Introduction
Post-traumatic stress disorder (PTSD) is a common
condition characterized by persistent mental disturbances following
a traumatic event. The incidence of PTSD has gradually increased,
as a result of frequent natural disasters such as earthquakes and
tsunamis and incidents of violence such as war, terrorist attacks.
and serious traffic accidents. It has been previously demonstrated
that extremely severe psychological trauma contributes considerably
to this disorder (1).
The main symptoms of PTSD include uncontrollable
re-experiencing of the trauma, escape behavior, and irritability
caused by hyperarousal (1). Deep
memories of events or situations that evoke intense fear in PTSD
patients may be related to over-activity of parts of the limbic
system, specifically the amygdala and hippocampus (2). The amygdala is an important component
of the human limbic system, responsible for emotional and
behavioral reactions and is considered to play a critical role in
panic reactions, particularly in terror symptoms (3–5). The
amygdala can be divided into three distinct subregions: The central
nucleus, the corticomedial nucleus, and the basolateral nucleus
(6), the largest of these three.
It is the key region for fear initiation and has been the focus of
significant attention. Vyas and co-workers reported that chronic,
unpredictable stress induces atrophy in bipolar neurons of the
basolateral amygdalae (7). We
focus here on observing changes of the basolateral nucleus.
Studies have revealed that the number of pyramidal
neuron dendrites of PTSD-like rats declined in the hippocampus, but
increased in the amygdalae (8).
Other studies have found that the amygdala is oversensitive in
PTSD, and its degree of activation is in direct proportion to
severity of PTSD symptoms (9).
This research clearly illustrates that changes in the amygdala play
a significant role in the morbidity of PTSD (10). The results demonstrated that the
amygdalae are consistently hyperactivated bilaterally in PTSD
patients (11). Kühn did not find
amygdala volume differences (12),
but other studies have found that the volume of the amygdalae of
PTSD patients shrink, and the degree of volume change correlates
positively with the severity of the PTSD symptoms (13,14).
One of the main mechanisms for maintenance of tissue
homeostasis is the control and regulation of apoptosis (15). Evidence suggests that the reduction
of volume in the amygdala of PTSD patients may be related to nerve
cell apoptosis (16), and the
death and loss of neurons in the amygdala is a generally observed
phenomenon in PTSD (17). However,
the molecular mechanisms underlying this phenomenon remain elusive.
Single-prolonged stress (SPS) is one of the animal models proposed
for PTSD (18). The SPS rat shows
enhanced inhibition of the hypothalamo-pituitary-adrenal (HPA)
axis, which has been frequently demonstrated in patients with PTSD.
It has been reported that SPS induces apoptosis of the hippocampus
and amygdala in the rat model by regulating
Ca2+-calmodulin (CaM)-Ca2+/CaM-dependent
protein kinase II (CaMKII) signaling pathways (19–21).
Our previous studies have demonstrated that apoptosis of neurons is
mainly carried out via mitochondria-related apoptosis factors, such
as caspase-9, caspase-3 and cytochrome c, but the genes Bcl-2 and
Bax also play a significant role (22,23).
The cell cycle refers to the process in which
eukaryotic cells carry out cell proliferation through mitosis. It
consists of four phases: Pre-DNA synthesis (G1), DNA synthesis (S),
post-DNA synthesis (G2), and the mitotic phase (M). The cell cycle
is complicated and delicate, requiring the participation of
multiple protein regulatory factors (24). These regulatory factors include
cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitor
proteins (CDKIs). During the process, cyclins and CDKs may form
specific compounds, which may further act on the specific
substrates, and carry out accurate regulation of the cell cycle
from beginning to end. Cyclin is the regulatory molecule, whereas
CDK is the catalytic subunit (25). Research has found that there are
corresponding cyclins and CDKs in each stage of the cell cycle
(26). CDK4 protein is expressed
substantially in the post-G1 phase, and combines with the cyclin D1
protein, which may finally promote the differentiation of cells
from G1 to S phase (27) and begin
the process of mitosis.
