Introduction
Emodin (C15H10O5;
Fig. 1A), as an active constituent
isolated from Chinese traditional herbs, has long been used as a
constituent of several prescriptions in traditional Chinese
medicine to treat and cure various diseases (1). Emodin has been shown to possess
several pharmacological effects, including antibacterial, antiviral
and anti-inflammatory effects (2).
Emodin has also been shown to inhibit cell growth in different
types of tumor cell, including human hepatocellular carcinoma
cells, human promyeloleukemic HL-60 cells, human cervical cancer
cells and Dalton's lymphoma in mice (3). The potency of emodin in inhibiting
cancer cell growth has been reported to be attributed primarily to
the induction of cell apoptosis (1). However, the detailed mechanism by
which Emodin exerts its anticancer effects remains to be fully
elucidated.
As a stress inducible effector, the tumor suppressor
gene p53 exerts important effects on the regulation of cellular
processes, including cell cycle arrest, apoptosis, autophagy,
necroptosis, cell metabolism and senescence (4,5).
Under cellular stresses, p53 is stabilized and binds to the
promoters of target genes, which results in the transcriptional
activation of downstream effectors (6,7).
However, p53 is the most frequently mutated tumor suppressor gene
in human cancer, and clinical studies have demonstrated that ~50%
of tumor cells lack functional p53 (8). P73, a p53 family member, shares
significant homology with p53; it is localized to chromosome
1p36.2–3, a region frequently deleted in several tumor types in
humans (9,10). The p73 gene encodes two isoforms:
Transcriptionally active full-length p73 (TAp73) and NH2-terminally
truncated dominant-negative p73 (∆Np73), which are expressed from
two different promoters, p73-promoter-1 and p73-promoter-2
(11,12). TAp73 shares significant similarity
with p53, including binding with p53 DNA target sites,
transactivation of p53-target genes, and induction of cell cycle
arrest and apoptosis; however, the ∆Np73 isoform has opposite
effects, as it can inhibit the functions of TAp73 and p53 by
competing for binding to the p53/TAp73 DNA target sequence or by
forming oligomers with p53/TAp73 (13). Compared with p53, p73 gene
mutations are rarely found in human tumors, suggesting the
existence of other molecular mechanisms, which may be responsible
for the regulation of p73 during oncogenesis.
The proto-oncoproteins MDM2 and MDMX are negative
regulators of p53 family members (14). These two proteins contain p53
binding domains, which can bind with p53, export it from the
nucleus to the cytosol, and target it for degradation on the 26S
proteasome or repress p53 transcriptional targets (15). Although MDM2 and MDMX can also bind
with p73 and inhibit P73-transcriptional activity, they do not
ubiquitinate p73 (16,17). Therefore, the overexpression of
MDM2 or MDMX can lead to suppression of p53/p73 tumor suppressor
activities. Additionally, the inhibition of MDM2/MDMX through small
interfering (si)RNA or specific inhibitors sensitizes cancer cells
to radiotherapy and chemotherapy (18,19),
suggesting the MDM2 and MDMX may be therapeutic targets for use in
cancer therapies.
In addition to MDM2 family members, other ubiquitin
E3 ligases, including ubiquitin-like protein containing PHD and
RING domains 1 (UHRF1), can also target p53 and p73 ubiquitination,
and suppress p53-dependent transactivation and cell apoptosis in
response to DNA damage signals (20,21).
Conversely, p53 and p73 can regulate the expression of UHRF1
(22,23). UHRF1 contains several domains,
including the N-terminal ubiquitin-like domain (NIRF), plant
homeodomain, Set and Ring Associated domain (also known as the YGD
domain), and C-terminal RING finger. It can form complexes with
histones and non-histone proteins, including promyelocytic leukemia
and DNA methyltransferase (DNMT) 1, and exert its E3 ligase
activities (24,25). In addition to UHRF1, the UHRF
family contains three other members, NIRF, ICBP55 (also known as
UHRF3) and ICBP87 (also known as UHRF3), all of which have similar
domains to those of UHRF1 (26),
suggesting they can also perform UHRF1-like biological functions.
However, the activities of NRIF, ICBP55 and ICBP87 remain to be
elucidated.
Earlier studies have reported that emodin has
antitumor activity against Dalton's lymphoma in vivo
(27), however, the detailed
mechanisms by which emodin induces apoptosis remain to be
elucidated. The present study aimed to analyze the mechanisms
underlying the response to emodin treatment. Using lymphoma Raji
cells, an emodin-induced cell proliferating inhibition model was
established and the possible underlying mechanisms were
investigated. It was found that emodin downregulated the expression
of UHRF1, which in turn suppressed p73 promoter 2 activity through
the upregulation of DNMT3A, and increased TAp73/∆Np73 and cell
apoptosis.
