Introduction
Translocations are the most frequent structural
aberration in the human genome, with an incidence of 0.178%
(1). Carriers of balanced
chromosome rearrangement exhibit an increased risk of abortion
and/or a chromosomally unbalanced child (2). The type of unbalanced translocation
is dependent upon the mode of segregation. A 2:2 segregation event
may result in gametes with partial trisomy/monosomy of the
chromosomes involved in the translocation (3). Conventional cytogenetic analysis is
unable to detect small rearrangements due to its low resolution.
The wide use of whole-genome array-based comparative genomic
hybridization (aCGH) techniques has allowed for the detection of
submicroscopic chromosomal aberrations and the establishment of
genotype-phenotype correlations, by delineating at high resolution
the regions involved in genomic copy number variations. The present
study assessed a pediatric patient with partial trisomy 4p and
partial monosomy 20q, resulting from a 2:2 segregation of a
maternal balanced t(4;20) translocation. The karyotype of
the proband exhibited 46, XY, add(20) (q13.3) and the aCGH analysis
identified partial trisomy of the short arm of chromosome 4 and
partial monosomy of distal 20q. The patient exhibited typical
trisomy 4p and monosomy 20q features, including intellectual
disability, delayed speech, tall stature, seizures and facial
dysmorphism. The present study supports the use of aCGH as the
first-tier cytogenetic diagnostic tool for patients with
unexplained delays in development, intellectual disability or
multiple congenital anomalies.
Case report
Clinical features
The patient was a 6-year-old male born to a
33-year-old father and a 32-year-old mother via vaginal delivery at
39 gestational weeks. During pregnancy, no specific problems were
identified. The birth weight was 3.8 kg (50th centile), birth
length was 53 cm (>75th centile) and occipital frontal
circumference was 36 cm (>95th centile). The Apgar scores were 9
and 10 at 1 and 5 min, respectively. Seizures began at ~2 months of
life. The proband began to walk at 2 years 4 months of age. At 4
years of age the proband had minimal expressive language and
minimal social interactions. The patient presented to Henan
Provincial People's Hospital (Zhengzhou, China) at age 6 years, due
to intellectual disability, delayed speech, tall stature, seizures,
delayed fine and gross motor skills and facial dysmorphism
(macrocephaly, prominent nasal bridge, low-set ears, epicanthus).
The parents had previously experience two spontaneous abortions
occurring at 7 and 11 weeks of gestation, respectively, although
product of conception (POC) samples were not analyzed.
Cytogenetic and aCGH analysis
Peripheral blood samples were obtained from the
proband and the parents for examination of chromosomes by metaphase
G-banding and aCGH. The Henan Provincial People's Hospital Ethics
Committee approved the sample collection procedures and the family
gave written informed consent. Standard procedures were used
isolate the genomic DNA of the proband and the parents from whole
blood using the QIAamp DNA Mini kit (Qiagen GmbH, Hilden, Germany),
according to the manufacturer's protocol. DNA was assayed for
quantity and purity using the NanoDrop ND-2000 Spectrophotometer
(Thermo Fisher Scientific, Inc., Wilmington, DE, USA). aCGH
analysis was performed using Agilent 4×180 K commercial arrays
(Agilent Technologies, Inc., Santa Clara, CA, USA), which consist
of 110,712 oligonucleotide probes and 59,647 single nucleotide
polymorphism probes, to evaluate the entire genome with an
effective backbone resolution of ~25.3 kb [5 kb in International
Standards for Cytogenomic Arrays (ISCA) regions]. A total of 1,500
ng of experimental and gender-matched reference DNA (Promega
Corporation, Madison, WI, USA) was digested with AluI and
RsaI restriction endonucleases (Promega Corporation) and
fluorescently-labeled with cyanine 5-dUTP and cyanine 3-dUTP,
respectively. Labeled experimental and reference DNA was purified,
combined, denatured and hybridized to the microarrays in a rotating
oven (20 rpm) at 67°C for 24 h. Data were analyzed using
Cytogenomics 2.9 software (Agilent Technologies, Inc.). The
Cytogenomics 2.9 software, via the Aberration Detection Method-2
algorithm with a sensitivity threshold of 6.0 and a data filter,
identified aberrations and rejected those did not include at least
three probes with a log2 set of 0.25. All quality control metrics
were passed. The copy number variations were compared with the
Database of Genomic Variants, Database of Chromosomal Imbalance and
Phenotype in Humans using Ensembl Resources (DECIPHER v9.10;
decipher.sanger.ac.uk), the ClinGen
Dosage Sensitivity Map (http://www.ncbi.nlm.nih.gov/projects/dbvar/clingen),
the RefSeqGene database (https://www.ncbi.nlm.nih.gov/refseq/rsg/), OMIM
(http://omim.org/) and the relevant publications in
PubMed (http://www.ncbi.nlm.nih.gov/pubmed). Genome
coordinates among different assemblies were converted to Hg19 using
the LiftOver tool (genome.ucsc.edu/cgi-bin/hgLiftOver).
