Introduction
Inflammation has been implicated in the
pathophysiology of type 2 diabetes. During insulitis, activated
macrophages and T-cells release cytokines, including interleukin
(IL) 1β, tumor necrosis factor (TNF)-α and interferon (IFN)-γ,
close to β-cells, contributing to β-cell dysfunction and death
(1,2). Additionally, there is evidence that
elevated levels of IL1β, IL6, monocyte chemotactic protein 1 and
C-reactive protein are predictive of type 2 diabetes (3). Previous findings suggest that islet
β-cells secrete cytokines by themselves, which induce inflammatory
responses. In this process, thioredoxin-interacting protein (TXNIP)
is induced by endoplasmic reticulum (ER) stress through the protein
kinase RNA-like endoplasmic reticulum kinase (PERK) and
inositol-requiring enzyme 1 (IRE1) pathways, which activates the
production of IL1β by the NOD-like receptor (NLRP) 3/caspase1
inflammasome, and mediates ER stress-mediated β-cell death
(4).
Autophagy is a major pathway for the delivery of
proteins, organelles, lipids, DNA and RNA to lysosomes, where they
are to be degraded and recycled in the vacuole. Autophagy provides
pools of raw material for anabolic processes and drives a
continuous flow of materials in a degradation-regeneration cycle
within the cell (5).
Paradoxically, autophagy is considered to be a cell-fate decision
maker and may lead to a form of non-apoptotic cell death, which is
termed type 2 programmed cell death (6). Therefore, whether autophagy protects
cells from diverse types of injuries or promotes cell death may
depend on either the cellular or the environmental context.
At present, it is known that autophagy is induced by
ER stress in mammalian cells and in plants (7,8).
Although the molecular mechanism linking ER stress and autophagy
remains to be elucidated, the process of cleavage and lipidation of
microtubule-associated protein 1 light chain 3 (LC3) into LC3-II is
reportedly mediated by the phosphorylation of PERK/eukaryotic
translation initiation factor 2A (eIF2a) (6). No autophagosomes were observed in an
ire1β-knockout mutant when treated with ER stress agents,
suggesting that the ER stress sensor, IRE1βb is required for ER
stress-induced autophagy (8,9).
All of the above factors may be involved in
diabetes. In the present study, the potential involvement of
autophagy in the inflammatory response was investigated in INS-1
cells. In addition, whether IRE-1-pathway-mediated ER stress is
involved in the associated between inflammation and autophagy in
diabetes was examined.
Materials and methods
Cell culture
INS-1 cells from rats (cells were provided by the
Department of Endocrinology, The Second Affiliated Hospital of
Harbin Medical University, Heilongjiang, China) were passaged in
RPMI-1640 medium (Hyclone; GE Healthcare Life Sciences, USA),
supplemented with 10% (v/v) fetal bovine serum (FBS; Sijiqing,
Hangzhou, China), in a humidified atmosphere containing 95% air and
5% CO2 (10). The cells
were treated with 100 ng/ml lipopolysaccharide (LPS) to activate ER
stress-induced inflammatory cytokines, including IL1β and
caspase-1. 4-phenylbutyric acid (PBA; 2.5 mmol/l; pH 7.4; Sigma;
Merck Millipore, Darmstadt, Germany) was used to inhibit the IRE-1
pathway and mediate ER stress, and 3-methyadenine (3-MA; 5 mmol/l;
Sigma; Merck Millipore) was used to inhibit autophagy (10,11).
Cell Counting kit-8 (CCK8) viability
assay
The viability of cells was assessed using CCK8
(Dojindo Laboratories, Kumamoto, Japan) according to the
manufacturer's protocol. CCK8 is more sensitive than
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay
(10). The INS-1 cells were plated
in 96-well plates at a density of 5×107 l−1
and treated with 10 ml CCK8 at 37°C for 1 h. Absorbance was
measured at 450 nm using a microplate reader (10).