Materials and methods
Chemicals
Emodin,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) and dimethyl sulfoxide (DMSO) were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). RPMI-1640,
Lipofectamine™ 2000, Opti-MEM I medium, TRIzol RNA purification kit
and SYBR green I mix, for quantitative polymerase chain reaction
(qPCR) analysis, were obtained from Invitrogen; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Fetal bovine serum (FBS) was
from Hyclone; GE Healthcare Life Sciences (Logan, UT, USA). Protein
isolation and BCA protein quantification kits were supplied by
Beyotime Institute of Biotechnology (Nanjing, China). The Annexin
V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis
detection kit was obtained from MultiSciences Biotechnology
(Hangzhou, China). The siRNA sequence targeting UHRF1 (cat. no.
sc-270551) was a product of Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). P73-Luc-1, p73-Luc-2 reporter constructs, and the
negative control plasmid were obtained from GeneChem Co., Ltd.
(Shanghai, China). All specific primers for specific genes and the
PrimeScript RT Reagent kit were purchased from Takara Biotechnology
Co., Ltd. (Dalian, China).
Antibodies
Primary antibodies against active-caspase 3 (cat.
no. 9661L), active-caspase 9 (cat. no. BS7070), active-poly
(ADP-ribose) polymerase (PARP; cat. no. AB3620), p53 (cat. no.
AP0104), β-actin (cat. no. BS2237) and UHRF1 (cat. no. MB0055) were
purchased from Bioworld Technology, Inc. (St. Louis Park, MN, USA).
Primary antibodies against ICBP 55 (cat. no. BM1924), ICBP 87 (cat.
no. BM2090) and NIRF (cat. no. BM2989) were supplied by Wuhan
Boster Biological Technology, Ltd. (Wuhan, China). Primary
antibodies against MDM2 (cat. no. sc-812), MDM4 (cat. no.
sc-14740), TAp73 (cat. no. sc-9651), ∆Np73 (cat. no. sc-70966),
DNMT1 (cat. no. sc-271729), DNMT2 (cat. no. sc-271513), DNMT3A
(cat. no. sc-10232), DNMT3B (cat. no. sc-393845) and DNMT3L (cat.
no. sc-10239) were all products of Santa Cruz Biotechnology, Inc.
IRDye-conjugated anti-rabbit secondary antibody (cat. 611-744-127),
IRDye-conjugated anti-mouse secondary antibody (cat. 610-145-121)
and IRDye-conjugated anti-goat secondary antibody (cat.605-744-002)
were all supplied by Rockland Immunochemicals Inc. (Limerick, PA,
USA).
Cell culture and treatment
The Raji cells were obtained from the Cell Bank of
the Type Culture Collection of the Chinese Academy of Science
(Shanghai, China). The Raji cells were maintained in DMEM
supplemented with 10% (v/v) FBS and grown at 37°C with a humidified
5% CO2 atmosphere.
Cell proliferation assay
The Raji cells were plated at 1×104 cells per well
in a 96-well plate. Following culture for ~24 h, the Raji cells
were treated with or without different concentrations of Emodin
including 6.25, 12.5, 25 and 50 µM for 4, 8, 12, 24 and 48 h, and
cultured at 37°C with a humidified 5% CO2 atmosphere. At
the end of the experiment, medium was replaced with 100 µl (500
µg/ml) MTT solution and incubated for 4 h at 37°C. The MTT solution
was then removed, and 150 µl DMSO was added to dissolve the
formazan crystals. The absorbance at 570 nm for each well was read
on a microtiter plate reader (BioTek Instruments, Inc., Winooski,
VT, USA). The results were determined as the percentage of the
control group.
Flow cytometry
The Raji cells were plated at 2×105 cells
per well in a 6-well plate. After 24–48 h, the Raji cells were
treated with or without emodin at concentrations of 6.25, 12.5, 25
and 50 µM for 24 h. At the end of the exposure, the Raji cells were
collected and washed twice in PBS, an then suspended in 0.5 ml
binding buffer containing 5 µl annexin V-FITC and 10 µl PI, and
incubated in the dark at room temperature for 30 min. The samples
were then analyzed using a FACScan flow cytometer (BD Biosciences,
Hercules, CA, USA), and 1×104 events were collected,
recorded on a dot plot, and analyzed using ModFit software version
2.0 (Verity Software House, Inc., Topsham, ME, USA).
Western blot analysis
The Raji cells were treated with or without emodin
at concentrations of 6.25, 12.5, 25 and 50 µM for 24 h and, at the
end of exposure, the Raji cells were harvested for protein
extraction. The whole-cell lysate was extracted using a protein
isolation kit and protein content was quantified using a BCA
protein quantification kit. The lysate proteins (~45 µl; 30 µg)
were denatured at 96°C for 5 min following mixing with 5 µl
SDS-loading buffer. The proteins were separated on a 10% SDS-PAGE
gel and transferred onto polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were then blocked
with TBS containing 5% BSA at 4°C for 1 h, and then exposed to the
specific primary antibodies at 4°C overnight. This was followed by
incubation with the corresponding IRDye-conjugated secondary
antibodies (1:5,000 dilution for all) at room temperature for 1 h.