Results
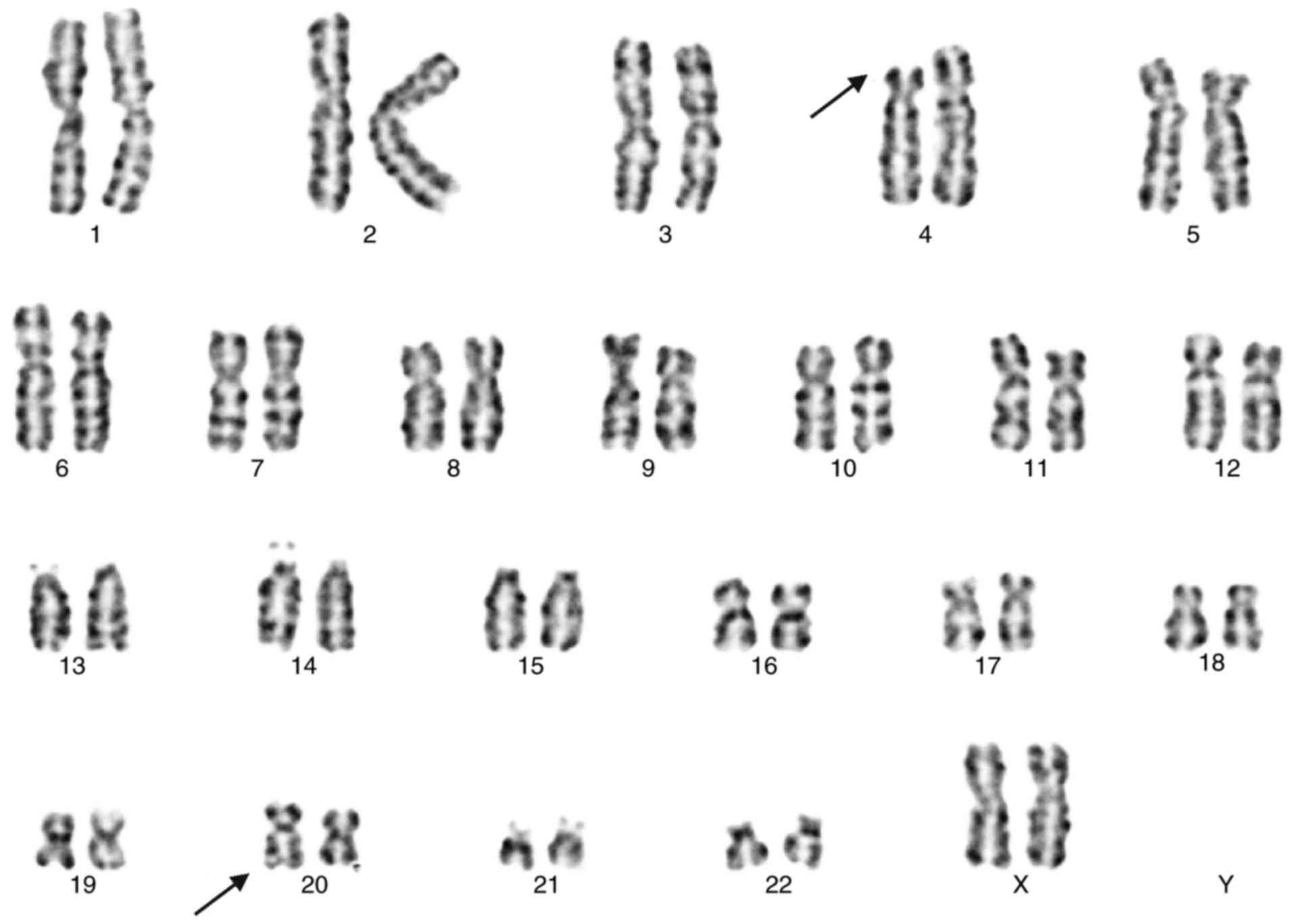
The karyotype of the proband (Fig. 1) exhibited 46, XY, add(20) (q13.3).
