Introduction
Acute liver injury or acute liver failure is a rare,
but life-threatening, disorder associated with high morbidity and
mortality (1). Extensive
hepatocyte cell death is typically observed in the pathogenesis of
acute liver injury (2). The
creation of animal models with acute liver injury through exposure
to lipopolysaccharide (LPS) and D-galactosamine (GalN) is an
extensively applied approach in rodents; the efficacy of combined
treatment with these agents in inducing hepatotoxicity is greater
as compared with that induced by either drug alone (3,4).
However, the pathogenesis of liver damage induced by
co-administration of LPS and GalN has not yet been fully
elucidated.
Hepatocyte apoptosis and necrosis is a predominant
feature during acute liver injury (5). In addition to apoptosis and necrosis,
it has previously been demonstrated that autophagy may be induced
in hepatocytes upon LPS/GalN-induced liver injury (6). Autophagy is known to exhibit a
pivotal role in maintaining cellular homeostasis through
self-digestion of long-lived proteins or damaged organelles under
stimuli (7). During induction of
autophagy, cytosolic components are encapsulated in a phagophore,
which forms a double-membrane-bound vesicle termed an
autophagosome. This newly formed autophagosome fuses with a
lysosome, allowing degradation of encapsulated products, whereby
molecules and proteins may be recycled to improve cell survival. It
has been suggested that autophagy not only modulates normal liver
function, but also participates in the pathogenesis of various
liver disorders (8). A previous
study demonstrated that autophagy is closely associated with
endoplasmic reticulum (ER) stress during the development of
steatohepatitis in diabetic mice (9). The expression of ER stress-signaling
components, including CCAAT/enhancer binding protein
(C/EBP)-homologous protein (CHOP; a pro-apoptotic transcription
factor), are positively associated with autophagic activity in
liver tissues (9). In accordance
with the results from the present study, it has previously been
demonstrated that CHOP contributes to hepatocyte death during acute
liver injury (10).
Given that ER stress is a potent trigger for
induction of autophagy (11,12),
it is possible that acute liver injury may lead to ER stress, which
subsequently activates autophagy. To investigate this hypothesis, a
murine model of acute liver injury was created by intraperitoneally
injecting LPS and GalN. Expression of autophagy and
ER-stress-associated proteins were examined at indicated time
points following the onset of acute liver injury.
Materials and methods
Reagents
Lipopolysaccharide was obtained from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). GalN was provided by Chongqing
Medical University, (Chongqing, China). Anti-microtubule-associated
protein 1 light chain (LC)-3, anti-Beclin-1, anti-CHOP, and
anti-β-actin primary antibodies were all purchased from Cell
Signaling Technology, Inc., (Danvers, MA, USA). Horseradish
peroxidase (HRP)-conjugated secondary antibodies were purchased
from Wuhan Boster Biological Technology Co., Ltd (Wuhan,
China).
Animals and experimental
assignment
A total of 34 male Kunming mice (age, 8–10 weeks;
weight, 25±5 g) were obtained from the Laboratory Animal Center,
Shanxi Medical University (Taiyuan, China) [SCXK (Jin) 2009-0001].
Mice were housed together and acclimatized at 18–26°C at a constant
humidity of 65–80% under standard lighting conditions (12/12 h
light/dark) for 1 week and then randomly assigned to 4 groups:
Control (n=8), 1-h liver injury (n=8), 3-h liver injury (n=8), and
6-h liver injury (n=10). Liver injury in test mice was induced by
an intraperitoneal injection of 0.2 ml normal saline containing 5
µg LPS and 0.02 g GalN, whereas controls were administered 0.2 ml
normal saline by intraperitoneal injection. Mice had free access to
food and water. This study was approved by the ethics committee of
Shanxi Medical University.
Sample collection
At 1, 3, or 6 h following LPS/GalN injection, blood
samples were collected from the eye vein into heparinized tubes,
and mice were sacrificed via cervical dislocation. The left lobe of
the liver was removed from each mouse and tissue samples were
processed by fixation in 10% formaldehyde solution for 24–48 h at
room temperature, washed with sterile normal saline, wrapped with
sterile tinfoil, frozen in liquid nitrogen, and stored at −70°C in
a refrigerator until further analysis.
