL, Ruan XZ, Powis SH, Moorhead JF and
Varghese Z: Anti-atherosclerotic effects of sirolimus on human
vascular smooth muscle cells. Am J Physiol Heart Circ Physiol.
292:H2721–H2728. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mathis AS, Jin S, Friedman GS, Peng F,
Carl SM and Knipp GT: The pharmacodynamic effects of sirolimus and
sirolimus-calcineurin inhibitor combinations on macrophage
scavenger and nuclear hormone receptors. J Pharm Sci. 96:209–222.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu M, Kang X, Xue H and Yin H: Toll-like
receptor 4 is up-regulated by mTOR activation during THP-1
macrophage foam cells formation. Acta Biochim Biophys Sin
(Shanghai). 43:940–947. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thomson AW, Turnquist HR and Raimondi G:
Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol.
9:324–337. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sordi V, Bianchi G, Buracchi C, Mercalli
A, Marchesi F, D'Amico G, Yang CH, Luini W, Vecchi A, Mantovani A,
et al: Differential effects of immunosuppressive drugs on chemokine
receptor CCR7 in human monocyte-derived dendritic cells: Selective
upregulation by rapamycin. Transplantation. 82:826–834. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trogan E, Feig JE, Dogan S, Rothblat GH,
Angeli V, Tacke F, Randolph GJ and Fisher EA: Gene expression
changes in foam cells and the role of chemokine receptor CCR7
during atherosclerosis regression in ApoE-deficient mice. Proc Natl
Acad Sci USA. 103:3781–3786. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feig JE, Pineda-Torra I, Sanson M, Bradley
MN, Vengrenyuk Y, Bogunovic D, Gautier EL, Rubinstein D, Hong C,
Liu J, et al: LXR promotes the maximal egress of monocyte-derived
cells from mouse aortic plaques during atherosclerosis regression.
J Clin Invest. 120:4415–4424. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takeda-Watanabe A, Kitada M, Kanasaki K
and Koya D: SIRT1 inactivation induces inflammation through the
dysregulation of autophagy in human THP-1 cells. Biochem Biophys
Res Commun. 427:191–196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zeng HT, Fu YC, Yu W, Lin JM, Zhou L, Liu
L and Wang W: SIRT1 prevents atherosclerosis via liver-X-receptor
and NF-κB signaling in a U937 cell model. Mol Med Rep. 8:23–28.
2013.PubMed/NCBI
|
|
22
|
Zhang S, Cai G, Fu B, Feng Z, Ding R, Bai
X, Liu W, Zhuo L, Sun L, Liu F and Chen X: SIRT1 is required for
the effects of rapamycin on high glucose-inducing mesangial cells
senescence. Mech Ageing Dev. 133:387–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Medvedik O, Lamming DW, Kim KD and
Sinclair DA: MSN2 and MSN4 link calorie restriction and TOR to
sirtuin-mediated lifespan extension in saccharomyces cerevisiae.
PLoS Biol. 5:e2612007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghosh HS, McBurney M and Robbins PD: SIRT1
negatively regulates the mammalian target of rapamycin. PLoS One.
5:e91992010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cui MZ: Lysophosphatidic acid effects on
atherosclerosis and thrombosis. Clin Lipidol. 6:413–426. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lamming DW and Sabatini DM: A Central role
for mTOR in lipid homeostasis. Cell Metab. 18:465–469. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma KL, Liu J, Wang CX, Ni J, Zhang Y, Wu
Y, Lv LL, Ruan XZ and Liu BC: Activation of mTOR modulates SREBP-2
to induce foam cell formation through increased retinoblastoma
protein phosphorylation. Cardiovasc Res. 100:450–460. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fueller M, Wang DA, Tigyi G and Siess W:
Activation of human monocytic cells by lysophosphatidic acid and
sphingosine-1-phosphate. Cell Signal. 15:367–375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bot M, Bot I, Lopez-Vales R, van de Lest
CH, Saulnier-Blache JS, Helms JB, David S, van Berkel TJ and
Biessen EA: Atherosclerotic lesion progression changes
lysophosphatidic acid homeostasis to favor its accumulation. Am J
Pathol. 176:3073–3084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ghosh HS, McBurney M and Robbins PD: SIRT1
negatively regulates the mammalian target of rapamycin. PLoS One.
5:e91992010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu J, Dang Y, Ren YR and Liu JO:
Cholesterol trafficking is required for mTOR activation in
endothelial cells. Proc Natl Acad Sci USA. 107:4764–4769. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gareus R, Kotsaki E, Xanthoulea S, van der
Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de
Winther MP and Pasparakis M: Endothelial cell-specific NF-κB
inhibition protects mice from atherosclerosis. Cell Metab.
8:372–383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead
JF and Varghese Z: Inflammatory stress exacerbates lipid
accumulation in hepatic cells and fatty livers of apolipoprotein E
knockout mice. Hepatology. 48:770–781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-κB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Salminen A, Kauppinen A, Suuronen T and
Kaarniranta K: SIRT1 longevity factor suppresses NF-κB-driven
immune responses: Regulation of aging via NF-kappaB acetylation?
Bioessays. 30:939–942. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cohen HY, Miller C, Bitterman KJ, Wall NR,
Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R and Sinclair
DA: Calorie restriction promotes mammalian cell survival by
inducing the SIRT1 deacetylase. Sci. 305:390–392. 2004. View Article : Google Scholar
|
|
37
|
Blagosklonny MV: Calorie restriction:
decelerating mTOR-driven aging from cells to organisms (including
humans). Cell Cycle. 9:683–688. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma L, Dong W, Wang R, Li Y, Xu B, Zhang J,
Zhao Z and Wang Y: Effect of caloric restriction on the SIRT1/mTOR
signaling pathways in senile mice. Brain Res Bull. 116:67–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|