Annexin V/propidium iodide (PI)
staining
Cell death was determined using flow cytometry. The
INS-1 cells were stained with Annexin V and PI using the
Annexin-V-FLUOS staining kit (Roche Diagnostics, Basel,
Switzerland) in accordance with the manufacturer's protocol.
Briefly, the cells were plated in 24-well plates at a density of
1.2×105 cells per cm2, treated with lysosomal
proteases and stained with an incubation buffer containing Annexin
V and PI for 10 min at 15–25°C in the dark. Images of the apoptotic
cells were captured using a fluorescence microscope (Nikon Eclipse
TE2000-U; Nikon Corporation, Tokyo, Japan) with an excitation
wavelength in the range of 450–500 nm and detection in the range of
515–565 nm (12).
Western blot analysis
Cells were washed with PBS and lysed in lysis buffer
[62.5 mM Tris-HCl (pH 6.8), 2% SDS, 5% BME, 1% TritonX-100, 1 mM
EDTA, 1 mM EGTA, 10 mM DTT and 1 mm Na3VO4).
The lysates were then incubated on ice for 30 min and centrifuged
at 8,000 × g for 5 min at 4°C. Protein concentrations were
determined using the Bicinchoninic Acid method. Equal quantities of
protein (50 µg) were resolved by 10–15% SDS-PAGE, transferred onto
polyvinylidine difluoride membranes and blocked with 5% skim milk
at room temperature for 1 h. The membranes were incubated with
specific primary antibodies overnight at 4°C. The following
antibodies were used at a 1:1,000 dilution (unless otherwise
indicated): LC3 (Cell Signaling Technology, Inc., Danvers, MA, USA;
cat. no. 2775), IL1β (Cell Signaling Technology, Inc.; cat. no.
2002), caspase-1 (Abcam, Cambridge, UK; cat. no. 2225), C/EBP
homologous protein (CHOP; Cell Signaling Technology, Inc.; cat. no.
2895) and β-actin (Cell Signaling Technology, Inc.; cat. no. 8475).
The membranes were washed with PBS-0.1% Tween-20, then incubated
with horseradish peroxidase-conjugated secondary antibodies
(1:5,000; Sigma; Merck Millipore) at room temperature for 1 h
(10). Protein bands were detected
using Enhanced chemiluminescence (Beyotime Institute of
Biochemistry, Haimen, China). Immunoblots were quantified by
densitometric analysis using Image Lab software v2.0.1 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The quantification of
protein phosphorylation was normalized to the corresponding total
protein expression. The relative expression level of a certain
protein was normalized to β-actin and experiments were repeated in
triplicate.
RNA analysis
To analyze mRNA expression, total RNA was extracted
using TRNzol reagent (Tiangen Biotech Co., Ltd., Beijing, China;
cat. no. DP405-02) according to the manufacturer's instructions.
The purity and integrity of the RNA were examined spectroscopically
prior to obtaining cDNA using the Nanodrop 2000. Reverse
transcription was performed using the PrimeScript™ RT reagent kit
with gDNA Eraser system (Takara Bio, Inc., Otsu, Japan; cat. no.