Histological examination
For histological examination, tissue samples were
prepared for hematoxylin and eosin (HE) staining; briefly, samples
were dehydrated in an ethanol series, cleared in xylene and
embedded in paraffin. Paraffin-embedded tissues were sectioned in
an automatic tissue processor (Leica Microsystems GmbH, Wetzlar
Germany) into 4–5 µm-thick slices and subjected to HE staining. For
HE staining, samples were stained with hematoxylin for 1–2 min at
room temperature. After washing, slices were stained with eosin for
30–60 sec at room temperature. Micrographs under ×400 magnification
were randomly selected and captured using a light microscope (Nikon
Corporation, Tokyo, Japan).
Determination of serum alanine
aminotransferase levels
Serum was obtained from blood samples by
centrifugation at 3,000 × g for 10 min at 4°C and was evaluated for
serum alanine aminotransferase (ALT) activity using a commercial
kit (cat. no. C009-1) according to the manufacturer's protocol
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Western blot analysis
A total of 200 µg liver tissue was removed from each
mouse in the study groups and ground in liquid nitrogen. Protein
was extracted using lysis buffer containing 50 mM Tris-HCl (pH
7.5), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1%
SDS, 1% NP-40, 1 µg/ml leupeptin, 1 µg/ml aprotinin and 1 mM PMSF.
The protein concentration was determined using Coomassie Brilliant
Blue assay (Fluka Chemie GmbH, Buchs, Switzerland). Proteins were
denatured by boiling and 40 µg denatured proteins were loaded on
and separated by 8% (for Beclin 1) or 12% (for LC3) SDS-PAGE, prior
to transfer to nitrocellulose membranes. Membranes were blocked in
5% non-fat dry milk in Tris-buffered saline containing Tween-20
(TBS-T; 0.1% Tween-20) for 2 h at room temperature, and then
incubated with primary antibodies for overnight at 4°C. Primary
antibodies used include rabbit monoclonal anti-LC3 (1:1,000; cat.
no. 13118), rabbit monoclonal anti-Beclin-1 (1:1,000; cat. no.
3495), rabbit monoclonal anti-CHOP (1:1,000; cat no. 5554) and
rabbit monoclonal anti-β-actin (1:1,000; cat. no. 4970) antibodies.
Following washing, membranes were incubated with goat anti-rabbit
HRP-conjugated secondary antibodies (1:4,000; cat no. BA1003) for 2
h. Immunobands were visualized via exposure to an x-ray beam in a
dark room, the x-ray film was scanned by a gel imaging system
(JD801; Jiangsu JEDA Science-Technology Development Co., Ltd.,
China), and the densitometric values of the bands were quantified
using ImageJ software version 1.47e (National Institutes of Health,
Bethesda, MD, USA). The housekeeping protein β-actin was used as an
internal control.
Statistical analysis
Data were analyzed by SPSS software, version 21 (IBM
Corp, Armonk, NY, USA) and results are presented as the mean ±
standard error of the mean. Statistical significance was examined
using one-way analysis of variance followed by a post hoc least
significant difference test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
LPS/GalN induces liver injury in
mice
The present study firstly established a rodent model
of liver injury by injecting LPS and GalN intraperitoneally. In the
6-h liver injury group, 1 mouse died of liver failure at 5.5 h
following model establishment and, therefore, blood and tissue
samples of this mouse were not collected for analysis. All other
study animals survived until analysis. Gross examination of livers
revealed a deep red color and a soft, smooth surface in normal
livers (Fig. 1). No obvious
difference in gross appearance of liver between 1-h and 3-h liver
injury groups and the control group was evident. However, livers in
the 6-h liver injury group appeared dark red and had a slightly
roughened surface. HE-stained liver specimens did not manifest any
histological abnormalities in control, 1-h, or 3-h liver injury
mice (Fig. 2); however, evident
histological alterations with irregularly distributed hepatocytes
and abnormal cell morphology were observed in liver tissues of the
6-h liver injury mice. In accordance with these observations, the
serum ALT level was significantly elevated at 6 h following LPS and
GalN administration (786.93±51.55 U/l; P<0.001 compared with
control), although no significant difference was detected in the
1-h or 3-h liver injury groups compared with control (control,
46.23±8.17; 1-h liver injury, 48.07±9.33; 3-h liver injury,
93.49±16.37 U/l; Fig. 3). These
results demonstrated that co-administration of LPS and GalN in mice
effectively induced liver injury that evidently occurred 6 h
following drug injection.