The karyotype of the mother (Fig.
2) indicated a balanced translocation karyotype: 46, XX,
t(4;20) (p15.2; q13.1). The father exhibited a normal male
karyotype (data not shown). The aCGH analysis demonstrated a 12.8
Mb terminal duplication at 4p16.3-p15.33 (72,447–12,900,236) (hg19)
(Fig. 3) and a 1.2 Mb terminal
deletion at 20q13.33 (61,722,950–62,908,674) (hg19) (Fig. 4) in the proband, while no
duplication or deletions were detected in the parents.
Discussion
The present case report demonstrated that the
proband carried an unbalanced translocation inherited from a
balanced translocation carrier mother, which resulted in partial
trisomy for 4p (spanning ~12.8 Mb) and partial monosomy for 20q
(spanning ~1.2 Mb). Karyotyping did not reliably detect the
unbalanced rearrangement. The aCGH analysis identified genetic
anomalies in the patient. Although the clinical features of the two
variants have been separately described in the literature (4,5),
there are no published cases illustrating the two variants
occurring in the same patient. To the best of our knowledge, the
present study is the first report of an unbalanced translocation
involving chromosomes 4p and 20q.
Trisomy 4p syndrome was first reported as a distinct
clinical entity ~40 years ago (6).
This syndrome is characterized by intellectual disability, delayed
speech, facial dysmorphism (prominent nasal bridge, and low-set and
malformed ears) and, in certain cases, overgrowth and macrocephaly
(7–9) (Table
I). However, diagnosis is not definitive, since the phenotypic
features are variable and not unique to trisomy 4p. A number of
trisomy 4p cases occur as a result of unbalanced meiotic
segregation from a parental balanced translocation and may
consequently be accompanied by monosomy of the partner chromosome,
which may contribute to the phenotype (10). The variable size of the duplicated
4p segment and the phenotypic features make the clinical diagnosis
of trisomy 4p syndrome difficult.
 | Table I.Clinical features of the proband
compared with previous cases of 4p duplication and 20q
deletion. |
Table I.
Clinical features of the proband
compared with previous cases of 4p duplication and 20q
deletion.
| Author | Aberration | Other chrom osomal
aberration | Age diagnosis | Sex | Intellectual
disability | Delayed speech | Growth delay | Over growth | Seizures | Hypotonia | Cardiac defects | Psy chomotor
developmental defects | Microcephaly | Microcephaly | Brachycephaly | Bitem
Micrognathia | Promporal
narrowing | inentnasal
bridge | Lowset ears | Malformedears | Thin upper Epica
Epicanthus | lipvermilion | Other phenotypes | (Refs.) |
|---|
| Schönewolf Greulich
et al | arr[hg19] 4p16.3
73,645–3,072, 968)x3 | − | 6 years | Female | + | + | − | + | − | − | − | − | − | + | − | − | − | + | + | + | − | + | Proximal placement of
thumbs; small eyes | (7) |
| Iype et al;
case III:4 | arr[hg19] 4p16.3–16.1
(0–10,290, 552)x3 | arr[hg19]
3p26.3(0–2,118, 422)x1 | 40 years | Female | + | + | − | − | − | + |
| + | − | − | + |
| − | + | + | + | − | + | Campodactyly;
strabismus; cleft lip; gum hypertrophy | (8) |
| Hannes et
al | arr[hg19] 4p16.3
(1,458, 385–1907, 425)x3 | − | 11 months | Male | + | + | + | − | + | + | − | + | − | − | − | − | + | − | + | + | + | − | Malformation of right
hand (shortened fingers); glaucoma of the left eye short neck | (9) |
| Mefford et
al | arr[hg19] 20q13
(chr20:60, 700,000–62,435, 964)x1 | − | 7 years | Male | + | + | + | − | + | + | − | − | + | − | − | − | − | − | − | − | − | − | Blind | (12) |
| Marques et
al | arr[hg19] 20q13.33
61,643,144–63,003,805)x1 | arr[hg19] arr17q25.3
(78,952, 204–81, 060,886) ×3 | 6 years | Male | + | + | + | − | + | + | + | + | + | − | − | − | + | + | − | − | + | − | Short neck;
syndactyly; asymmetric legs | (13) |
| Traylor et al;
case 2 | arr 20q13.33 60,760,
865–62,379, 119)x1 | − | 4 years | Female | − | + | + | − | + | − | − | + | − | − | − | − | + | − | − | − | − | − | − | (14) |
| Wu et
al | arr[hg19]
4p16.3-p15.33 (72,447–12,900, 236)x3 | arr[hg19] 20q13.33
(61,722, 950–62, 908,674) ×1 | 6 years | Male | + | + | − | + | + | + | − | + | − | + | − | − | − | + | + | − | + | − | − | Present study |
In the present study, the duplication in
4p16.3p15.33 observed in the proband overlapped with 122 RefSeq
genes, including 20 morbid genes in the database of OMIM. It is
difficult to confirm that one specific gene is responsible for the
specific phenotype. Notably, certain parameters of the features
concerning growth in 4p duplication (macrocephaly, overgrowth and
tall stature) are opposed to those of 4p deletion (microcephaly,
small for gestational age and delayed growth) (11). The mirror phenotypes may result
from reciprocal deletion/duplication in chromosomal regions
containing dosage-sensitive genes. Therefore, the ClinGen Dosage
Sensitivity Map was searched, and no genes with evidence of
triplosensitive phenotypes were identified. In the DECIPHER
database, the haploinsufficiency scores of the genes fibroblast
growth factor receptor 3 (FGFR3; OMIM, 134934), huntingtin
(OMIM, 613004), macrophage erythroblast attacher (OMIM, 606801),
msh homeobox 1 (MSX1; OMIM, 142983) were 6.40, 7.59, 7.12
and 1.21%, respectively; this indicated that these genes were more
likely to exhibit haploinsufficiency and dosage sensitivity.
Regarding the growth alterations and musculoskeletal
malformations, the gene FGFR3, which regulates the growth of
bone, may be a candidate gene for the anomalous growth in patients
with 4p duplication. Mutations in the gene FGFR3 are
associated with 14 human disorders, and skeletal malformations
represent the principal clinical presentations. Mutations in the
gene MSX1 are associated with ectodermal dysplasia 3 (Witkop
type), orofacial cleft 5 and tooth agenesis (with or without
orofacial cleft). Therefore, MSX1 may be the candidate gene
for facial dysmorphism in patients with 4p duplication.
The phenotype of the present patient was modified by
the 1.2Mb terminal deletion 20q. Terminal deletions of the long arm
of chromosome 20 have been reported previously in a number of
patients, with phenotypes including neonatal or infantile seizures,
intellectual disability, language deficits and behaviors
characteristic of autism spectrum disorder (12–14)
(Table I). Similar to the
previously-described trisomy 4p syndrome, the variable size of the
deleted 20q segment and the phenotypic features make it difficult
to identify one gene to be responsible for the specific phenotype.
In the present study, the deletion in 20q13.33 described in the
proband overlapped 37 RefSeq genes, including 7 OMIM morbid genes.
Potassium voltage-gated channel family Q member 2 (KCNQ2;
OMIM, 602235) exhibited haploinsufficiency phenotypes of benign
familial neonatal seizures in the ClinGen Dosage Sensitivity Map.
KCNQ2 encodes a subunit of the voltage-gated potassium
channel, and mutations have been observed in patients with benign
familial neonatal seizures and unexplained neonatal epileptic
encephalopathy (15). Myelin
transcription factor 1 (MYT1; OMIM, 600379) may affect
myelination and the regulation of neural differentiation (16,17),
while mutations in cholinergic receptor nicotinic α4 subunit
(CHRNA4; OMIM, 118504) have been observed to be associated
with autosomal dominant nocturnal frontal lobe epilepsy (18). Thus, KCNQ2, MYT1 and
CHRNA4 may be the candidate genes for seizures and delayed
cognitive development in patients with 20q terminal deletion.
The parents of the proband experience two
spontaneous abortions at 7 and 11 weeks of gestation, respectively,
although POC samples were not analyzed. A total of ~50% of
first-trimester miscarriages result from fetal chromosomal
abnormalities (19). Using aCGH to
analyze POC samples may determine possible genetic causes of
miscarriage and predict the recurrence risk for subsequent
pregnancies. Since an increased rate of miscarriage exists in
couples with balanced chromosomal rearrangements, cytogenetic
analysis and/or aCGH analysis for all POC samples may be
recommended.
In conclusion, the present case report described a
partial 4p duplication and partial monosomy of 20q in a patient
with the majority of the typical phenotypes of 4p duplication and
20q deletion (intellectual disability, delayed speech, tall
stature, seizures and facial dysmorphism). The use of aCGH may
facilitate a sensitive and powerful approach towards the diagnosis
of submicroscopic unbalanced genomic rearrangements. FGFR3
and MSX1 may be the important genes for 4p duplication, and
KCNQ2, MYT1 and CHRNA4 may be the important
genes for 20q terminal deletion. Additional studies may help to
refine the relevant genes associated with the variable clinical
features.
Acknowledgements
The present study used data generated by the
DECIPHER community. A full list of centers who contributed to the
generation of the data is available from decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding
for the DECIPHER project was provided by the Wellcome Trust
(London, UK).
References
|
1
|
De Braekeleer M and Dao TN: Cytogenetic
studies in male infertility: A review. Hum Reprod. 6:245–250. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sheth FJ, Liehr T, Kumari P, Akinde R,
Sheth HJ and Sheth JJ: Chromosomal abnormalities in couples with
repeated fetal loss: An Indian retrospective study. Indian J Hum
Genet. 19:415–422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jalbert P, Sele B and Jalbert H:
Reciprocal translocations: A way to predict the mode of imbalanced
segregation by pachytene-diagram drawing. Hum Genet. 55:209–222.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gérard-Blanluet M, Romana S, Munier C, Le
Lorc'h M, Kanafani S, Sinico M, Touboul C, Levaillant JM, Haddad B,
Lopez N, et al: Classical West ‘syndrome’ phenotype with a
subtelomeric 4p trisomy. Am J Med Genet A. 130A:1–302. 2004.
View Article : Google Scholar
|
|
5
|
Okumura A, Atsushi Ishii, Shimojima K,
Kurahashi H, Yoshitomi S, lmai K, Imamura M, Seki Y, Shimizu T
Toshiaki, Hirose S and Yamamoto T: Phenotypes of children with
20q13.3 microdeletion affecting KCNQ2 and CHRNA4. Epileptic Disord.
17:165–171. 2015.PubMed/NCBI
|
|
6
|
Wilson MG, Towner JW and Negus LD:
Wolf-Hirschhorn syndrome associated with an unusual abnormality of
chromosome no. 4. J Med Genet. 7:164–170. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schönewolf-Greulich B, Ravn K,
Hamborg-Petersen B, Brøndum-Nielsen K and Tümer Z: Segregation of a
4p16.3 duplication with a characteristic appearance, macrocephaly,
speech delay and mild intellectual disability in a 3-generation
family. Am J Med Genet A. 161A:–2362. 2013.
|
|
8
|
Iype T, Alakbarzade V, Iype M, Singh R,
Sreekantan-Nair A, Chioza BA, Mohapatra TM, Baple EL, Patton MA,
Warner TT, et al: A large Indian family with rearrangement of
chromosome 4p16 and 3p26.3 and divergent clinical presentations.
BMC Med Genet. 16:1042015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hannes F, Drozniewska M, Vermeesch JR and
Haus O: Duplication of the Wolf-Hirschhorn syndrome critical region
causes neurodevelopmental delay. Eur J Med Genet. 53:136–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim YH, Kim HS, Ryoo NH and Ha JS: Two
cases of partial trisomy 4p and partial trisomy 14q. Ann Lab Med.
33:69–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carmany EP and Bawle EV: Microduplication
of 4p16.3 due to an unbalanced translocation resulting in a mild
phenotype. Am J Med Genet A. 155A:1–824. 2011.PubMed/NCBI
|
|
12
|
Mefford HC, Cook J and Gospe SM Jr:
Epilepsy due to 20q13.33 subtelomere deletion masquerading as
pyridoxine-dependent epilepsy. Am J Med Genet A. 158A:1–3195. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marques F, Heredia R, de Oliveira C,
Cardoso MT, Mazzeu J and Pogue R: Partial trisomy 17q and partial
monosomy 20q in a boy with craniosynostosis. Am J Med Genet A.
167:412–416. 2015. View Article : Google Scholar
|
|
14
|
Traylor RN, Bruno DL, Burgess T, Wildin R,
Spencer A, Ganesamoorthy D, Amor DJ, Hunter M, Caplan M, Rosenfeld
JA, et al: A genotype-first approach for the molecular and clinical
characterization of uncommon de novo microdeletion of 20q13.33.
PLoS One. 5:e124622010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Weckhuysen S, Mandelstam S, Suls A,
Audenaert D, Deconinck T, Claes LR, Deprez L, Smets K, Hristova D,
Yordanova I, et al: KCNQ2 encephalopathy: Emerging phenotype of a
neonatal epileptic encephalopathy. Ann Neurol. 71:15–25. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kroepfl T, Petek E, Schwarzbraun T,
Kroisel PM and Plecko B: Mental retardation in a girl with a
subtelomeric deletion on chromosome 20q and complete deletion of
the myelin transcription factor 1 gene (MYT1). Clin Genet.
73:492–495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Romm E, Nielsen JA, Kim JG and Hudson LD:
Myt1 family recruits histone deacetylase to regulate neural
transcription. J Neurochem. 93:1444–1453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Z, Wang L, Wang C, Chen Q, Zhai Q,
Guo Y and Zhang Y: Mutational analysis of CHRNB2, CHRNA2 and CHRNA4
genes in Chinese population with autosomal dominant nocturnal
frontal lobe epilepsy. Int J Clin Exp Med. 8:9063–9070.
2015.PubMed/NCBI
|
|
19
|
Schaeffer AJ, Chung J, Heretis K, Wong A,
Ledbetter DH and Martin C Lese: Comparative genomic
hybridization-array analysis enhances the detection of aneuploidies
and submicroscopic imbalances in spontaneous miscarriages. Am J Hum
Genet. 74:1168–1174. 2004. View
Article : Google Scholar : PubMed/NCBI
|