RR047B) according to the manufacturer's instructions. The reactions
were set up with the following master mix: 2.0 µl gDNA Eraser
Buffer, 1.0 µl gDNA Eraser, 1 µg Total RNA and 6.0 µl RNase Free
H2O, which was incubated for 2 min at 42°C. This master
mix was then mixed with 1.0 µl PrimeScript RT Enzyme Mix I, 1.0 µl
RT Primer Mix, 4.0 µl PrimeScript Buffer 2 and 4.0 µl RNase Free
H2O, and incubated at 37°C for 15 min then at 85°C for 5
sec. For thermocycling reactions, the ABI prism 7500 sequencer
detection system was used. Using the SYBR® Premix Ex
Taq™ II (Tli RNaseH Plus) and the ROX plus reaction system (both
Takara Bio, Inc.; cat. no. RR82LR), the following thermocycling
conditions were applied: 95°C for 30 sec, then 45 cycles of 95°C
for 5 sec and 60°C for 40 sec. The relative quantity of each
transcript was calculated by a standard curve of quantification
cycle values for serial dilutions of a cDNA sample. PCR was
performed in triplicate for each sample. The following sets of
primers were used for PCR analysis: Rat actin forward
5′-GGAGATTACTGCCCTGGCTCCTA-3′ and reverse
5′-GACTCATCGTACTCCTGCTTGCTG-3′; rat binding immunoglobulin heavy
chain protein (Bip), forward 5′-GAATCCCTCCTGCTCCCCGT-3′ and reverse
5′-TTGGTCATTGGTGATGGTGATTTTG-3′, rat activating transcription
factor 4 (ATF4), forward 5′-TATGAGCCCTGAGTCCTACCTG-3′ and
5′-CTGCTGTCTTGTTTTGCTCCAT-3′. The primers used for analysis were
designed and synthesized by Invitrogen; Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). All experiments were performed in
triplicate and the relative mRNA levels were analyzed using the
2−ΔΔCq method (13).
Statistical analysis
The results are expressed as the mean ± standard