LPS/GalN induces autophagy induction
during early stage liver injury
In order to understand the potential involvement of
autophagy in liver injury, the present study determined the protein
expression of autophagy-associated proteins at different stages of
liver injury. The conversion from cytosolic (LC3-I) to a
lipid-bound form (LC3-II) of LC3 is a reliable biomarker for
autophagy induction (13). LC3-II
was revealed to be weakly expressed in normal liver tissues
(Fig. 4). At 1 h following liver
injury, the LC3-II level was reduced. However, markedly upregulated
LC3-II expression was detected at 3 h following LPS and GalN
administration, and the level of LC3-II then declined. Beclin-1 is
required for autophagosome formation, which is the initial step of
autophagy (13). Consistent with
that of LC3-II expression, western blot analysis demonstrated that
Beclin-1 expression in liver tissues was reduced at 1 h, whereas it
was enhanced at 3 h following liver injury (Fig. 4). Levels of Beclin 1 reverted to
baseline 6 h following injury (Fig.
4). These findings indicated that liver injury induced by LPS
and GalN administration results in initial autophagy inhibition
followed by activation, which precedes severe liver injury.
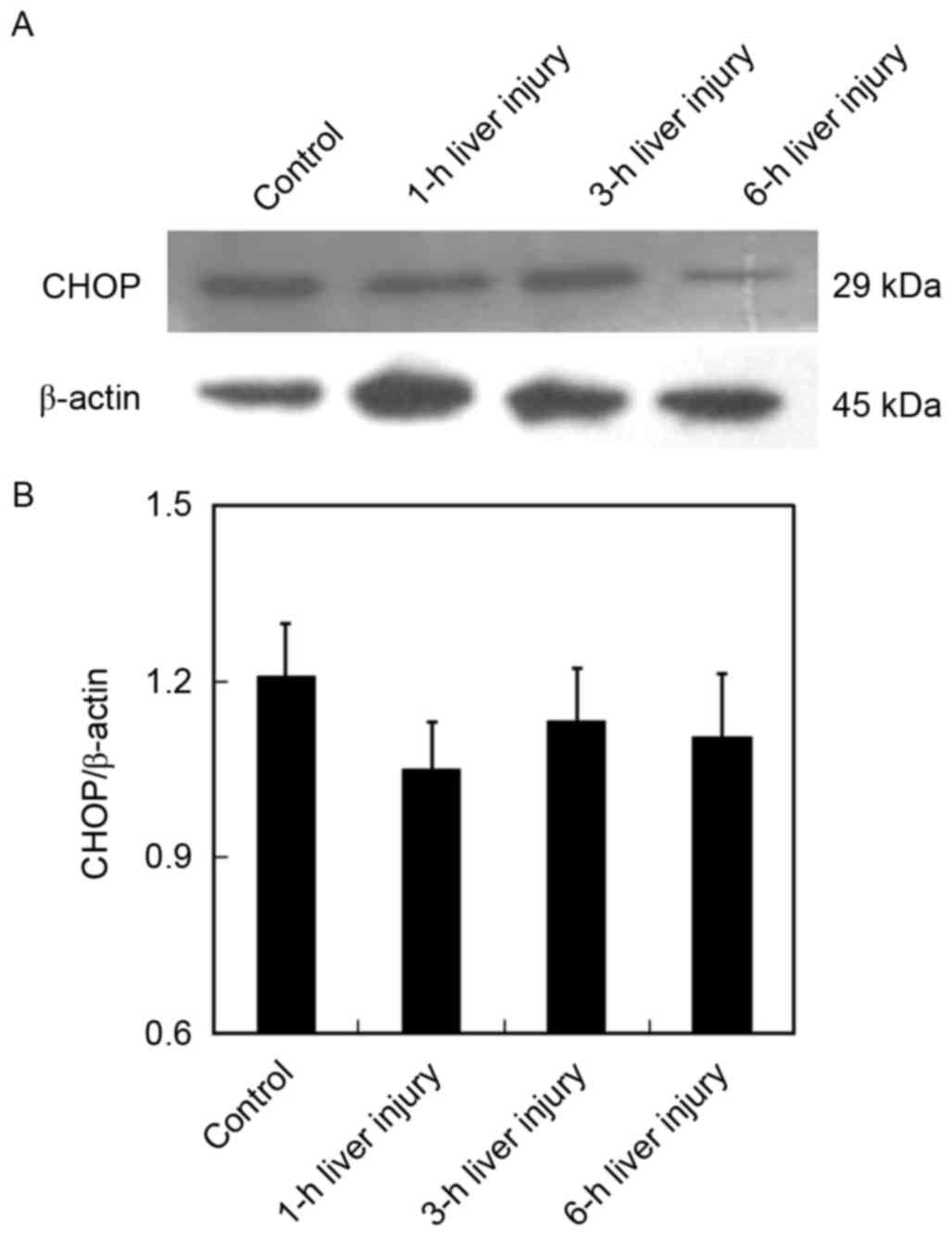
LPS/GalN upregulates CHOP expression
during early stage of liver injury
The expression of CHOP is evidence of highly induced
ER stress and is, therefore, recognized as a reliable biomarker for
the perturbation of the ER (14).
It has previously been demonstrated that CHOP contributes to
hepatotoxicity (15); therefore,
the present study investigated the role of CHOP in LPS/GalN-induced
liver damage. Although no significant difference was observed,
protein expression of CHOP was slightly reduced in the liver at 1 h
following injury, compared with in healthy liver tissues (Fig. 5). However, a marked upregulation of
CHOP was observed at 3 h following co-administration of LPS and
GalN, which reverted to baseline levels at later stages of liver
injury (6 h). These data suggested that ER stress occurs
concomitantly with autophagy induction, both of which are initiated
during early-stage liver injury.
Discussion
By creating a mouse model of acute liver injury, the
present study investigated the potential involvement of autophagy