The proteins were visualized using the Odyssey Infrared Imaging
system with Odyssey v1.2 (LI-COR Biosciences, Lincoln. NE, USA).
The relative expression levels of target proteins were normalized
to the intensities of β-actin.
Total RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
The Raji cells were treated with or without emodin
at concentrations of 6.25, 12.5, 25 and 50 µM for 24 h. At the end
of the experiment, the Raji cells were harvested for RNA extraction
using the TRIzol RNA purification kit. First-strand cDNA synthesis
was performed using the PrimeScript RT Reagent kit according to the
manufacturer's protocol. Amplification was performed using a 7500
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.), using the SYBR Green RT-PCR kit, according to the
manufacturer's protocol [95°C for 10 sec and 56°C for 30 sec (45
cycles)]. Relative mRNA expressions were calculated using the
2−ΔΔCq method and normalized to the internal reference
gene β-actin (28).
Luciferase reporter assay
The p73 promoter 1 and promoter 2 sequences,
synthesized by TransheepBio-Tech Co., Ltd. (Shanghai, China), were
cloned into the pGL3-Luc vector (cat. no. 1741; Promega
Corporation, Madison, WI, USA) using a Universal GenomeWalker kit
supplied by Seebio Biotech (Shanghai) Co., Ltd. (cat. no. 638904;
Shanghai, China), according to the manufacturer's protocol. The
Raji cells were transfected with the pGL3-Luc vector or a negative
vector (pRNL-TK plasmid) using Lipofectamine™ 2000 transfection
reagent according to the manufacturer's protocol. Following
transfection for 24 h, the Raji cells were treated with or without
emodin at concentrations of 6.25, 12.5, 25 and 50 µM for 24 h. At
the end of the exposure, the Raji cells were harvested for mRNA
isolation, following which cDNA amplification was performed at 95°C
for 10 sec and 55°C for 30 sec (40 cycles), and RT-qPCR analysis
were performed using a two-step method.
UHRF1 siRNA transfection
The Raji cells were plated into 6-well plates at a
density of 5×105 cells per well. Following incubation
for 24 h, the cells were harvested and diluted to a density of
8×105/ml with DMEM. The Lipofectamine™ 2000 transfection
reagent and the UHRF1 siRNA sequence were diluted with Opti-MEM I
medium, the final concentrations were 5% and 50 nM, respectively.
The above reagents were incubated at 37°C for 10 min. The two
reagents were then mixed and incubated at room temperature for an
additional 20 min. Finally, the transfection mixture was added to
the 6-well plates. The cell suspensions were overlaid onto the
transfection mixture. Following incubation for 4 h, the medium was
removed and the cells were cultured with fresh DMEM containing 10%
FBS.
Results
Emodin decreases the viability of Raji
cells
The inhibitory effects of emodin (0, 6.25, 12.5, 25
and 50 µM) on Raji cell viability were evaluated at 4, 8, 12, 24
and 48 h post-treatment. As shown in Fig. 1B, compared with the control groups
(untreated groups), significant inhibition of cell viabilities were
induced by 6.25–50 µM emodin at 24 and 48 h (P<0.05), whereas 25
and 50 µM emodin significantly decreased Raji cell viabilities at
4, 8 and 12 h. However, no significant cytotoxic effects were found
in the 6.25 and 12.5 µM emodin-treated groups of Raji cells over
4–12 h (P>0.05).
Emodin induces the apoptosis of Raji
cells
To investigate whether the emodin-induced decrease
in cell viability in Raji cells was attribute to apoptosis, the
cell death was detected using flow cytometry. As shown in Fig. 2A, compared with the control group,
6.25–50 µM emodin treatment for 24 h significantly increased the
apoptosis of Raji cells (P<0.05); these results were further
confirmed by the activation of apoptotic effectors caspase 3, PARP
and caspase 9, compared with the control group, as shown in
Fig. 2B. Treatment with 6.25–50 µM
emodin for 24 h significantly increased the protein levels of
active-caspase 3, active-PARP and active-caspase 9 (P<0.05).
Emodin induces an increase of
TAp73/∆Np73 in Raji cells through the downregulation of ∆Np73
To investigate whether the emodin-induced apoptosis
was associated with p53 or p73, the mRNA and protein expression
levels of p53 and p73 in Raji cells were examined using RT-qPCR and
western blot analyses. As shown in Fig. 3A, compared with the control group,
6.25–50 µM emodin treatment for 24 h significantly decreased the
mRNA levels of p73 (P<0.05), however, no change in the mRNA
level of p53 was observed in the emodin-treated groups (P>0.05).
Analysis of protein levels, as shown in Fig. 3B, showed that, compared with the
control group, the protein levels of ∆Np73, but not of p53 or
TAp73, were significantly reduced in the 6.25–50 µM emodin-treated
groups (P<0.05). The relative ratio of TAp73 and ∆Np73
(TAp73/∆Np73) was calculated and is summarized in Fig. 3C. Treatment with 6.25–50 µM emodin
for 24 h increased the TAp73/∆Np73 ratio, compared with that in the
control group, and these changes were significant (P<0.05).