error of the mean. Comparisons of a single variable in >2 groups
were analyzed using one-way analysis of variance followed by
Tukey's multiple comparison tests using GraphPad Prism (GraphPad
Software, Inc., La Jolla, CA, USA). Statistical analysis was
performed using the paired and unpaired t-test between two groups
using SPSS 12.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
LPS induces the production of IL1β,
inflammasome activation and apoptosis in INS-1 cells
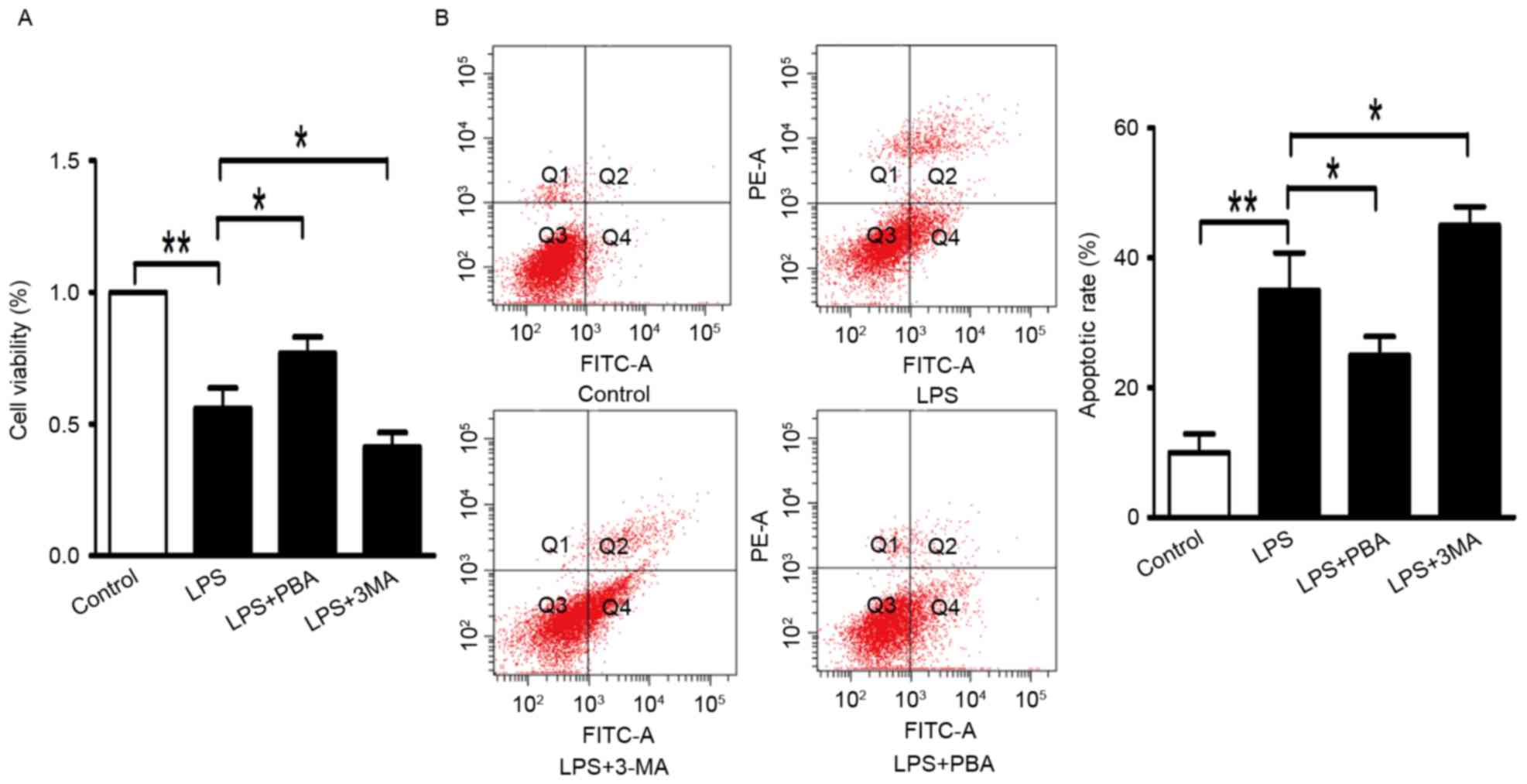
The INS-1 cells were incubated with 100 ng/ml LPS in
RPMI-1640 for 24 h. As shown in Fig.
1A, the viability of the INS-1 cells analyzed using the CCK8
assay was reduced to 64% with LPS treatment, compared with that in
the control group. Apoptosis of cells in the LPS group was
increased by 26.8%, determined using an Annexin V-FITC/PI
quantification assay (Fig.
1B).
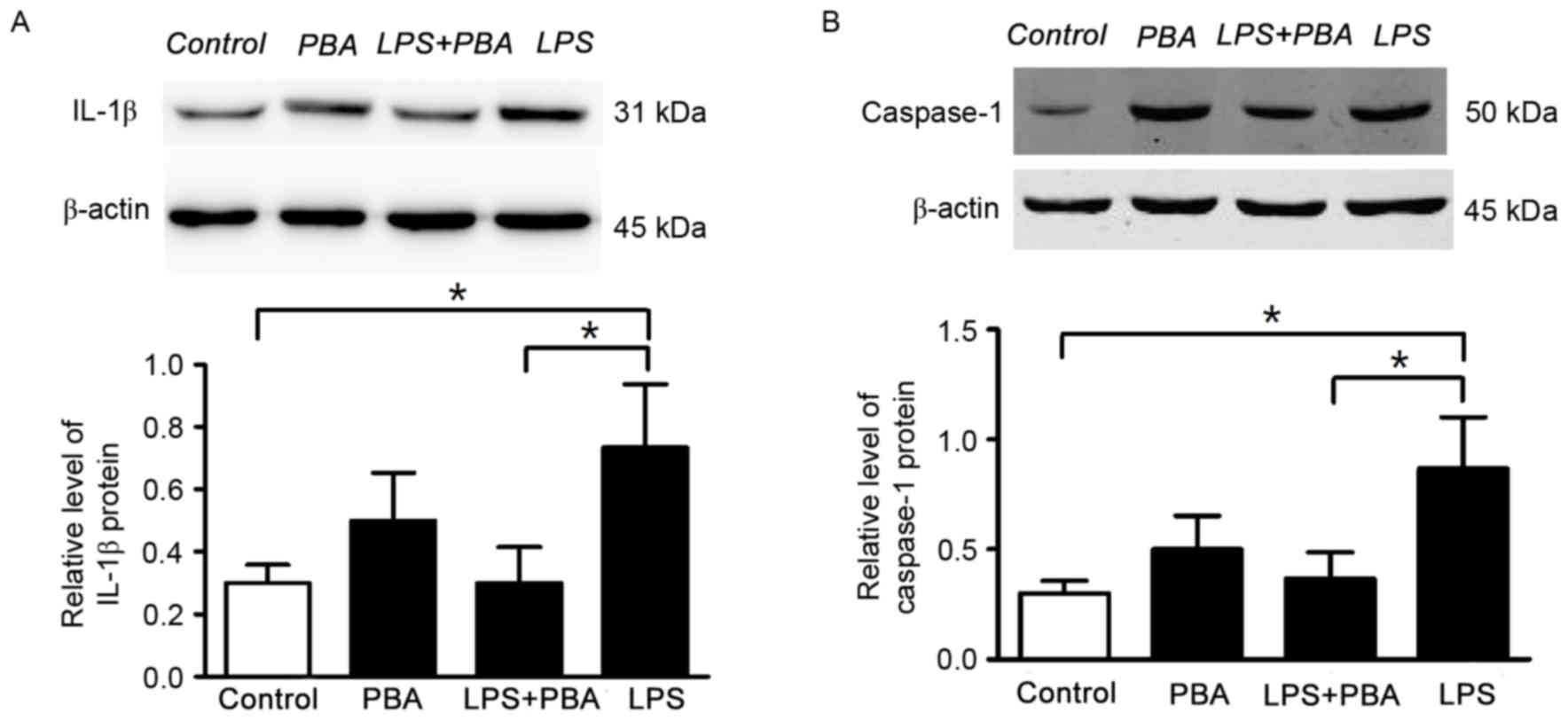
PBA attenuates LPS-induced production
of IL1β and enhances survival of in INS-1 cells
The INS-1 cells were treated with LPS and PBA. PBA
is a low-molecular-weight compound, which is known to reduce the
load of unfolded proteins in the ER by facilitating folding
capacity and trafficking of mutant proteins out of the ER. Ozcan
et al (14) and Tang et
al (15) demonstrated that PBA
decreases the activation of IRE-1, but not of the PERK or ATF6
pathways. On the basis of previous experimental analysis, the
present study used PBA as a chemical chaperone to inhibit IRE-1
pathway-mediated ER stress. The survival of INS-1 cells was
improved to 78% (Fig. 1A), and the
percentage of apoptotic cells was reduced to 21.2% (Fig. 1B) following treatment with LPS and
PBA (5 mmol/l) for 24 h. In the LPS+PBA-treated INS-1 cells, the
protein expression levels of IL-1β (Fig. 2A) and caspase-1 (Fig. 2B) were reduced, compared with those
in the groups treated with LPS alone. These results suggested that
the modulation of ER stress reduced the production of inflammatory
cytokine (IL-1β and improved INS-1 cell survival.
IRE-1-pathway-mediated ER stress is
involved in the process of inflammation and autophagy
As LC3B-II is a key protein associated with
autophagy, the conversion of LC3B-I to LC3B-II was examined in the
present study. There were fewer autophagic vacuoles (AVs; Fig. 3A, blue arrowheads), and a number of
swelling mitochondria (Fig. 3A,
green arrowheads) detected in the LPS+PBA group, determined using
EM. In addition, the LPS-induced LC3B-II protein levels were
reduced in the LPS+PBA group, compared with those in the
LPS-treated group (Fig. 3B). These
results suggested that IRE-1-pathway-mediated ER stress was
involved in the association between inflammation and autophagy, and
in promoting the processes of inflammation and autophagy.
Autophagy decreases INS-1 cell death
induced by the inflammatory response
To determine the role of autophagy in the
inflammatory response, the INS-1 cells were treated with autophagy
inhibitor (3-MA). INS-1 cell viability was significantly reduced to
42% by treatment with LPS+3-MA (Fig.
1A). 3-MA increased the apoptotic rate to 43.8% in the LPS+3-MA
group (Fig. 1B). Additionally, the
typical autophagosome (Fig. 3A,
red arrowhead) double-limiting membrane in the LPS-treated INS-1
cells was detected by EM. Double-membrane autophagic vesicles
containing cell organelles in the cytoplasm of INS-1 cells is an
integrated autophagosome, shown by ultrastructural image analysis.
There were numerous different periods of autophagosomes (Fig. 3A, red arrowheads) and
autophagolysosomes (ALs) in the LPS group. These results indicated
that autophagy had a protective effect on the LPS-induced
inflammatory response in INS-1 cells and was necessary to maintain
the normal architecture and function of INS-1 cells. The mRNA
expression levels of Bip and ATF4, and the protein expression of
CHOP were assessed following treatment with 3-MA. The mRNA levels
of Bip and ATF4, and the protein expression of CHOP were modestly
upregulated (Fig. 4A-C). These
data suggested that autophagy protected INS-1 cells by attenuating
excessive ER stress, and decreasing cell apoptosis mediated by
PERK/CHOP-mediated ER-stress and the production of inflammatory
cytokines (IL1β).
Discussion
Type 2 diabetes is closely associated with the
activation of inflammatory signaling pathways, resulting in a
marked increase in autophagy, in addition to abnormal cytokine
production (16). These results
suggested that pancreatic β cells can secrete cytokine IL1β, which
is involved in the inflammatory response and is detrimental to cell
survival. Autophagy is also involved in type 2 diabetes, however,
whether autophagy promotes INS-1 cell death or protects cells from
injuries remains to be elucidated. Therefore, INS-1 cells were
treated with autophagy inhibitor (3-MA). The further decrease in
proliferation and increased apoptosis of INS-1 cells suggested that
autophagy protected the cells from death.
Type 2 diabetes is associated with insulin
resistance, cell depletion, and immune attack, involving
lipotoxicity, glucotoxicity and inflammation (17). Several studies have demonstrated
that the intra-islet expression of inflammatory cytokines,
particularly IL-1β, contributes to the pathogenesis of type 2
diabetes. The most prominent feature of type 2 diabetes, compared
with type 1 diabetes, is that the islet inflammatory response is
‘low-grade’ and its role in the pathophysiology of type 2 diabetes
is somewhat controversial (18).
In the present study, pancreatic β cells secreted cytokines,
including IL1β, which was involved in the inflammatory response and
is detrimental to the survival of β-cells. These results are
consistent with a previous study by Böni-Schnetzler et al
(19). This previous study
differed in that the RNA from β-cells of individuals with type 2
diabetes were analyzed, and it was revealed that the mRNA
expression of IL1β was induced by high glucose and IL1β
autostimulation, and was decreased by the IL-1 receptor antagonist
IL-1Ra. Therefore, the production of inflammatory cytokines may be
a positive feedback mechanism, and the autostimulation of IL1β is
transient and nuclear factor-κB-dependent (19).
Clinical and experimental evidence suggests that ER
stress is a potent, evolutionarily conserved response to misfolded
proteins and cellular metabolic stress, which contributes to the
life-and-death decisions of β-cells during type 2 diabetes. The
inflammatory response is frequently triggered as a consequence of
ER stress, caused either by metabolic problems or by the
accumulation of misfolded proteins (20). Oslowski et al (4) showed that TXNIP is a critical
signaling node, which links ER stress to inflammation. TXNIP
induced by ER stress is under the control of PERK and IRE1
pathways, which induce the mRNA transcription of IL1β (4). IL1β is activated by the NLRP3
inflammasome and mediates ER stress-mediated β-cell death.
Activation induces oligomerisation of the NLRP3 inflammasome and
recruits ASC through a homotypic PYD-PYD interaction. The ASC then
recruits pro-caspase-1, leading to the autocatalytic activation of
caspase-1, and these active caspase-1 hetero-tetramers can convert
inactive pro-IL1β into their bioactive and secreted forms (3). The results of the present study
demonstrated that modulating ER and increase folding capacity can
reduce inflammatory cytokines (IL1β) and improve INS-1 cell
survival. The results also suggested that IRE-1-pathway-mediated ER
stress is a key molecule linking inflammation to autophagy, which
may contribute to the death and dysfunction of β-cells, caused by a
combination of glucotoxicity and lipotoxicity (21).
Autophagy is an evolutionary conserved process,
which has two effects on cell survival. The data presented in the
present study suggested that autophagy is a cell-protection
mechanism. However, Eskelinen and Saftig suggested that, when the
disposal mechanism is carried to excess, the cells are committed to
die, dependent on autophagy (22).
In mammalian cells, autophagy is activated by ER stress, with
IRE1-JNK as a link between the two processes, and the kinase
function of IRE1 is required for the induction of autophagy.
However, PERK and ATF6 pathways are not considered to be important
in the activation of autophagy following ER stress (7). In the present study, inhibiting IRE-1
mediated ER stress not only through reducing the protein expression
of caspase-1 and IL1β, but also by depressing the LC3-II/LC3-I
ratio, compared with levels in the LPS-treated group. There are two
possible reasons for these results. It may be that autophagy, which
is induced by inflammatory cytokines, is reduced, followed by
decreased expression levels of caspase-1 and IL1β. Alternatively,
it may be that the autophagy induced by IRE-1-mediated ER stress is
decreased. However, the production of inflammatory cytokines (IL1β)
is also mediated by the IRE-1 pathway. Therefore, the results of
the present study suggested that IRE1 is a link between autophagy
and ER stress in INS-1 cells. Additionally, Gonzalez et al
(6) suggested that LC3I can be
converted to the lipidated form (LC3-II), mediated by the
phosphorylation of PERK/eIF2α, and the connection between autophagy
and ER stress requires further investigation.
Inflammasome and autophagy are essential elements of
the innate immune system, and their disruption has been implicated
in the pathogenesis of type 2 diabetes. ER has previously been
tightly linked to autophagy and inflammation, and has been
considered as an intersection integrating multiple stress
responses, and these processes are associated with the pathogenesis
of diabetes mellitus and its complications. The results of the
present study showed that the IRE1-mediated ER stress pathway
activated the production of IL1β. Autophagy was also activated by
the IRE1 pathway, which suggested that IRE1-mediated ER stress is
essential for inflammatory cytokine secretion and the activation of
autophagy. The results of the present study and previous findings,
which link autophagy and inflammasome regulation, suggest that
autophagy cooperates with inflammation and apoptosis, and the
adaptive immune system, to orchestrate cellular homeostasis against
danger signals, including ER stress. The data obtained in the
present study, when combined with previous findings, indicate that
there is a tight link between ER stress and inflammation autophagy,
suggesting that a therapeutic strategy, which aims to target the
common molecular processes altered in β-cells, may be
effective.
In conclusion, the present study indicated that LPS
induced am NLRP3-dependent proinflammatory response via excessive
ER stress. Autophagy was shown to be important in ameliorating ER
stress and NLRP3-dependent inflammatory cytokine secretion, further
reducing INS-1 cell death. It was also shown that
IRE-1-pathway-mediated ER stress is a crucial link connecting
autophagy with the NLRP3-dependent inflammatory response.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81370929) and China
Diabetes Young Scientific Talent Research Funding (2016).
Glossary
Abbreviations
Abbreviations:
|
3-MA
|
3-methyadenine
|
|
ER
|
endoplasmic reticulum
|
|
PBA
|
4-phenylbutyric acid
|
|
NLRP
|
NOD-like receptor
|
|
LC3
|
light chain 3
|
|
LPS
|
lipopolysaccharide
|
|
CCK8
|
Cell Counting kit-8
|
|
AV
|
autophagic vacuoles
|
|
AL
|
autophagolysosomes
|
References
|
1
|
Eizirik DL and Mandrup-Poulsen T: A choice
of death-the signal-transduction of immune-mediated beta-cell
apoptosis. Diabetologia. 44:2115–2133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kaminitz A, Stein J, Yaniv I and Askenasy
N: The vicious cycle of apoptotic beta-cell death in type 1
diabetes. Immunol Cell Biol. 85:582–589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Donath MY and Shoelson SE: Type 2 diabetes
as an inflammatory disease. Nat Rev Immunol. 11:98–107. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oslowski CM, Hara T, O'Sullivan-Murphy B,
Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, et
al: Thioredoxin-interacting protein mediates ER stress-induced β
cell death through initiation of the inflammasome. Cell Metab.
16:265–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gonzalez CD, Lee MS, Marchetti P,
Pietropaolo M, Towns R, Vaccaro MI, Watada H and Wiley JW: The
emerging role of autophagyin the pathophysiology of diabetes
mellitus. Autophagy. 7:2–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y, Burgos JS, Deng Y, Srivastava R,
Howell SH and Bassham DC: Degradation of the endoplasmic reticulum
by autophagy during endoplasmic reticulum stress in Arabidopsis.
Plant Cell. 24:4635–4651. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang X, Srivastava R, Howell SH and
Bassham DC: Activation of autophagy by unfolded proteins during
endoplasmic reticulum stress. Plant J. 85:83–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yin J Jing, Bo Li Y, Cao M Ming and Wang
Y: Liraglutide improves the survival of INS-1 cells by promoting
macroautophagy. Int J Endocrinol. Metab. 11:184–190. 2013.
|
|
11
|
Akerfeldt MC, Howes J, Chan JY, Stevens
VA, Boubenna N, McGuire HM, King C, Biden TJ and Laybutt DR:
Cytokine-induced beta-cell death is independent of endoplasmic
reticulum stress signaling. Diabetes. 57:3034–3044. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan JY, Cooney GJ, Biden TJ and Laybutt
DR: Differential regulation of adaptive and apoptotic unfolded
protein response signalling by cytokine-induced nitric oxide
production in mouse pancreatic beta cells. Diabetologia.
54:1766–1776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M,
Vaillancourt E, Smith RO, Görgün CZ and Hotamisligil GS: Chemical
chaperones reduce ER stress and restore glucose homeostasis in a
mouse model of type 2 diabetes. Science. 313:1137–1140. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang C, Koulajian K, Schuiki I, Zhang L,
Desai T, Ivovic A, Wang P, Robson-Doucette C, Wheeler MB, Minassian
B, et al: Glucose-induced beta cell dysfunction in vivo in rats:
Link between oxidative stress and endoplasmic reticulum stress.
Diabetologia. 55:1366–1379. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kosacka J, Kern M, Klöting N, Paeschke S,
Rudich A, Haim Y, Gericke M, Serke H, Stumvoll M, Bechmann I, et
al: Autophagy in adipose tissue of patients with obesity and type 2
diabetes. Mol Cell Endocrinol. 409:21–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu H, Cao MM, Wang Y, Li LC, Zhu LB, Xie
GY and Li YB: Endoplasmic reticulum stress is involved in the
connection between inflammation and autophagy in type 2 diabetes.
Gen Comp Endocrinol. 210:124–129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Imai Y, Dobrian AD, Morris MA and Nadler
JL: Islet inflammation: A unifying target for diabetes treatment?
Trends Endocrinol Metab. 24:351–360. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Böni-Schnetzler M, Thorne J, Parnaud G,
Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC
and Donath MY: Increased interleukin (IL)-1beta messenger
ribonucleic acid expression in beta -cells of individuals with type
2 diabetes and regulation of IL-1beta in human islets by glucose
and autostimulation. J Clin Endocrinol Metab. 93:4065–4074. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quan W, Jo EK and Lee MS: Role of
pancreatic β-cell death and inflammation in diabetes. Diabetes Obes
Metab. 3:141–151. 2013. View Article : Google Scholar
|
|
22
|
Eskelinen EL and Saftig P: Autophagy: A
lysosomal degradation pathway with a central role in health and
disease. Biochim Biophys Acta. 1793:664–673. 2009. View Article : Google Scholar : PubMed/NCBI
|