and ER stress in injured liver tissues. The results demonstrated
that autophagy-associated proteins (LC3-II, Beclin 1) and the
ER-stress-associated protein CHOP were markedly upregulated 3 h
following liver injury, and preceded severe liver injury.
In the present study, acute liver injury in mice was
induced by intraperitoneal co-administration of LPS and GalN. LPS
constitutes the outer membrane of most gram-negative bacteria, and
is essential in mediating endotoxemia (16). Accumulating evidence suggests that
intraperitoneal LPS injection induces systemic inflammation and
contributes to hepatic dysfunction in rodent studies (17,18).
GalN, which is a hepatocyte-specific inhibitor of RNA synthesis,
mediates liver damage through specific induction of uridine
triphosphate deficiency in the liver (19). Furthermore, the induction of acute
liver injury by GalN administration has been demonstrated in
previous studies (20,21). Notably, treatment with GalN
sensitizes animals to LPS-induced hepatotoxicity (22); therefore, co-administration of LPS
and GalN has a widespread application in inducing acute liver
injury in mice. In the present study, LPS-GalN-induced liver injury
progressed to severe liver damage with enhanced cell death and
elevated serum ALT levels at 6 h following induction, although no
marked ALT or pathological alterations were observed in 1–3 h
following induction. These data are in agreement with a previous
report that demonstrated detectable liver injury at 4 h and
significant liver damage at 6 h following co-treatment with LPS and
GalN (23).
Autophagy is a lysosomal-dependent degradative
pathway whereby cells digest cytoplasmic components, damaged
organelles or long-lived proteins. In the present study, 3 h
following intraperitoneal injection of LPS-GaIN, considerable
upregulation of the autophagy-associated proteins LC3-II and Beclin
1 was observed in liver tissues, together with minimal detectable
liver injury. Therefore, autophagy may possibly serve as a
compensatory mechanism during early liver injury, and induction of
autophagy may improve hepatocyte survival by providing energy
against an adverse environment. To date, the precise role of
autophagy in acute liver injury remains to be fully elucidated.
Amir et al (23)
demonstrated acceleration of hepatocellular death following
LPS-GalN administration in autophagy-associated gene
Atg7-deficient mice. Conversely, Li et al (6) revealed that wortmannin, a potent
autophagy inhibitor, blocks autophagic activity by alleviating
LPS-GalN-induced acute liver injury in mice. These discrepancies
may be attributable to the differences between genetic manipulation
and pharmacological modulation. Nevertheless, the functional role
of autophagy in hepatic injury requires further investigation.
It has previously been demonstrated that ER stress
is associated with liver diseases (24,25),
and ER stress has been implicated in acetaminophen-induced acute
liver failure (26). Furthermore,
a marked elevation in protein expression of CHOP, the key molecule
in ER stress, has been observed in liver tissues of patients with
acute liver failure (10).
Sustained hepatocellular ER stress may ultimately lead to
hepatocyte death and contribute to liver injury (27). Inhibition of ER stress has been
indicated to attenuate liver damage in a rat model of acute liver
injury following 90% hepatectomy (28). Notably, ER stress is a
well-characterized inducer of autophagy (11,12),
implying the close association between ER stress and autophagy in
acute liver injury. The present study detected upregulated CHOP
expression with autophagy activation 3 h following LPS and GalN
co-administration. Therefore, LPS-GalN may induce ER stress in
liver tissues, subsequently inducing autophagy during early-stage
liver damage that may then counter further progression of liver
injury. Notably, 6 h following LPS-GalN injection, autophagy
induction and ER stress reverted to baseline levels, indicating
this self-protective mechanism may be insufficient for preventing
sustained liver damage. These data provide valuable insights into
understanding the involvement of autophagy and ER stress in the
development of acute liver failure.
In summary, the results of the present study reveal
that LPS-GalN efficiently induces acute liver injury in mice.
Furthermore, ER stress and autophagy induction occur in early-stage
liver damage, and this activation of autophagy may have a
compensatory role in liver injury.
Acknowledgements
The present study was supported by Fenyang College,
Shanxi Medical University (grant no. 1204).
References
|
1
|
McPhail MJ, Kriese S and Heneghan MA:
Current management of acute liver failure. Curr Opin Gastroenterol.
31:209–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuhla A, Thrum M, Schaeper U, Fehring V,
Schulze-Topphoff U, Abshagen K and Vollmar B: Liver-specific Fas
silencing prevents galactosamine/lipopolysaccharide-induced liver
injury. Apoptosis. 20:500–511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mignon A, Rouquet N, Fabre M, Martin S,
Pagès JC, Dhainaut JF, Kahn A, Briand P and Joulin V: LPS challenge
in D-galactosamine-sensitized mice accounts for caspase-dependent
fulminant hepatitis, not for septic shock. Am J Respir Crit Care
Med. 159:1308–1315. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galanos C, Freudenberg MA and Reutter W:
Galactosamine-induced sensitization to the lethal effects of
endotoxin. Proc Natl Acad Sci USA. 76:5939–5943. 1979; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malhi H, Gores GJ and Lemasters JJ:
Apoptosis and necrosis in the liver: A tale of two deaths?
Hepatology. 43 2 Suppl 1:S31–S44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y and Wang X, Wei Z, Mao H, Gao M, Liu
Y, Ma Y, Liu X, Guo C, Zhang L and Wang X: Pretreatment with
wortmannin alleviates lipopolysaccharide/d-galactosamine-induced
acute liver injury. Biochem Biophys Res Commun. 455:234–240. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vicinanza M, Korolchuk VI, Ashkenazi A,
Puri C, Menzies FM, Clarke JH and Rubinsztein DC: PI (5)P regulates
autophagosome biogenesis. Mol Cell. 57:219–234. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cursio R, Colosetti P, Codogno P, Cuervo
AM and Shen HM: The role of autophagy in liver diseases: Mechanisms
and potential therapeutic targets. Biomed Res Int. 2015:4805082015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang Q, Li Y, Liang T, Lu X, Zhang C, Liu
X, Jiang X, Martin RC, Cheng M and Cai L: ER stress and autophagy
dysfunction contribute to fatty liver in diabetic mice. Int J Biol
Sci. 11:559–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rao J, Zhang C, Wang P, Lu L, Qian X, Qin
J, Pan X, Li G, Wang X and Zhang F: C/EBP homologous protein (CHOP)
contributes to hepatocyte death via the promotion of ERO1α
signalling in acute liver failure. Biochem J. 466:369–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rashid HO, Yadav RK, Kim HR and Chae HJ:
ER stress: Autophagy induction, inhibition and selection.
Autophagy. 11:1956–1977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee WS, Yoo WH and Chae HJ: ER stress and
autophagy. Curr Mol Med. 15:735–745. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Feng Y, He D, Yao Z and Klionsky DJ: The
machinery of macroautophagy. Cell Res. 24:24–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uzi D, Barda L, Scaiewicz V, Mills M,
Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y,
Xavier R, et al: CHOP is a critical regulator of
acetaminophen-induced hepatotoxicity. J Hepatol. 59:495–503. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang X and Quinn PJ: Endotoxins:
Lipopolysaccharides of gram-negative bacteria. Subcell Biochem.
53:3–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
El-Tanbouly DM, Abdelsalam RM, Attia AS
and Abdel-Aziz MT: Pretreatment with magnesium ameliorates
lipopolysaccharide-induced liver injury in mice. Pharmacol Rep.
67:914–920. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao LN, Yan K, Cui YL, Fan GW and Wang YF:
Protective effect of Salvia miltiorrhiza and Carthamus tinctorius
extract against lipopolysaccharide-induced liver injury. World J
Gastroenterol. 21:9079–9092. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Keppler DO, Pausch J and Decker K:
Selective uridine triphosphate deficiency induced by
D-galactosamine in liver and reversed by pyrimidine nucleotide
precursors. Effect on ribonucleic acid synthesis. J Biol Chem.
249:211–216. 1974.PubMed/NCBI
|
|
20
|
Lu Y, Wang WJ, Song YZ and Liang ZQ: The
protective mechanism of schisandrin A in d-galactosamine-induced
acute liver injury through activation of autophagy. Pharm Biol.
52:1302–1307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Z, Zhao YC, Cheng Y, Jian GD, Pan MX
and Gao Y: Hybrid bioartificial liver support in cynomolgus monkeys
with D-galactosamine-induced acute liver failure. World J
Gastroenterol. 20:17399–17406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alcorn JM, Fierer J and Chojkier M: The
acute-phase response protects mice from D-galactosamine
sensitization to endotoxin and tumor necrosis factor-alpha.
Hepatology. 15:122–129. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Amir M, Zhao E, Fontana L, Rosenberg H,
Tanaka K, Gao G and Czaja MJ: Inhibition of hepatocyte autophagy
increases tumor necrosis factor-dependent liver injury by promoting
caspase-8 activation. Cell Death Differ. 20:878–887. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dara L, Ji C and Kaplowitz N: The
contribution of endoplasmic reticulum stress to liver diseases.
Hepatology. 53:1752–1763. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Malhi H and Kaufman RJ: Endoplasmic
reticulum stress in liver disease. J Hepatol. 54:795–809. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tai M, Zhang J, Song S, Miao R, Liu S,
Pang Q, Wu Q and Liu C: Protective effects of luteolin against
acetaminophen-induced acute liver failure in mouse. Int
Immunopharmacol. 27:164–170. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han CY, Lim SW, Koo JH, Kim W and Kim SG:
PHLDA3 overexpression in hepatocytes by endoplasmic reticulum
stress via IRE1-Xbp1 s pathway expedites liver injury. Gut.
65:1377–1388. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jia C, Dai C, Bu X, Peng S, Xu F, Xu Y and
Zhao Y: Co-administration of prostaglandin E1 with somatostatin
attenuates acute liver damage after massive hepatectomy in rats via
inhibition of inflammatory responses, apoptosis and endoplasmic
reticulum stress. Int J Mol Med. 31:416–422. 2013. View Article : Google Scholar : PubMed/NCBI
|