Emodin induces decreased protein
levels of UHRF1 in Raji cells
To investigate whether the emodin-induced changes in
the expression of ∆Np73 were associated with negative regulators of
p53 family members, the mRNA and protein expression levels of MDM2,
MDM4, ICBP55, ICBP87, NIRF and UHRF1 were examined using RT-qPCR
and western blot analyses. As shown in Fig. 4A, compared with the control group,
no significant changes were found in the mRNA levels of the target
genes in the 6.25–50 µM emodin-treated groups (P>0.05). By
contrast, as shown in Fig. 4B, the
protein levels of UHRF1 in the 6.25–50 µM emodin-treated groups
were decreased, compared with those in the control group, and these
changes were significant (P<0.05).
 | Figure 4.Emodin induces the downregulation of
protein levels of UHRF1. (A) mRNA levels of ICBP55, ICBP87, NIRF,
UHRF1, MDM2 and MDM4 in Raji cells were evaluated using reverse
transcription-quantitative polymerase chain reaction analysis
following treatment with or without 6.25, 12.5, 25 and 50 µM emodin
for 24 h. (B) Protein levels of ICBP55, ICBP87, NIRF, UHRF1, MDM2
and MDM4 in Raji cells were evaluated using western blot analysis
following treatment with or without 6.25, 12.5, 25 and 50 µM emodin
for 24 h. **P<0.01 vs. the untreated control groups. UHRF1,
ubiquitin-like protein containing PHD and RING domains 1. |
Emodin decreases p73 promoter 2
activity through the inhibition of UHRF1
To investigate the associations of UHRF1 and ∆Np73
in this emodin-treated cell model, the activities of p73 promoter 1
(p73-Luc-1) and p73 promoter 2 (p73-Luc-2) in the Raji cells, and
the UHRF1 siRNA-transfected Raji cells were examined using RT-qPCR
analysis. As shown in Fig. 5A,
compared with the control group, the activities of p73-Luc-2 in the
6.25–50 µM emodin-treated groups were significantly decreased
(P<0.05). These emodin-decreased p73-Luc-2 activities were
attenuated by UHRF1 siRNA transfection, as shown in Fig. 5B.
 | Figure 5.Emodin increases the transcriptional
activity of p73-Luc-2 through the inhibition of UHRF1. Raji cells
were co-transfected with p73-Luc-1 and p73-Luc-2 plasmids or
transfected with negative plasmid, following which stable
transfected cells were selected. (A) Stable transfected cells were
treated with or without 6.25, 12.5, 25 and 50 µM emodin for 24 h,
and transcriptional activities of p73-Luc-1 and p73-Luc-2 were
assessed using RT-qPCR analysis. (B) Stable transfected cells were
transfected with UHRF1 siRNA or negative siRNA sequence, and the
transcriptional activities of p73-Luc-1 and p73-Luc-2 were assessed
using RT-qPCR analysis following treatment with or without 6.25,
12.5, 25 and 50 µM emodin for 24 h. **P<0.01 vs. the untreated
control groups. UHRF1, ubiquitin-like protein containing PHD and
RING domains 1; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; siRNA, small interfering RNA; con,
control. |
UHRF1 inhibits p73-Luc-2 activity
through the upregulation of DNMT3A
It is well known that p73 promoter activities can be
directly regulated by methylases, therefore, the present study
examined the mRNA and protein levels of DNMT1, DNMT2, DNMT3A,
DNMT3B and DNMT3L using RT-qPCR and western blot analyses. As shown
in Fig. 6A, no significant change
in the mRNA levels of the target genes were found in the 6.25–50 µM
emodin-treated groups, compared with the control group (P>0.05);
however, as shown in Fig. 6B,
treatment with 6.25–50 µM emodin significantly increased the
protein levels of DNMT3A compared with that in the control group
(P<0.05). The associations between UHRF1 and DNMT3A were further
investigated, as shown in Fig. 6C and
D, and it was found that the emodin-induced upregulation of
DNMT3A and decrease of Raji cell viability were attenuated by the
knockdown of UHRF1.
 | Figure 6.Emodin-induced inhibition of Raji
cell proliferation is dependent on the UHRF1-DNMT3A pathway. (A)
mRNA levels of DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L in Raji
cells were evaluated using reverse transcription-quantitative
polymerase chain reaction following treatment with or without 6.25,
12.5, 25 and 50 µM Emodin for 24 h. (B) Protein levels of DNMT1,
DNMT2, DNMT3A, DNMT3B and DNMT3L in Raji cells was evaluated using
western blot analysis following treatment with or without 6.25,
12.5, 25 and 50 µM Emodin for 24 h; (C) Raji cells were transfected
with UHRF1 siRNA or negative siRNA sequence, and then treated with
or without 6.25, 12.5, 25 and 50 µM Emodin for 24 h. Protein levels
of DNMT3A were detected using western blot analysis; (D) Raji cells
were transfected with UHRF1 siRNA or negative siRNA sequence, and
then treated with or without 6.25, 12.5, 25 and 50 µM Emodin for 24
h. Cell proliferation ratio was evaluated using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
assay. *P<0.05 and **P<0.01 vs. the untreated control groups.
UHRF1, ubiquitin-like protein containing PHD and RING domains 1;
DNMT, DNA methyltransferase; siRNA, small interfering RNA; con,
control. |
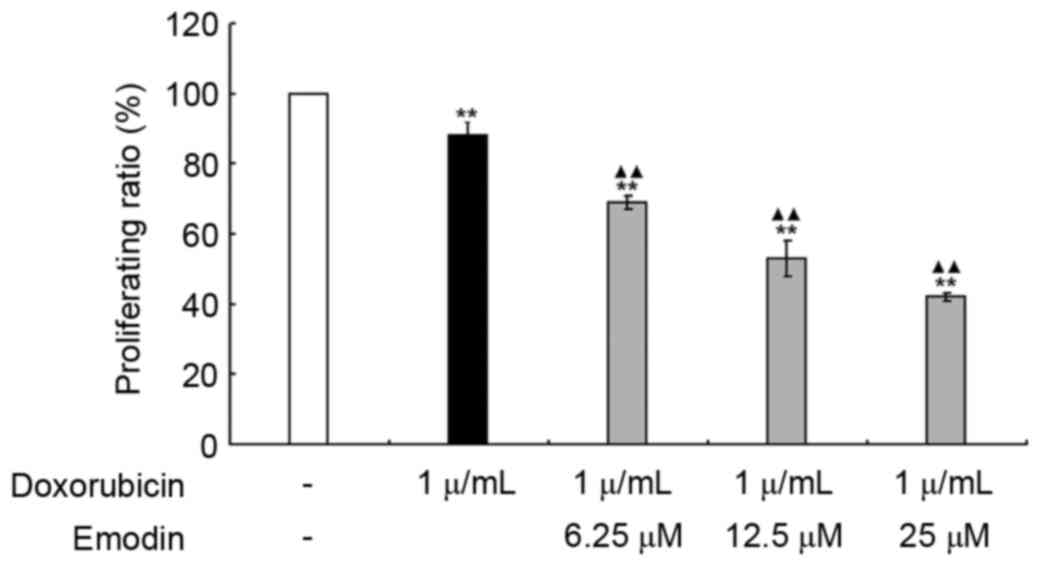
Emodin sensitizes doxorubicin-induced
Raji cell apoptosis
To further investigate whether emodin exerted a
possible synergistic effect in doxorubicin-induced Raji cell
apoptosis. The inhibitory effects of emodin (6.25, 12.5 and 25 µM)
and doxorubicin (1 µg/ml), alone or in combination were examined.
As shown in Fig. 7, compared with
the control group (untreated group), both emodin and doxorubicin
significantly decreased Raji cell viabilities (P<0.01); when the
concentrations of emodin and doxorubicin were added in combination,
the decrease in cell viability was more marked (P<0.01).
Discussion
Lymphoma is a blood cell tumor, which develops from
lymphatic cells. It is characterized by enlarged lymph nodes,
fever, drenching sweats, unintended weight loss, itching and
tiredness (29,30). Although lymphoma can be cured as it
is usually localized, it can be life threatening, with the majority
of victims being children or young adults. Current treatments for
lymphoma consist primarily of chemotherapy and radiation therapy;
however, their lack of specificity and the existence of toxicity to
normal cells are limitations of these methods. Therefore, the
identification of more effective anticancer agents to improve tumor
specificity and decrease toxicity to normal cells is urgently
required. Emodin is a natural compound derived from Chinese
traditional herbs, which has been found to induce apoptosis in
several human tumor cell lines in vitro (3). In agreement with these findings, the
present study found that emodin at concentrations of 6.25, 12.5, 25
and 50 µM markedly decreased the viability of Raji cells at 24 h
post-treatment. This was further confirmed using flow cytometry and
western blot analysis, which demonstrated that emodin
concentrations of 6.25, 12.5, 25 and 50 µM induced apoptosis of the
Raji cells, and increased the activation of caspase 3, caspase 9
and PARP following 24 h exposure.
p73 is a p53 family member and, unlike the p53 gene
which is expressed from a single transcript encoding a single
protein, p73 transcribes multiple isoforms (TAp73 and ∆Np73) due to
the utilization of two promoters. The overexpression of TAp73 can
induce the transactivation of p53-target genes, and inhibit cell
proliferation in a p53-like manner through cell apoptosis (9). The overexpression of ∆Np73 has been
found in various human cancer cell lines and tumor tissues
(10,11). It has been suggested that the ratio
between the levels of TAp73 and ∆Np73 (TAp73/∆Np73) is important in
regulating cell fate in response to anticancer agents during
chemotherapy (31,32). In the present study, the results
confirmed the effects of TAp73/∆Np73 on cell growth, which was
demonstrated by the concurrence of cell apoptosis and increase in
TAp73/∆Np73, the latter attributed to the downregulation of mRNA
levels of ∆Np73.