CDK4 and cyclin D1 are important cell cycle
regulators of the G1-S transition (28). Mature neural cells are the end cell
type formed after the differentiation of neuroepithelial stem cells
and have already lost the capacity for division and multiplication.
Deregulation of the cell cycle is associated with injury to the
central nervous system. When injured, neural cells maintain their
reactivation potential and upregulate signaling molecules
responsible for controlling the cell cycle, including cyclin D1 and
CDK4 (29,30). However, reactivation of the cell
cycle is disorganized and cannot control the complete division and
proliferation of cells. Consequently, the cell cycle stops at a
certain phase, arresting the cell cycle arrest and giving rise to
apoptosis (31,32). Studies have shown that Cyclin D1
and CDK4 present excessive expression of the neural cell in
ischemic brain injury, and this may give rise to neural cell
apoptosis (33,34).
This study describes further investigation into
whether SPS affects the expression of cyclin D1 and CDK4 in the
neural cells of the amygdala, and whether this alteration is
correlated with the progression of PTSD. Our results may provide
new insight for the treatment and prevention of PTSD.
Materials and methods
Experimental animals and grouping
The animal experiments were approved by the
Institutional Animal Care and Use Committee of China Medical
University. Male Wistar rats weighing 130 to 170 g and aged 8 to 10
weeks were subjected to a PTSD model using the SPS method. During
the experiment, the rats were housed in groups and were maintained
in a 12/12 h light/dark cycle, at environmental temperatures of
22–25°C, and fed and watered ad libitum. The rats (n=100)
were randomly divided into five groups of 20 rats per group: A
control group, SPS 1-day group (1 day after SPS exposure), SPS
4-day group (4 days after SPS exposure), SPS 7-day group (7 days
after SPS exposure), and SPS 14-day group (14 days after SPS
exposure). In each group of 20 rats, 5 were used for western
blotting, 5 for immunofluorescence studies, 5 for quantitative
polymerase chain reaction (qPCR), and 5 for transmission electron
microscopy (TEM). All experimental procedures were approved by the
Ethics Committee of China Medical University and conducted in
accordance with the Guidelines Principles on Animal Experimentation
for Laboratory Animal Science, China Medical University.
Modeling of PTSD rat: SPS
SPS is recognized internationally as a rat model for
PTSD research, and is described in the following steps (35). Rats were immobilized for 2 h,
followed immediately by a 20-min forced swim conducted in an
acrylic cylindrical tank filled with water (40 cm height, 25°C
water temperature). After recuperating for 15 min, rats were placed
in a shock chamber and anesthetized with ether, and then returned
to their home cages without any stimulation, and fed regularly
until sampling.
TEM
Following SPS, the rats were anesthetized with ether
and 2% pentobarbital. The rats were transcardially perfused with
250 ml cold phosphate-buffered saline (PBS) to remove intravascular
blood. Amygdala tissues were sampled and fixed with 2.5%
glutaraldehyde. The tissue samples were processed for regular TEM,
in semi-thin sections by standard techniques. The 70 nm sections
were positioned for observation and imaging under a transmission
electron microscope (JEOL-1200EX; JEOL, Tokyo, Japan).
Tissue preparation
Rats were deeply anesthetized, then perfused with
cold PBS and 4% paraformaldehyde (PFA). Amygdala tissue was
postfixed in 4% PFA for 3 h, and then immersed in Holt's liquid
(30% sucrose, 0.01 mol/l PBS preparation) until it reached the
bottom. Coronal sections were cut at a thickness of 10 µm using a
cryostat vibratome, and this cryopreservation used for the
immunofluorescence staining.
Immunofluorescence double
staining
The section was dried for 2 h and rinsed with PBS
three times for 5 min, washed for 10 min with 0.3% Triton liquid,
and dried with absorbent paper to remove the Triton liquid.
Sections were blocked in 5% bovine serum albumin in PBS at room
temperature for 20 min, and incubated with a primary antibody
against CDK4 (1:200 in PBS) (ImmunoWay Biotechnology Co., Newark,
DE, USA) overnight at 4°C, with PBS used as a negative control.
Following three washes in PBS, sections were incubated with
anti-rabbit IgG (1:50) labeled with Cy3 (Nanjing KeyGen Biotech.
Co., Ltd., Nanjing, China) and anti-rabbit IgG (1:50) labeled with
4′,6-diamidino-2-phenylindole (DAPI; Nanjing KeyGen Biotech. Co.,
Ltd.), respectively, and maintained in darkness overnight at 4°C.
They were then rinsed with PBS liquid three times for 5 min, and
closed with glycerinum. Images were acquired using a Nikon laser
scanning confocal microscope with EZ-C1 3.70 FreeViewer image
analysis software (Nikon, Tokyo, Japan). The positive cell
fluorescence strength of CDK4 and the positive nucleus fluorescence
strength of DAPI staining of the 5 groups were measured.
Fluorescence strength was indicated as AU/µm2. Five
slides were randomly selected from each group; in each, 5 visual
fields (×40) in the amygdala were randomly selected. For every
animal, approximately 100 cells were counted.
Detection of cyclin D1 and CDK4 with
western blotting
Rats were deeply anesthetized, intracardially
perfused with cold PBS, and the amygdala tissue of the controls and
the SPS group at each time point was rapidly removed. Then the
basolateral amygdalae were dissected according to the atlas of
Paxinos and Watson (36). The
tissue was homogenized in lysis buffer containing protease
inhibitors with further sonication and centrifugation at 12,000
rev/min for 20 min. Total protein content of the supernatant was
quantified using a bicinchoninic acid (BCA) assay kit. An amount of
50 µg of the total protein was electrophoresed in 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel
and transferred to a polyvinylidene fluoride (PVDF) membrane.
Monoclonal antibody cyclin D1 (sc-735; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) at 1:600 dilution and rabbit polyclonal
antibody CDK4 (sc-260; Santa Cruz Biotechnology, Inc.) also at
1:600 dilution were used as primary antibodies. Following three
washes in Tris-buffered saline with Tween-20, the membranes were
incubated with IgG labeled with horseradish peroxidase (HRP). The
HRP-IgG antibody (Hebei Bio-High Technology Development Co.,
Shijiazhuang, China) exposure (1:1,000) continued for 2 h at room
temperature. Immunoreactivity was visualized with an enhanced
chemiluminescence reagent kit. The integrated density value was
calculated with a Fluorchem V 2.0 system and normalized to the
values for glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Wuhan
Boster Biological. Technology, Ltd., Wuhan, China).
Detection of cyclin D1 mRNA, and CDK4
mRNA using reverse transcription (RT)-qPCR
After anesthesia with 2% pentobarbital, amygdala
tissue was isolated from the cerebral hemisphere of rates in the
control group and the SPS group. Total RNA was isolated from the
amygdala tissue by Trizol assay. First-strand cDNA synthesis and
amplification were performed according to a kit from Takara Bio,
Inc. (Otsu, Japan). An amount of 2 µl cDNA was removed for PCR
amplification; the amplification reaction system is shown in
Table I and the primer sequences
are shown in Table II (Shenggong
Biotech Co., Shanghai, China). Amplification conditions were as
follows: Denaturing at 95°C for 30 sec; amplification at 95°C for 5
sec, and heating at 60°C for 20 sec. Forty cycles were performed
with the solution at a temperature of 95°C for 10 sec, and 65°C for
10 sec, and cooled 40°C for 30 sec.
 | Table I.Amplification reaction system. |
Table I.
Amplification reaction system.
| Reagent | Volume |
|---|
| SYBR | 10.0 µl |
| PCR forward
primer | 0.4 µl |
| PCR reverse
primer | 0.4 µl |
| cDNA | 2.0 µl |
|
dH2O | 7.2 µl |
| Total | 20.0 µl |
| Table II.Primers for reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Primers for reverse
transcription-quantitative polymerase chain reaction.
| Name size (bp) | Primer | Product size
(bp) |
|---|
| Cyclin D1 |
|
Sense |
5′-GAGACCATTCCCCTGACTGC-3 | 79 |
|
Antisense |
5′-CCATTTGCAGCAACTCCTCG-3′ |
|
| CDK4 |
|
Sense |
5′-GGAGGCCTTTGAACATCCCA-3 | 182 |
|
Antisense |
5′-ACTGGCGCATCAGATCCTTA-3 |
|
| GAPDH |
|
Sense |
5′-ACTTTGGCATCGTGGAAGGG-3′ | 264 |
|
Antisense |
5′-ACTTGGCAGGTTTCTCCAGG-3′ |
|
All quantifications were normalized to an endogenous
GAPDH control. Cyclin D1 mRNA and CDK4 mRNA expression levels were
validated by using the Real-Time fluorescence qPCR instrument
LightCycler 480, according to the manufacturer's instructions.
After the end qPCR amplification, Cq value of the cyclin D1/CDK4
and GAPDH in control group and the tested group were outputted.
Normalize the Cq value of the target gene with the Cq value of the
GAPDH gene with the formula: ΔCq=Cq (Goal)-Cq (GAPDH). And then
normalize the ΔCq value of the test group with the ΔCT value of the
control group with ΔΔCq=ΔCq (tested group)-ΔCq (control group).
Next, calculate the ratio of expression levels of the tested group
which is relative to the control group with 2−ΔΔCq.
Finally, input the results of 2−ΔΔCq of all groups into
the spss statistical software to draw the chart. The relative
expression level between treatments was standardization of the
target mRNA. We repeated the experiment three times and obtained
similar results.
Statistical analysis
All of the results were analyzed with SPSS 18.0
statistical software (SPSS, Inc., Chicago, IL, USA), and data were
presented as mean ± SEM. Multiple comparisons were carried out
using one-way analysis of variance followed by the Tukey post hoc
test; results of the two groups were compared with the Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
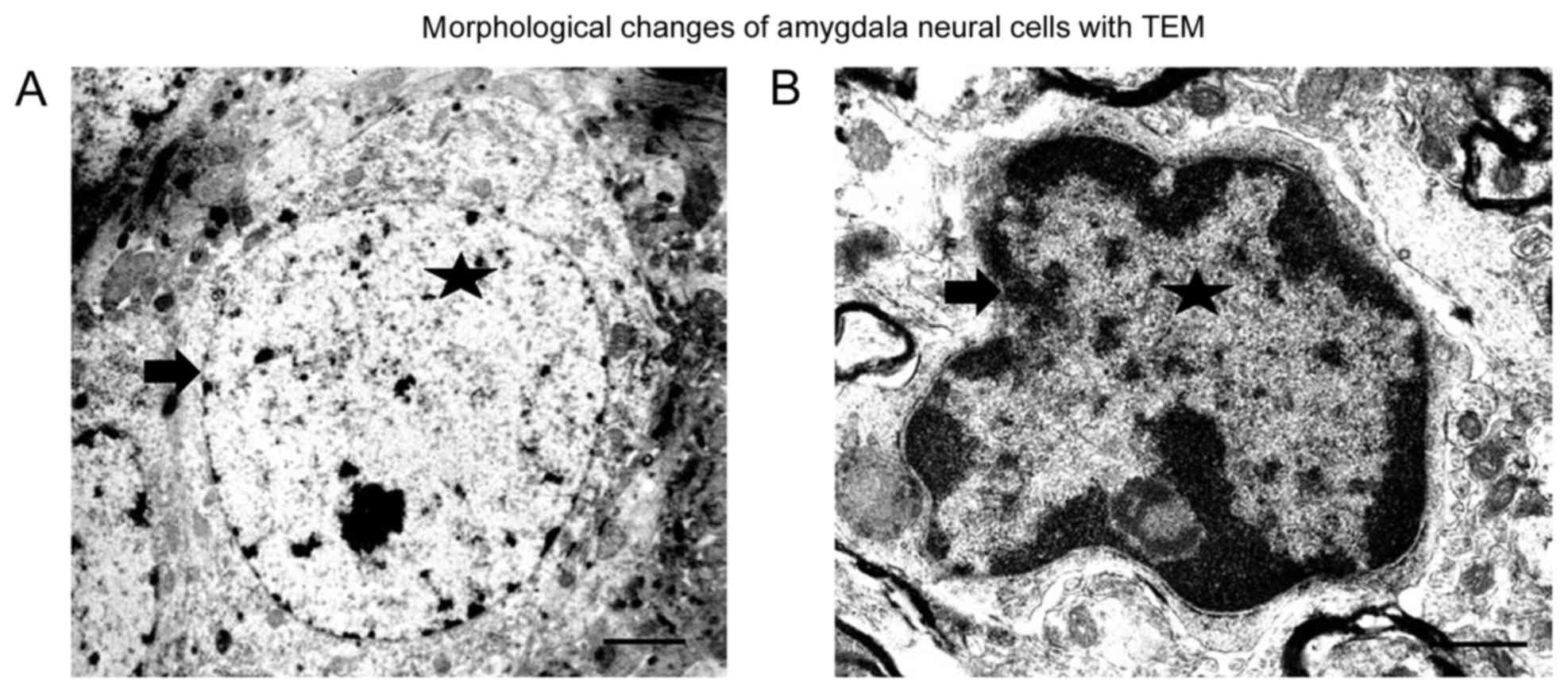
Observation of morphological changes
of amygdala cells with TEM
In the amygdalae of the control group, evenly
distributed chromatin in the nuclear and complete nuclear membrane
was observed (Fig. 1A). After SPS
stimulation, the amygdala neural cells became smaller, and the
chromatin concentrated around the nuclear membrane. The nuclear
membrane was damaged, especially in the SPS 7-day rats (Fig. 1B). Our previous study found there
were different sizes of bubbles in the cytoplasm in the hippocampal
cells, but we did not observe presence of bubbles in the cells from
the amygdala.
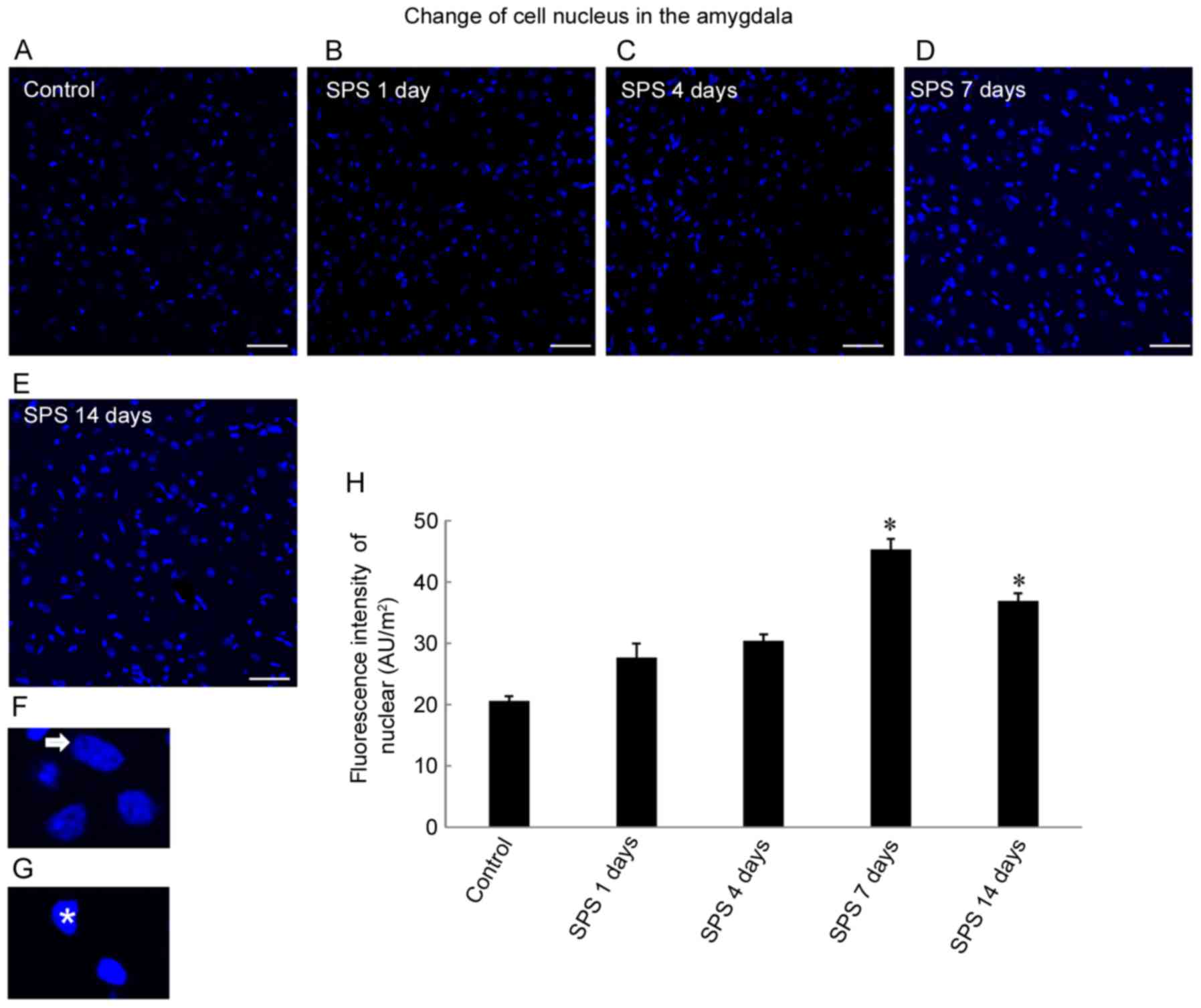
Cell nuclear changes
DAPI is used to stain nuclei so that DNA can be
visualized. We found differences in
4′,6-diamidino-2-phenylindole-stained nuclear fluorescence staining
intensities among the control and SPS groups (Fig. 2A-E). SPS increased staining
intensity in cell nuclei, and reached a peak after 7 days
(P<0.05; Fig. 2H). The
magnification images showed normal cellular nuclei (Fig. 2F, arrowhead) and abnormal cell
nuclei (Fig. 2G, arrow). Nuclei of
normal cells showed low-intensity staining, but the abnormal cells
were smaller and had higher intensity staining because of condensed
chromatin and nuclear pycnosis that were visualized by electron
microscopy (Fig. 1A and B).
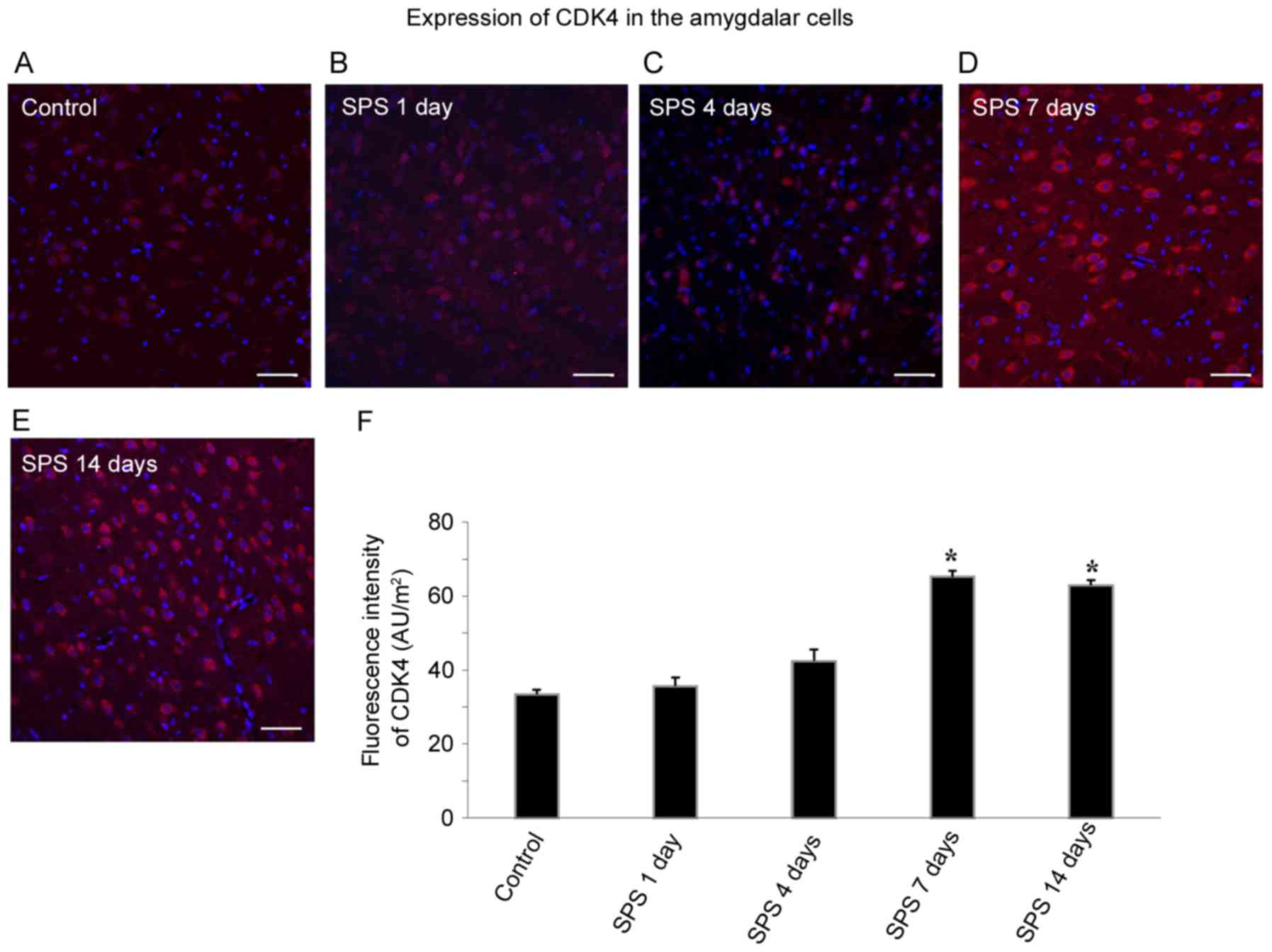
Detection of CDK4 using
immunofluorescence
Amygdala cell CDK4 was labeled with Cy3 fluorescent
staining, and the CDK4 positive cells had red fluorescence.
Apoptosis in the amygdala was measured by co-staining of CDK4 (red)
and DAPI (blue) after SPS stimulation. With high-intensity CDK4
staining of the cytoplasm, the DAPI staining of the
CDK4-immunoreactivity (ir)-positive cells was barely visible,
indicating that CDK4 was only expressed in the non-apoptotic cells,
with none in apoptotic cells. As shown in Fig. 3A, CDK4-ir was relatively weak, and
there were few positive cells in the control group. At 1 and 4 days
after SPS exposure, CDK4-ir did not show significant change in the
amygdala cells. However, SPS significantly increased CDK4
expression at 7 days after SPS (Fig.
3D, n=5, F=32.56, P<0.05) and 14 days after SPS (Fig. 3E, n=5, F=16.91, P<0.05). The
differences in fluorescence intensity between the SPS group at 7
and 14 days, and the control group were statistically significant
(P<0.05; Fig. 3F).
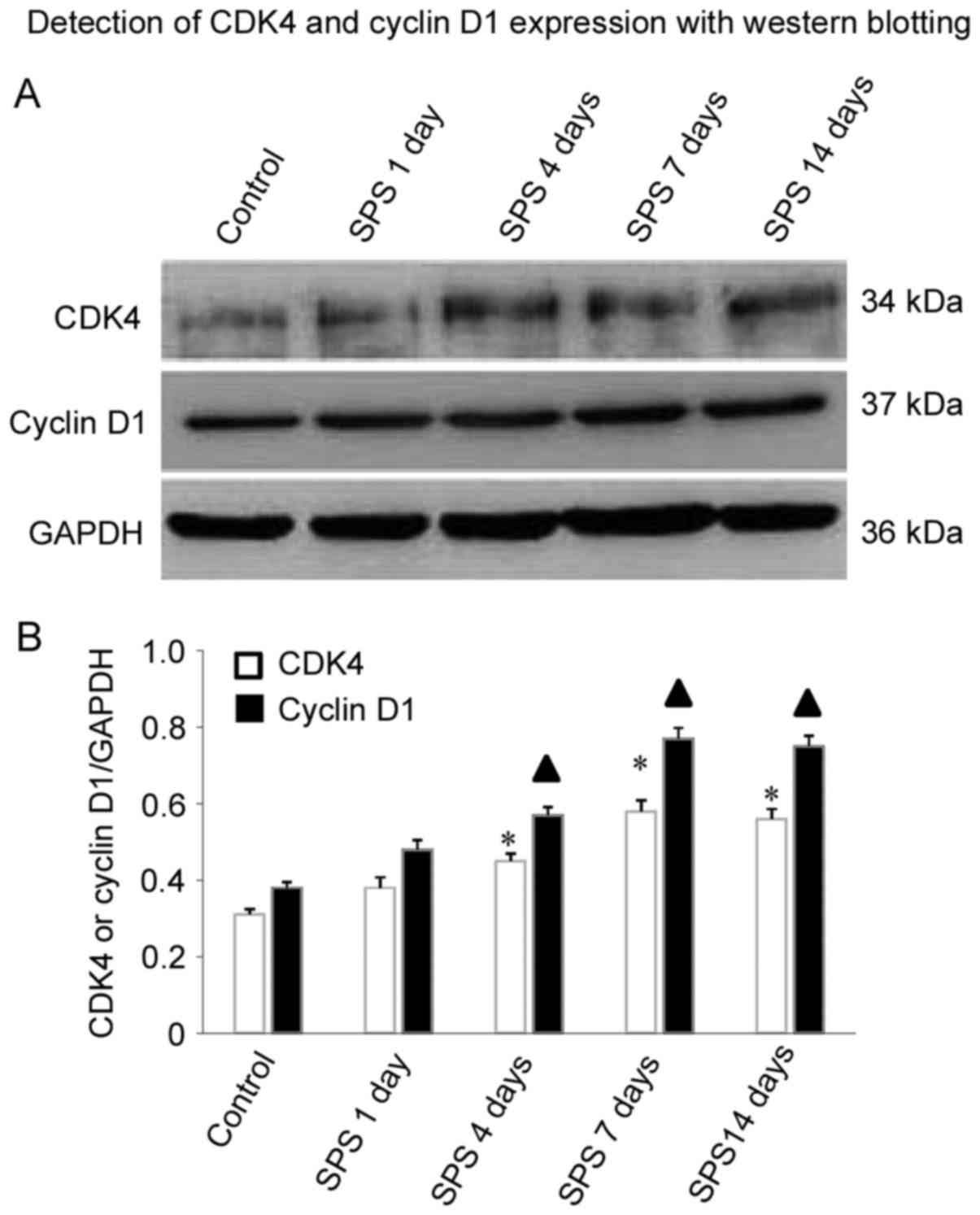
Detection of cyclin D1 and CDK4
expression with western blotting
As shown in Fig.
4A, cyclin D1 protein levels were increased in the amygdala
cells, compared with controls at 1, 4 and 7 days after SPS
stimulation. Interestingly, cyclin D1 protein levels increased with
the duration of SPS stimulation. Cyclin D1 significantly increased
at 4 days (n=5, F=14.32, P<0.05) and reached a peak at 7 days
(n=5, F=30.21, P<0.05), and maintained a relatively high level
at 14 days (n=5, F=23.67, P<0.05) in comparison with control
group. