Introduction
Traumatic brain injury (TBI), a form of acquired
brain injury, is one of the leading causes of death in children and
adults worldwide. It causes long-term disability with a broad
spectrum of symptoms, including headache, confusion, dizziness,
blurred vision or tired eyes, ringing in the ears, bad taste in the
mouth, fatigue or lethargy, a change in sleep patterns, behavioral
or mood changes, and trouble with memory, concentration, attention,
or thinking (1). TBI is documented
to have detrimental effects on the central nervous system (CNS)
metabolism, including the depression of mitochondrial oxidative
phosphorylation. Studies have also provided evidence that TBI can
suppress the activity of the cytochrome oxidase (COX) and
inhibition of COX is an important aspect of trauma pathology
(2–4).
Sodium azide (NaN3) is a colorless,
explosive, and highly toxic salt that is soluble in water. Its
principal toxic action is in inhibiting the function of COX in the
mitochondrial electron transport chain (5). The tissue-specific inhibition of COX
by NaN3 could serve as a useful research tool for the
evaluation of cell death in vivo and in vitro. A
growing body of evidence suggests that neuronal cell death serves a
pivotal role in the TBI process (6–8).
Autophagy is known as one of the critical cellular homeostatic
mechanisms. Dysregulation of autophagy contributes to neuronal cell
death following TBI (9). Previous
studies from our group have demonstrated that autophagic cell death
could be induced by TBI and therefore, this pathway may serve as a
target for future treatments (10,11).
Another study has reported that NaN3 could lead to
apoptosis in primary cortical neuronal cells (12). Evidence is accumulating that
non-apoptotic cell death is associated with neuronal damage induced
by NaN3 in primary cortical neuron cultures (13). However, whether NaN3 may
induce autophagic cell death remains poorly understood.
Research into the biology of hydrogen sulfide
(H2S) over the last decade has exponentially increased
our understanding of the way in which this gasotransmitter
influences physiological and pathophysiological processes in a wide
range of biological systems (14).
H2S has long been hypothesized to be an environmental
pollutant which is a colorless, flammable, water-soluble gas
characterized by a peculiar smell of rotten eggs (15). H2S is also produced
endogenously in mammals, including humans. In particular,
cystathionine-β-synthase (CBS) in the central nervous system and
cystathionine-γ-lyase (CSE) in the cardiovascular system are the
key enzymes mostly responsible for the endogenous generation of
H2S (16).
3-mercaptopyruvate sulfurtransferase (3-MST) is also known to be a
significant producer of endogenous H2S in the brain
(17). H2S has recently
been regarded as a novel gasotransmitter, possessing very important
physiological and pharmacological functions in the brain: enhancing
N-methyl-D-aspartate receptor-mediated responses, facilitating the
induction of hippocampal long-term potentiation, and inhibiting
synaptic transmission in the hippocampus (16,18).
However, it is not known whether H2S participates in
cell physiology and autophagic pathways in the neuronal cells
treated with NaN3.
In the present study, the features of the neuronal
damage induced by NaN3 treatment were investigated.
Then, the potential neuroprotective activity of H2S and
its effect on autophagic cell death were investigated following
NaN3 treatment in neuron-like rat pheochromocytoma
(PC12) cells. This is the first study demonstrating that
NaN3 can induce autophagic cell death in PC12 cells and
that H2S can suppress this effect. The present findings
may help to gain a better insight into the physiological functions
of H2S in the normal and injured conditions and its
association with the cellular and molecular mechanisms underlying
nervous system lesion and repair.
Materials and methods
Cell culture
Rat pheochromocytoma PC12 cells were obtained from
the Shanghai Institute of Cell Biology, Chinese Academy of Sciences
(Shanghai, China). PC12 cells were grown on polystyrene tissue
culture dishes in Dulbeccos modified Eagles medium (DMEM)
containing 10% horse serum (both from Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 5% fetal bovine serum (FBS;
Sijiqing Biological Engineering Materials Co., Ltd., Hangzhou,
China), supplemented with 2 mmol/l glutamine, 100 µg/ml
streptomycin and 100 U/ml penicillin (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C with 95% air-5% CO2. Prior to
differentiation, the medium was changed twice a week and the cells
were subcultured at a ratio of 1:4 once a week. For
differentiation, the cells were washed and incubated in fresh
medium containing nerve growth factor (NGF; final concentration of
50 ng/ml) for 48 h at 37°C in a cell incubator. The addition and
concentration of NGF in the media was maintained throughout all
experiments and cell were used between passages 3–8.
Cell injury model
For simulating injury, the DMEM medium was removed,
PC12 cells were washed twice with glucose-free Earles balanced salt
solution (pH 7.5), and changed to glucose-free DMEM medium without
FBS prior to treatment. Then neurotoxic damage was induced by
adding the indicated concentrations of NaN3 for
different periods of time in the cultured cells. Cells were
preincubated with the indicated concentrations of sodium
hydrosulfide (NaHS), as a donor of H2S, for 30 min prior
to NaN3 treatment and maintained throughout the entire
experiment. NaHS was dissolved in saline and was freshly prepared
just before use. The stock solutions were directly added into the
bath solution to achieve the final concentration. Control cultures
were maintained in DMEM medium for the same duration and were left
untreated. The concentrations of all reagents were maintained
throughout the injury period.
Determination of cell viability
The viability of PC12 cells was determined by Cell
Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan), according to the manufacturers instructions. PC12
cells were cultured in 96-well plates at 37°C under an atmosphere
of 5% CO2 and 95% air. At the end of treatment, CCK-8
reagent (10 µl) was added to each well of the plates and then the
plates were incubated at 37°C for 3–4 h in the incubator.
Absorbance at a wavelength of 450 nm was measured with a microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA). The mean
optical density (OD) of 6 replicate wells in each of the indicated
groups were calculated, and the cell viability was expressed as a %
of the control. All experiments were performed in triplicate and
repeated three independent times.
Nuclear staining for assessment of
cell death
Chromosomal condensation and morphological changes
in the nucleus of PC12 cells were observed by DAPI and propidium
iodide (PI) staining. The PC12 cells were fixed with 4%
paraformaldehyde for 10 min. Following three rinses with PBS, the
cells were stained with 10 µg/ml DAPI for 10 min. PI (10 mg/ml;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was diluted in 0.9%
NaCl to a working concentration of 40 µg/ml. For detection of
PI-labeled cells, cells were fixed in 100% ethanol for 10 min at
room temperature, then were stained with PI for 30 min at room
temperature, washed 3 times in PBS and coverslipped with Permount
(Biomeda Corporation, Burlingame, CA, USA), and photographed on a
Nikon Eclipse Ti-S fluorescence microscope (Nikon Corporation,
Tokyo, Japan) using excitation/emission filters at 568/585 nm for
PI. Viable cells exhibited a normal nucleus size and uniform
fluorescence in the DAPI channel, whereas dead cells exhibited
PI/DAPI double positive staining and condensed nuclei.
Colocalization and morphometric measurements were performed using
ImageJ software, version 1.6 (National Institutes of Health,
Bethesda, MD, USA). To quantify the immunoreacted cells, the
fluorescence intensity was measured in 10 randomly selected images.
Data were obtained from at least three independent experiment.
Immunofluorescence analysis
PC12 cells in 24-well plates were fixed with 4%
paraformaldehyde for 15 min at room temperature and washed thrice
with PBS for 10 min. The cells were then blocked with 5% donkey
serum (Gibco; Thermo Fisher Scientific, Inc.) with 0.3% Triton
X-100 and 5% bovine serum albumin (BSA) for 2 h at room
temperature. Cells were incubated with rabbit polyclonal primary
antibodies targeting microtubule-associated protein 1A/1B-light
chain 3 (LC3; cat. no. ab48394; 1:100; Abcam, Cambridge, UK)
overnight at 4°C, followed by a mixture of fluorescein
isothiocyanate-conjugated secondary antibodies (1:200; cat. no.
131699; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA,
USA) for 2 h at room temperature. Following three washes with PBS
for 10 min each, 40 µg/ml PI was added for 30 min at room
temperature. Following three further washes with PBS for 10 min
each, the coverslips were mounted using Antifade Mounting Medium
(Beyotime Institute of Biotechnology, Haimen, China) and observed
with an Eclipse Ti-S fluorescence microscope (Nikon Corporation).
Colocalization and morphometric measurements were performed using
ImageJ software, version 1.6. To quantify the immunoreacted cells,
the fluorescence intensity was measured in 10 randomly selected
images. Data were obtained from at least three independent
experiment.
Western blot analysis
The cells were homogenized in lysis buffer (1%
NP-40, 50 mmol/l Tris PH 7.5, 5 mmol/l EDTA, 1% SDS, 1% sodium
deoxycholate, 1% Triton X-100, 1 mmol/l phenylmethanesulfonyl
fluoride, 10 µg/ml aprotinin, and 1 µg/ml leupeptin) and the
lysates were centrifuged at 15,000 × g for 20 min at 4°C. Following
determination of protein concentration with a Bradford assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), 50 µg of total
protein was subjected to 12% SDS-polyacrylamide gel
electrophoresis. The separated proteins were transferred to a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA) by a transfer apparatus at 90 V for 1 h. The membrane was then
blocked with 5% non-fat milk for 2 h at room temperature and
incubated overnight at 4°C with primary antibody against CBS
(1:200; cat. no. sc-67154; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), 3-MST (1:400; cat. no. sc-374326; Santa Cruz
Biotechnology, Inc.), LC3 (1:3,000; cat. no. ab48394; Abcam,
Cambridge, UK), Beclin-1 (1:500; cat. no. mb0030; Bioworld
Technology, Inc., St. Louis Park, MN, USA), sequestosome 1 (also
known as P62; 1:1,000; cat. no. ab56416; Abcam), or β-actin
(1:10,000; cat. no. jla20; Merck KGaA). Following incubation with
horseradish peroxidase-conjugated secondary antibodies (A0208,
A0216; Beyotime Institute of Biotechnology, Haimen, China), protein
signals were visualized using an enhanced chemiluminescence (ECL)
system (cat. no. 32106; Pierce; Thermo Fisher Scientific, Inc.).
For semiquantitative analysis, protein bands detected by ECL were
scanned into Adobe Photoshop CS6 (Adobe Systems, Inc., San Jose,
CA, USA) and analyzed using ImageJ software, version 1.6.
Statistical analysis
All statistical analyses were conducted with SPSS
statistical software version 16.0 (SPSS, Inc., Chicago, IL, USA).
Data are expressed as means ± standard error of the mean. The
statistical significance of differences between groups was
determined by one-way analysis of variance followed by Tukeys post
hoc multiple comparison tests, or Student t-test for two means
comparisons. P<0.05 was considered to indicate a statistically
significant difference. Each experiment consisted of at least three
replicates per condition.
Results
NaN3 causes cytotoxicity to
PC12 cells
The effect of NaN3 on the viability of
PC12 cells was measured by CCK-8 assay following treatment with a
range of concentrations from 5 to 100 mmol/l and for different
durations (1, 3, 6, 12, 18 and 24 h). As presented in Fig. 1, treatment of PC12 cells with
NaN3 for 12 h at concentrations from 20 to 100 mmol/l
resulted in a concentration-dependent reduction of cell viability.
Further analysis demonstrated that the cell survival decrease
caused by treatment with 30 mmol/l NaN3 was also
time-dependent (Fig. 2).
NaN3 induces autophagic
cell death in PC12 cells
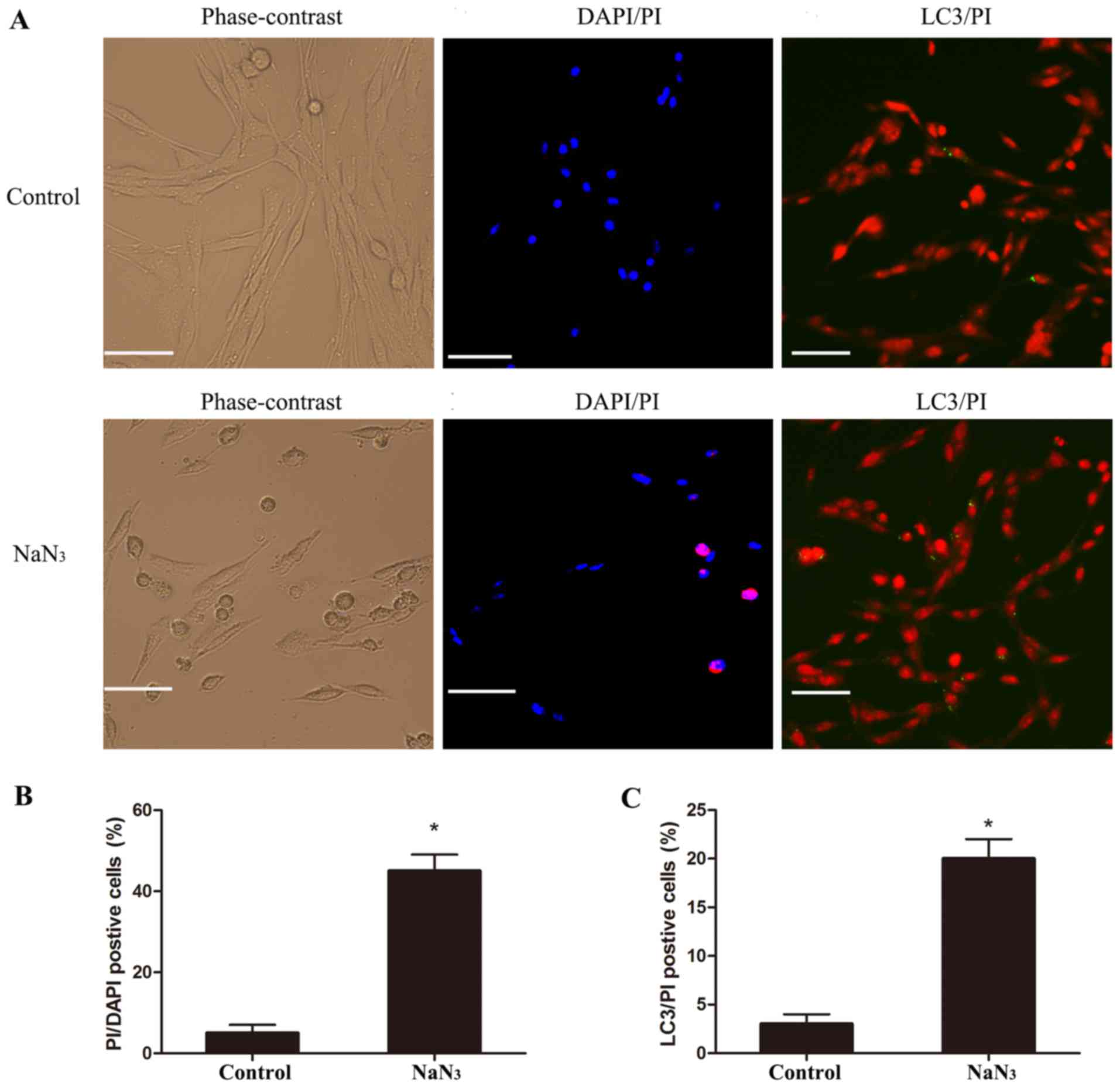
The cell morphology of the treated PC12 cells was
analyzed by PI/DAPI staining in order to evaluate cell death. By
phase-contrast microscopy, PC12 cells treated with 30 mmol/l
NaN3 for 12 h appeared more round to oval in shape,
compared with the long spindle-like shape of untreated control PC12
cells (Fig. 3A). Following DAPI/PI
double staining, the nuclei of control cells appeared round to
oval, with a separate pattern of blue fluorescence (DAPI) and red
fluorescence (PI). Upon NaN3 treatment, nuclei became
increasingly bright, decreased in size, and condensed into round
bodies (Fig. 3A). To distinguish
which type of cell death was induced by NaN3 treatment,
neuronal cultures were examined following staining for PI and for
the autophagy marker LC3. The results indicated that LC3/PI
double-positive cells existed in both the control and
NaN3-injured group (Fig.
3A). Quantification of the microscopy images confirmed that
treatment of PC12 cells with NaN3 resulted in a
significant increase in both the number of PI/DAPI-positive cells
(Fig. 3B) and the number of PI/LC3
double-positive cells (Fig. 3C),
compared with control, suggesting that NaN3 induced
autophagic cell death in PC12 cells.
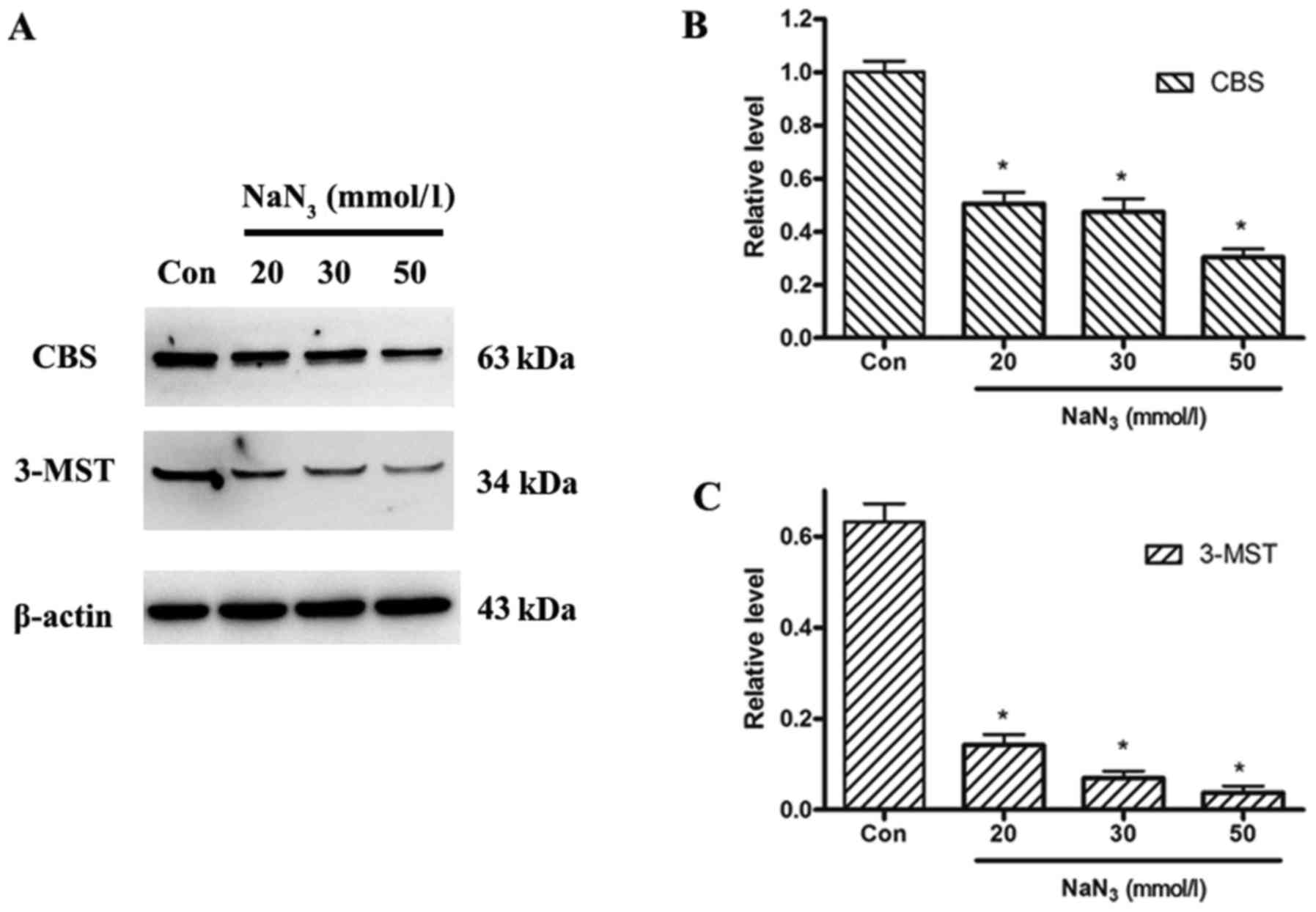
Effect of NaN3 on CBS and
3-MST protein expression in PC12 cells
To examine the expression levels of the endogenous
H2S-producing enzymes, expression of CBS and 3-MST
proteins was analyzed by western blotting in control untreated
cells and cells treated with different concentrations of
NaN3. The expression levels of both CBS and 3-MST
proteins decreased in a dose-dependent manner following
NaN3 treatment compared with control (Fig. 4).
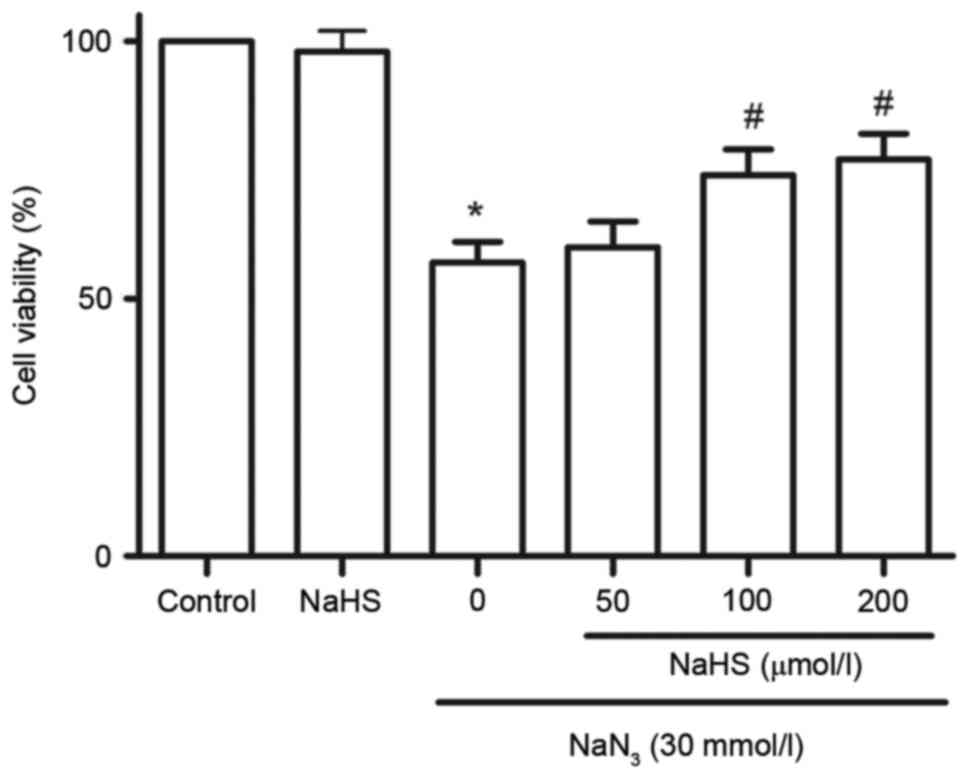
H2S protects PC12 cells
against NaN3-induced cytotoxicity
To investigate the effect of H2S on
NaN3-induced cytotoxicity, cell viability was analyzed
by CCK-8 assay. As illustrated in Fig.
5, treatment with NaN3 at concentrations of 30
mmol/l for 12 h significantly attenuated cell viability. The
cytotoxic effects of NaN3 on PC12 cells were
significantly blocked by pretreatment with 100 and 200 µmol/l NaHS
for 30 min (Fig. 5). At 200
µmol/l, NaHS alone did not affect the viability of PC12 cells
(Fig. 5).
H2S suppresses
NaN3-induced autophagic cell death in PC12 cells
Next, the effects of H2S on
NaN3-induced autophagic cell death were assessed, by
examining the protein expression levels of LC3, Beclin-1 and P62
with western blot analysis (Fig.
6A). Treatment of PC12 cells with 30 mmol/l NaN3 for
12 h significantly increased the protein expression levels of LC3
(Fig. 6B) and Beclin-1 (Fig. 6C), but decreased P62 expression
(Fig. 6D) compared with control
untreated cells. However, Pretreatment with 200 µmol/l NaHS
significantly abolished the sodium azide-induced decrease of P62
expression and the increase of LC-3 and Beclin-1 expression
(Fig. 6). These results indicated
that H2S was able to block the NaN3-elicited
downregulation of P62 expression and upregulation of LC3 and
Beclin-1 expression.
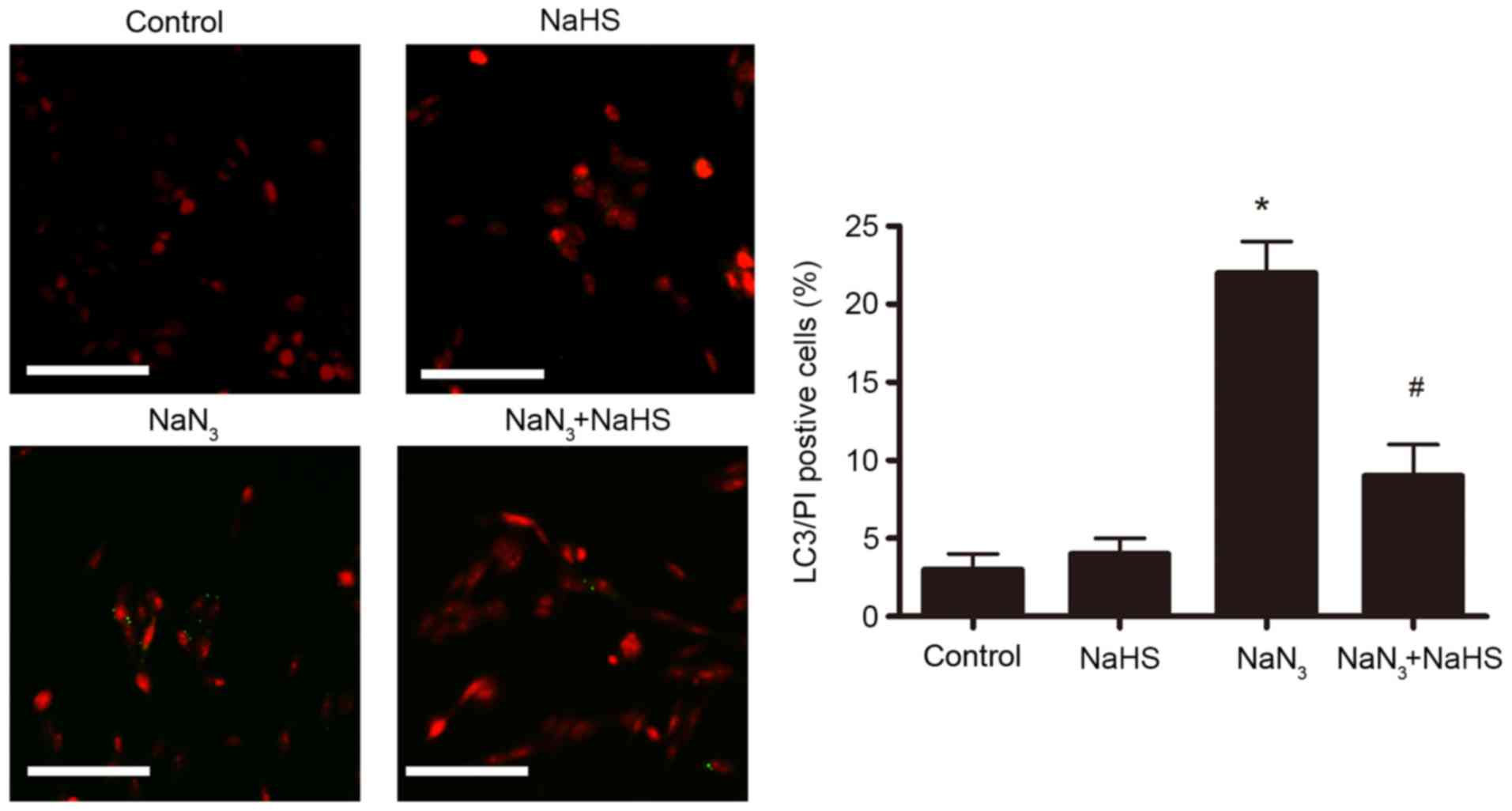
The effect of NaHS pretreatment on autophagic cell
death in PC12 cells was further examined by LC3/PI double staining
and microscopy analysis. As illustrated in Fig. 7, following treatment with
NaN3 (30 mmol/l, 12 h), the number of LC3/PI
double-positive cells significantly increased compared with the
control untreated group. Pretreatment with NaHS (200 µmol/l)
dramatically ameliorated this NaN3-induced increase of
PI/LC3 double-positive cells (Fig.
7), suggesting that H2S protected the cells from
autophagic cell death. The control and NaHS-treated groups
displayed few LC3/PI double-positive cells (Fig. 7).
Discussion
The present study examined the cell damage occurring
in cultured PC12 cells exposed to NaN3. Due to the
ability of NaN3 to induce cell death through inhibition
of the electron transfer between COX and oxygen, this in
vitro model represents an interesting tool in neurotoxicity
studies. In the present study, the possible molecular mechanisms
underlying the neuroprotective effects of H2S to protect
against NaN3-induced neuron cell injury were
investigated. The results demonstrated a concentration-dependent
loss of cell viability induced by NaN3. To explore
whether autophagic cell death was induced by NaN3,
double immunofluorescence staining was performed for the autophagy
marker LC3 and for PI. Microscopy analysis indicated that LC3
positive staining was partly colocalized with PI (a cell death
marker), implying that a proportion of dying cells were undergoing
autophagy, which is one of the mechanisms of
NaN3-induced neurotoxicity. This finding is the first
report of NaN3 inducing autophagic cell death in PC12
cells. In addition, exposure of PC12 cells to NaN3
downregulated the expression of the endogenous H2S
synthases (CBS and 3-MST) in a concentration-dependent manner,
suggesting that H2S was involved in the pathophysiology
of NaN3-induced cell injury. Furthermore, NaHS, a
H2S donor was demonstrated to prevent the
NaN3-exerted upregulation of LC3 and Beclin-1 expression
and downregulation of P62 expression. A significant reduction in
the number of LC3/PI double-positive cells was also observed
following pretreatment with NaHS, suggesting that H2S
may have yielded a protective effect against
NaN3-mediated autophagic cell death. Taken together, the
present findings suggest that H2S may be an important
protective factor against NaN3-induced neurotoxicity by
modulating the autophagic cell death pathway.
NaN3 is a highly reactive white
crystalline powder used in industry, which is also used as a
preservative in aqueous laboratory reagents and biologic fluids,
and as a fuel in automobile airbag gas generates (19). It is also a broad-spectrum biocide
used in research and agriculture. NaN3, as a COX
inhibitor, has been extensively considered as a useful tool to
study different pathological conditions. Mitochondrial energy
metabolism has been hypothesized to be a determining element to
interpret impaired neuron function, reduced molecular turnover, and
enhanced cell death (20,21). Inhibition of mitochondrial COX has
been reported to induce cell death in a variety of cells.
Programmed cell death can be classified into apoptosis, necrosis,
and autophagic cell death, and emerging evidence suggests that all
three may be important modes of cell death in neural
stem/progenitor cells (22).
Previous studies suggested that NaN3 could induce
neuronal apoptosis and necrosis, which was associated with the
mitochondrial pathway (11,13).
The role of NaN3 in apoptosis and necrosis has been the
subject of extensive investigation, however its role in autophagic
cell death remains poorly understood (23–25).
PC12 cells, which are generally considered to have
neuronal-like characteristics, appear to be more sensitive to
NaN3 compared with other neural tumor cell lines. To
induce hypoxia/hypoglycemia or oxidative stress, NaN3
concentrations used in PC12 cells range from 1 mM to 10 mM
(26,27). In order to induce autophagic cell
death, high concentrations of NaN3 were employed in the
present study. Increased autophagy is observed in several
experimental injury models (28,29).
However, it is not known whether the role of autophagy is
protective or detrimental in neural cell injury. It is possible
that the role of autophagy following cell injury is dependent upon
the cells capacity to respond in relation to the cumulative burden
of damaged or dysfunctional macromolecules and organelles. If the
increase in autophagic capacity is insufficient, augmenting
autophagy would likely be beneficial. The increase in autophagic
capacity is in excess, and inhibiting autophagy may be beneficial.
Thus, the role of autophagy may be dictated by whether or not it
can meet intracellular demands. Examining cell viability is
important in order to evaluate if the cells are still
physiologically responsive, or if they are likely to be entering
cell death. Therefore, in the present study the overall toxic
effects of NaN3 were evaluated by monitoring cell
viability in PC12 cells following treatment. Under more severe
stress conditions (30 mM NaN3), when PC12 cell viability was
severely affected, an accumulation of autophagic cell death was
observed.
The concept of autophagic cell death was first
established based on observations of increased autophagic markers
in dying cells (30). LC3, an
autophagosomal ortholog of yeast Atg8, is one of the most
reliable markers in the study of autophagy induction (7). Beclin-1, the mammalian orthologue of
yeast Atg6, has a central role in autophagy (31). More recently, autophagy flux has
been assessed in several injury models based on the levels of the
autophagic substrate protein P62 (32). P62 is an adaptor protein that
directs ubiquitinated cargo to autophagosomes for degradation. As
P62 is degraded along with its cargo, when autophagy flux is
increased, its protein levels decrease; conversely, when autophagy
flux is inhibited, P62 levels increase (33). The present results demonstrated
that treatment with NaN3 significantly increased the
amount of LC3 and Beclin-1 expression but decreased P62 expression,
suggesting that autophagy flux was increased in the
NaN3-induced injury in vitro model. To
investigate whether an increase in autophagy was beneficial or
detrimental, fluorescence microscopy analysis of LC3/PI double
staining was performed in PC12 cells. The results indicated that
LC3-positive cells were partly colocalized with PI, implying that a
proportion of dying cells were undergoing some degree of autophagy.
The nuclei of LC3/PI-positive cells appeared round, which is
consistent with autophagic cell death, and they were neither
shrunken nor fragmented as is observed in apoptotic nuclei,
demonstrating that NaN3 could induce autophagic cell
death. The present results demonstrated that NaN3
induced autophagic cell death, which is consistent with prior
reports that NaN3 induces non-apoptotic cell death
(13).
Selvatici et al (13) reported that mitochondrial
dysfunction induced by NaN3 provides a common platform
for investigating the mechanisms of neuronal injury, useful for
screening potential protective agents against neuronal death.
H2S has increasingly been recognized as an important
signaling molecule of comparable importance to nitric oxide and
carbon monoxide in mammalian systems (34). In addition to its function as a
signal molecule, H2S also functions as a cytoprotectant
in neurons and cardiac muscle (17). The neuroprotective properties of
H2S have been the focus of extensive research for
decades (35). Previous reports
from our group demonstrated that endogenous H2S is
involved in neuronal autophagy in TBI mice (10,36,37).
Endogenous H2S is generated by three distinct enzymatic
pathways mediated by CSE, CBS and 3-MST (18). CBS and 3-MST are expressed in the
brain, while CSE is widely located in other organs (38). Miyamoto et al (39) reported that CSE is not expressed in
PC12 cells and that the 3-MST pathway is primarily responsible for
H2S production in PC12 cells. In order to explore
whether H2S was involved in NaN3-induced
autophagy, the expression of endogenous H2S synthases,
CBS and 3-MST, was analyzed by western blotting. Exposure of PC12
cells to NaN3 downregulated the expression of CBS and
3-MST in a concentration-dependent manner, suggesting that
H2S may be involved in NaN3-induced cell
injury. In addition, pretreatment with NaHS, a donor for
H2S, reduced the number of LC3/PI double-positive cells,
suggesting that H2S may have protective effects against
NaN3-mediated autophagic cell death.
The potential neuroprotective mechanism of
H2S may be that it reduces oxidative stress. Biochemical
studies have demonstrated that NaN3 inhibits COX and
interferes with cellular respiration (40). The mitochondrial respiratory chain
is one of the most important sites of reactive oxygen species (ROS)
production under physiological and pathological conditions
(41). ROS generation is
considered as a cause of cell death mediated by mitochondrial
electron transport chain inhibitors in dopaminergic neuronal cells
(42). Oxidative stress caused by
overproduction of ROS is detrimental and one of the etiological
factors of many neurologic diseases (43). H2S protects various
tissues and organs from oxidative stress and also scavenges ROS
(18,44,45).
H2S also upregulates the transcription of antioxidant
genes to exert its cytoprotective effect. The kelch-like ECH
associated protein 1 (Keap1)-nuclear factor E2-related factor 2
(Nrf2) pathway is the major regulator of cytoprotective responses
to oxidative and electrophilic stress (46). The translocation of Nrf2 to the
nucleus has been suggested as a molecular mechanism for the
H2S-mediated protection (47,48).
Further study is necessary to clarify the mechanisms underlying the
protective effects of H2S on the Keap1-Nrf2 pathway in a
NaN3-induced cell injury model.
Although H2S showed a promising
neuroprotective effect in NaN3-mediated cell injury in
the present study, further research is underway to identify
selective autophagy regulators that may serve as potential targets
for treatment. In summary, the present results suggested an active
role for H2S in the process of autophagy induced by
NaN3. This is the first report of autophagic cell death
in PC12 cells induced by NaN3. The present data may
provide a potential novel pathway to elucidate the underlying
molecular and cellular mechanisms of CNS following inhibition of
COX and potential novel strategies for the treatment of CNS
diseases. Further studies, such as examining other H2S
biosynthesis enzymes of the trans-sulfuration pathway, other signal
transduction pathways involved in the H2S protective
effect, and the functional consequences of H2S on
cellular autophagy and its downstream signaling targets, will
strengthen these conclusions.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81601306, 81301039,
81530062, 81271379 and 81172911); China Postdoctoral Science
Foundation Funded Project (grant no. 2015M570476); the Priority
Academic Program Development of Jiangsu Higher Education
Institutions (PAPD); Jiangsu Talent Youth Medical Program (grant
no. QNRC2016245); Shanghai Key Lab of Forensic Medicine (grant no.
KF1502); Key Laboratory of Evidence Science (China University of
Political Science and Law), Ministry of Education (grant no.
2016KFKT05); and the Suzhou Science and Technology Development
Project (grant no. SYSD2015119).
References
|
1
|
Mittenberg W, DiGiulio DV, Perrin S and
Bass AE: Symptoms following mild head injury: Expectation as
aetiology. J Neurol Neurosurg Psychiatry. 55:200–204. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harris LK, Black RT, Golden KM, Reeves TM,
Povlishock JT and Phillips LL: Traumatic brain injury-induced
changes in gene expression and functional activity of mitochondrial
cytochrome C oxidase. J Neurotrauma. 18:993–1009. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gu YL, Zhang LW, Ma N, Ye LL, de X Wang
and Gao X: Cognitive improvement of mice induced by exercise prior
to traumatic brain injury is associated with cytochrome c oxidase.
Neurosci Lett. 570:86–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huttemann M, Lee I, Kreipke CW and Petrov
T: Suppression of the inducible form of nitric oxide synthase prior
to traumatic brain injury improves cytochrome c oxidase activity
and normalizes cellular energy levels. Neuroscience. 151:148–154.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bennett MC, Mlady GW, Kwon YH and Rose GM:
Chronic in vivo sodium azide infusion induces selective and stable
inhibition of cytochrome c oxidase. J Neurochem. 66:2606–2611.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fayaz SM, Kumar VS Suvanish and Rajanikant
GK: Necroptosis: Who knew there were so many interesting ways to
die? CNS Neurol Disord Drug Targets. 13:42–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park Y, Liu C, Luo T, Dietrich WD,
Bramlett H and Hu B: Chaperone-mediated autophagy after traumatic
brain injury. J Neurotrauma. 32:1449–1457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheng G, Kong RH, Zhang LM and Zhang JN:
Mitochondria in traumatic brain injury and mitochondrial-targeted
multipotential therapeutic strategies. Br J Pharmacol. 167:699–719.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarkar C, Zhao Z, Aungst S, Sabirzhanov B,
Faden AI and Lipinski MM: Impaired autophagy flux is associated
with neuronal cell death after traumatic brain injury. Autophagy.
10:2208–2222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang M, Shan H, Chang P, Wang T, Dong W,
Chen X and Tao L: Hydrogen sulfide offers neuroprotection on
traumatic brain injury in parallel with reduced apoptosis and
autophagy in mice. PLoS One. 9:e872412014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Cheng XR, Hu JJ, Sun L and Du GH:
Neuroprotective Effects of hyperoside on sodium azide-induced
apoptosis in PC12 cells. Chin J Natural Med. 9:450–455. 2011.
|
|
12
|
Grammatopoulos TN, Morris K, Bachar C,
Moore S, Andres R and Weyhenmeyer JA: Angiotensin II attenuates
chemical hypoxia-induced caspase-3 activation in primary cortical
neuronal cultures. Brain Res Bull. 62:297–303. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Selvatici R, Previati M, Marino S, Marani
L, Falzarano S, Lanzoni I and Siniscalchi A: Sodium azide induced
neuronal damage in vitro: Evidence for non-apoptotic cell death.
Neurochem Res. 34:909–916. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang CW and Moore PK: H2S synthesizing
enzymes: Biochemistry and molecular aspects. Handb Exp Pharmacol.
230:3–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bhatia M: Role of hydrogen sulfide in the
pathology of inflammation. Scientifica (Cairo).
2012:1596802012.PubMed/NCBI
|
|
16
|
Kimura H: Hydrogen sulfide and
polysulfides as biological mediators. Molecules. 19:16146–16157.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kimura H: Hydrogen sulfide: Its
production, release and functions. Amino Acids. 41:113–121. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kimura H: Physiological roles of hydrogen
sulfide and polysulfides. Handb Exp Pharmacol. 230:61–81. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang S and Lamm SH: Human health effects
of sodium azide exposure: A literature review and analysis. Int J
Toxicol. 22:175–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ott M, Gogvadze V, Orrenius S and
Zhivotovsky B: Mitochondria, oxidative stress and cell death.
Apoptosis. 12:913–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yin F, Boveris A and Cadenas E:
Mitochondrial energy metabolism and redox signaling in brain aging
and neurodegeneration. Antioxid Redox Signal. 20:353–371. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ryu JR, Hong CJ, Kim JY, Kim EK, Sun W and
Yu SW: Control of adult neurogenesis by programmed cell death in
the mammalian brain. Mol Brain. 9:432016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sato E, Suzuki T, Hoshi N, Sugino T and
Hasegawa H: Sodium azide induces necrotic cell death in rat
squamous cell carcinoma SCC131. Med Mol Morphol. 41:211–220. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Inomata K and Tanaka H: Protective effect
of benidipine against sodium azide-induced cell death in cultured
neonatal rat cardiac myocytes. J Pharmacol Sci. 93:163–170. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lutton JD, Moonga BS and Dempster DW:
Osteoclast demise in the rat: Physiological versus degenerative
cell death. Exp Physiol. 81:251–260. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amador FC, Henriques AG, da Cruz E Silva
OA and da Cruz E Silva EF: Monitoring protein phosphatase 1 isoform
levels as a marker for cellular stress. Neurotoxicol Teratol.
26:387–395. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Satpute R, Bhattacharya R, S Kashyap R, J
Purohit H, Y Deopujari J, M Taori G and F Daginawala H: Antioxidant
potential of Fagonia arabica against the chemical ischemia-induced
in PC12 cells. Iran J Pharm Res. 11:303–313. 2012.PubMed/NCBI
|
|
28
|
Smith CM, Chen Y, Sullivan ML, Kochanek PM
and Clark RS: Autophagy in acute brain injury: Feast, famine, or
folly? Neurobiol Dis. 43:52–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu L, Sun T, Xin F, Cui W, Guo J and Hu
J: Nerve growth factor protects against alcohol-induced
neurotoxicity in PC12 cells via PI3K/Akt/mTOR pathway. Alcohol
Alcohol. 52:12–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen HM and Codogno P: Autophagic cell
death: Loch Ness monster or endangered species? Autophagy.
7:457–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lipinski MM, Wu J, Faden AI and Sarkar C:
Function and mechanisms of autophagy in brain and spinal cord
trauma. Antioxid Redox Signal. 23:565–577. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gotor C, Garcia I, Crespo JL and Romero
LC: Sulfide as a signaling molecule in autophagy. Autophagy.
9:609–611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kimura H: Hydrogen sulfide: Production,
release, and functions. Nihon Yakurigaku Zasshi. 139:6–8. 2012.(In
Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang M, Shan H, Chang P, Ma L, Chu Y,
Shen X, Wu Q, Wang Z, Luo C, Wang T, et al: Upregulation of 3-MST
relates to neuronal autophagy after traumatic brain injury in mice.
Cell Mol Neurobiol. 37:291–302. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang M, Shan H, Wang Y, Wang T, Liu W,
Wang L, Zhang L, Chang P, Dong W, Chen X and Tao L: The expression
changes of cystathionine-b-synthase in brain cortex after traumatic
brain injury. J Mol Neurosci. 51:57–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen WL, Niu YY, Jiang WZ, Tang HL, Zhang
C, Xia QM and Tang XQ: Neuroprotective effects of hydrogen sulfide
and the underlying signaling pathways. Rev Neurosci. 26:129–142.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miyamoto R, Otsuguro K, Yamaguchi S and
Ito S: Contribution of cysteine aminotransferase and
mercaptopyruvate sulfurtransferase to hydrogen sulfide production
in peripheral neurons. J Neurochem. 130:29–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang ZY, Chen B, Zhao DJ and Kang L:
Functional modulation of mitochondrial cytochrome c oxidase
underlies adaptation to high-altitude hypoxia in a Tibetan
migratory locust. Proc Biol Sci. 280:201227582013; View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sena LA and Chandel NS: Physiological
roles of mitochondrial reactive oxygen species. Mol Cell.
48:158–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Han M and Im DS: Effects of mitochondrial
inhibitors on cell viability in U937 monocytes under glucose
deprivation. Arch Pharm Res. 31:749–757. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nagpure BV and Bian JS: Brain, learning,
and memory: Role of H2S in neurodegenerative diseases. Handb Exp
Pharmacol. 230:193–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Whiteman M, Armstrong JS, Chu SH, Jia-Ling
S, Wong BS, Cheung NS, Halliwell B and Moore PK: The novel
neuromodulator hydrogen sulfide: An endogenous peroxynitrite
‘scavenger’? J Neurochem. 90:765–768. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kimura Y, Goto Y and Kimura H: Hydrogen
sulfide increases glutathione production and suppresses oxidative
stress in mitochondria. Antioxid Redox Signal. 12:1–13. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kansanen E, Kuosmanen SM, Leinonen H and
Levonen AL: The Keap1-Nrf2 pathway: Mechanisms of activation and
dysregulation in cancer. Redox Biol. 1:45–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Calvert JW, Jha S, Gundewar S, Elrod JW,
Ramachandran A, Pattillo CB, Kevil CG and Lefer DJ: Hydrogen
sulfide mediates cardioprotection through Nrf2 signaling. Circ Res.
105:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu J, Wu J, Sun A, Sun Y, Yu X, Liu N,
Dong S, Yang F, Zhang L, Zhong X, et al: Hydrogen sulfide decreases
high glucose/palmitate-induced autophagy in endothelial cells by
the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 6:332016.
View Article : Google Scholar : PubMed/NCBI
|