Abnormal DNA hypermethylation catalyzed by DNMTs is
critical in regulating gene transcription, gene imprinting and X
chromosome inactivation (33,34).
The transcription of p73 can be mediated by its two promoters,
which locate at CpG islands (35,36).
The methylation of cytosine residues of the CpG region results in
silencing of the expression of p73 (36,37).
A previous study reported that p73 was aberrantly methylated in
~30% of cases of primary acute lymphoblastic leukemia and Burkitt's
lymphomas, however, no p73 methylation was detected in normal
tissues (38,39). In the present study, a significant
association was found between the decreased mRNA expression of
∆Np73 and the downregulation in the activity of p73 promoter 2,
suggesting the modification of p73 promoter 2.
The DNMT superfamily contains five members,
including DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L (40). DNMT1 is primarily responsible for
maintaining the methylation status of the genome; DNMT2 encodes a
protein responsible for the methylation of tRNA; and DNMT3A, DNMT3B
and DNMT3L catalyze the addition of methyl groups to cytosine
residues of CpG nucleotides (40).
It was previously reported that DNMT1 can regulate p73 promoter
methylation in the overexpression of rTCS in Caski cells (41). In the cell model used in the
present study, the emodin-induced decrease in the activity of p73
promoter 2 concurred with an increase in the levels of DNMT3A,
suggesting an association between p73 promoter methylation and
DNMT3A. However, the detailed mechanism for the DNMT3A methylation
of p73 requires further investigation.
UHRF1, as an oncogenic factor, is expressed at high
levels in various types of cancer (26). It can transmit DNA methylation
information from parental cells to daughter cells by discriminating
hemimethylated DNA, recruiting DNMT1, and replicating cell nuclear
antigen to the correct location to methylate newly synthesized DNA
sequences (24,42). UHRF1 can also bind with histone H3,
which is tri-methylated at lysine 4 and 9, and maintain the later
heterochromatin status (42–44).
Therefore, UHRF1 is able to recognize DNA methylation and histone
methylation, physically linking these two epigenetic markers. Of
note, the present study found that emodin induced the
downregulation of UHRF1 and upregulation of DNMT3A, rather than
DNMT1. To determine the contribution of the downregulation of UHRF1
to the upregulation of DNMT3A, the present study inhibited the
expression of UHRF1 using siRNA in Raji cells. It was found that
the UHFF1 siRNAs effectively knocked down the expression of UHRF1,
and significantly attenuated the emodin-induced upregulation of
DNMT3A. This suggested the existence of associations between UHFF1
and DNMT3A. However, the detailed mechanism underlying the
upregulation of DNMT3A remains to be fully elucidated.
In conclusion, the present study demonstrated that
emodin treatment resulted in Raji cell proliferation arrest and
apoptosis through an increase in the UHRF1-DNMT3A-TAp73/∆Np73
pathways. These results suggested that emodin may be used as an
anticancer agent to treat the overexpression of UHRF1 in tumors.
Further investigations are required to confirm the biological
functions of emodin in tumor therapy using animal models.
Acknowledgements
The present study was sponsored by Clinical Medical
College of Fujian Medical University.
Glossary
Abbreviations
Abbreviations:
|
TAp73
|
transcriptionally active full-length
p73
|
|
∆Np73
|
NH2-terminally truncated
dominant-negative p73
|
|
UHRF1
|
ubiquitin-like protein containing PHD
and RING domains 1
|
|
DNMT3A
|
DNA methyltransferase 3A
|
References
|
1
|
Zheng ZH, Hu JD, Chen YY, Lian XL, Zheng
HY, Zheng J and Lin MH: Effect of emodin on proliferation
inhibition and apoptosis induction in leukemic K562 cells. Zhongguo
Shi Yan Xue Ye Xue Za Zhi. 17:1434–1438. 2009.(In Chinese).
PubMed/NCBI
|
|
2
|
Lai MY, Hour MJ, Wing-Cheung Leung H, Yang
WH and Lee HZ: Chaperones are the target in aloe-emodin-induced
human lung nonsmall carcinoma H460 cell apoptosis. Eur J Pharmacol.
573:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lai JM, Chang JT, Wen CL and Hsu SL:
Emodin induces a reactive oxygen species-dependent and ATM-p53-Bax
mediated cytotoxicity in lung cancer cells. Eur J Pharmacol.
623:1–9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pastor DM, Irby RB and Poritz LS: Tumor
necrosis factor alpha induces p53 up-regulated modulator of
apoptosis expression in colorectal cancer cell lines. Dis Colon
Rectum. 53:257–263. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang SX, Steinberg SM, Nguyen D and Swain
SM: p53, HER2 and tumor cell apoptosis correlate with clinical
outcome after neoadjuvant bevacizumab plus chemotherapy in breast
cancer. Int J Oncol. 38:1445–1452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goel A, Fuerst F, Hotchkiss E and Boland
CR: Selenomethionine induces p53 mediated cell cycle arrest and
apoptosis in human colon cancer cells. Cancer Biol Ther. 5:529–535.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tu SP, Chi AL, Ai W, Takaishi S,
Dubeykovskaya Z, Quante M, Fox JG and Wang TC: p53 inhibition of
AP1-dependent TFF2 expression induces apoptosis and inhibits cell
migration in gastric cancer cells. Am J Physiol Gastrointest Liver
Physiol. 297:G385–G396. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang L, Zhou Y, Li Y, Zhou J, Wu Y, Cui Y,
Yang G and Hong Y: Mutations of p53 and KRAS activate NF-κB to
promote chemoresistance and tumorigenesis via dysregulation of cell
cycle and suppression of apoptosis in lung cancer cells. Cancer
Lett. 357:520–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Machado-Silva A, Perrier S and Bourdon JC:
p53 family members in cancer diagnosis and treatment. Semin Cancer
Biol. 20:57–62. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Basu S and Murphy ME: p53 family members
regulate cancer stem cells. Cell Cycle. 15:1403–1404. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Becker K, Pancoska P, Concin N, Heuvel K
Vanden, Slade N, Fischer M, Chalas E and Moll UM: Patterns of p73
N-terminal isoform expression and p53 status have prognostic value
in gynecological cancers. Int J Oncol. 29:889–902. 2006.PubMed/NCBI
|
|
12
|
Castillo J, Goñi S, Latasa MU, Perugorría
MJ, Calvo A, Muntané J, Bioulac-Sage P, Balabaud C, Prieto J, Avila
MA and Berasain C: Amphiregulin induces the alternative splicing of
p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular
tumors. Gastroenterology. 137:1805–15, e1–4. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marrazzo E, Marchini S, Tavecchio M,
Alberio T, Previdi S, Erba E, Rotter V and Broggini M: The
expression of the DeltaNp73beta isoform of p73 leads to
tetraploidy. Eur J Cancer. 45:443–453. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mendoza M, Mandani G and Momand J: The
MDM2 gene family. Biomol Concepts. 5:9–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Momand J, Villegas A and Belyi VA: The
evolution of MDM2 family genes. Gene. 486:23–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Madhumalar A, Lee HJ, Brown CJ, Lane D and
Verma C: Design of a novel MDM2 binding peptide based on the p53
family. Cell Cycle. 8:2828–2836. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Johnson J, Lagowski J, Lawson S, Liu Y and
Kulesz-Martin M: p73 expression modulates p63 and Mdm2 protein
presence in complex with p53 family-specific DNA target sequence in
squamous cell carcinogenesis. Oncogene. 27:2780–2787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo W, Ahmed KM, Hui Y, Guo G and Li JJ:
siRNA-mediated MDM2 inhibition sensitizes human lung cancer A549
cells to radiation. Int J Oncol. 30:1447–1452. 2007.PubMed/NCBI
|
|
19
|
Yu H, Zou Y, Jiang L, Yin Q, He X, Chen L,
Zhang Z, Gu W and Li Y: Induction of apoptosis in non-small cell
lung cancer by downregulation of MDM2 using pH-responsive
PMPC-b-PDPA/siRNA complex nanoparticles. Biomaterials.
34:2738–2747. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma J, Peng J, Mo R, Ma S, Wang J, Zang L,
Li W and Fan J: Ubiquitin E3 ligase UHRF1 regulates p53
ubiquitination and p53-dependent cell apoptosis in clear cell Renal
Cell Carcinoma. Biochem Biophys Res Commun. 464:147–153. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alhosin M, Abusnina A, Achour M, Sharif T,
Muller C, Peluso J, Chataigneau T, Lugnier C, Schini-Kerth VB,
Bronner C and Fuhrmann G: Induction of apoptosis by thymoquinone in
lymphoblastic leukemia Jurkat cells is mediated by a p73-dependent
pathway which targets the epigenetic integrator UHRF1. Biochem
Pharmacol. 79:1251–1260. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Colley SM, Iyer KR and Leedman PJ: The RNA
coregulator SRA, its binding proteins and nuclear receptor
signaling activity. IUBMB Life. 60:159–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dai C, Shi D and Gu W: Negative regulation
of the acetyltransferase TIP60-p53 interplay by UHRF1
(ubiquitin-like with PHD and RING finger domains 1). J Biol Chem.
288:19581–19592. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang CC and Cohn MA: UHRF1 is a sensor
for DNA interstrand crosslinks. Oncotarget. 7:3–4. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Delagoutte B, Lallous N, Birck C, Oudet P
and Samama JP: Expression, purification, crystallization and
preliminary crystallographic study of the SRA domain of the human
UHRF1 protein. Acta Crystallogr Sect F Struct Biol Cryst Commun.
64:922–925. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bronner C, Achour M, Arima Y, Chataigneau
T, Saya H and Schini-Kerth VB: The UHRF family: Oncogenes that are
drugable targets for cancer therapy in the near future? Pharmacol
Ther. 115:419–434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Singh KB and Trigun SK: Apoptosis of
Dalton's lymphoma due to in vivo treatment with emodin is
associated with modulations of hydrogen peroxide metabolizing
antioxidant enzymes. Cell Biochem Biophys. 67:439–449. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Poupot M, Pont F and Fournié JJ: Profiling
blood lymphocyte interactions with cancer cells uncovers the innate
reactivity of human gamma delta T cells to anaplastic large cell
lymphoma. J Immunol. 174:1717–1722. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gujral S, Polampalli SN, Badrinath Y,
Kumar A, Subramanian PG, Nair R, Gupta S, Sengar M and Nair C:
Immunophenotyping of mature B-cell non Hodgkin lymphoma involving
bone marrow and peripheral blood: Critical analysis and insights
gained at a tertiary care cancer hospital. Leuk Lymphoma.
50:1290–1300. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Emmrich S, Wang W, John K, Li W and Pützer
BM: Antisense gapmers selectively suppress individual oncogenic p73
splice isoforms and inhibit tumor growth in vivo. Mol Cancer.
8:612009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lau LM, Wolter JK, Lau JT, Cheng LS, Smith
KM, Hansford LM, Zhang L, Baruchel S, Robinson F and Irwin MS:
Cyclooxygenase inhibitors differentially modulate p73 isoforms in
neuroblastoma. Oncogene. 28:2024–2033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Graça I, Sousa EJ, Baptista T, Almeida M,
Ramalho-Carvalho J, Palmeira C, Henrique R and Jerónimo C:
Anti-tumoral effect of the non-nucleoside DNMT inhibitor RG108 in
human prostate cancer cells. Curr Pharm Des. 20:1803–1811. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hopfer O, Komor M, Koehler IS, Freitag C,
Schulze M, Hoelzer D, Thiel E and Hofmann WK: Aberrant promotor
methylation in MDS hematopoietic cells during in vitro lineage
specific differentiation is differently associated with DNMT
isoforms. Leuk Res. 33:434–442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu K, Zhan M and Zheng P: Loss of p73
expression in six non-small cell lung cancer cell lines is
associated with 5′CpG island methylation. Exp Mol Pathol. 84:59–63.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watanabe T, Huang H, Nakamura M,
Wischhusen J, Weller M, Kleihues P and Ohgaki H: Methylation of the
p73 gene in gliomas. Acta Neuropathol. 104:357–362. 2002.PubMed/NCBI
|
|
37
|
Lai J, Nie W, Zhang W, Wang Y, Xie R, Wang
Y, Gu J, Xu J, Song W, Yang F, et al: Transcriptional regulation of
the p73 gene by Nrf-2 and promoter CpG methylation in human breast
cancer. Oncotarget. 5:6909–6922. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rizzo MG, Giombini E, Diverio D, Vignetti
M, Sacchi A, Testa U, Lo-Coco F and Blandino G: Analysis of p73
expression pattern in acute myeloid leukemias: Lack of DeltaN-p73
expression is a frequent feature of acute promyelocytic leukemia.
Leukemia. 18:1804–1809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kawano S, Miller CW, Gombart AF, Bartram
CR, Matsuo Y, Asou H, Sakashita A, Said J, Tatsumi E and Koeffler
HP: Loss of p73 gene expression in leukemias/lymphomas due to
hypermethylation. Blood. 94:1113–1120. 1999.PubMed/NCBI
|
|
40
|
El-Osta A: DNMT cooperativity-the
developing links between methylation, chromatin structure and
cancer. Bioessays. 25:1071–1084. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Peng DF, Kanai Y, Sawada M, Ushijima S,
Hiraoka N, Kitazawa S and Hirohashi S: DNA methylation of multiple
tumor-related genes in association with overexpression of DNA
methyltransferase 1 (DNMT1) during multistage carcinogenesis of the
pancreas. Carcinogenesis. 27:1160–1168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J,
Koseki H and Wong J: UHRF1 targets DNMT1 for DNA methylation
through cooperative binding of hemi-methylated DNA and methylated
H3K9. Nat Commun. 4:15632013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sharif J, Muto M, Takebayashi S, Suetake
I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T,
Okamura K, et al: The SRA protein Np95 mediates epigenetic
inheritance by recruiting Dnmt1 to methylated DNA. Nature.
450:908–912. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hu L, Li Z, Wang P, Lin Y and Xu Y:
Crystal structure of PHD domain of UHRF1 and insights into
recognition of unmodified histone H3 arginine residue 2. Cell Res.
21:1374–1378. 2011. View Article : Google Scholar : PubMed/NCBI
|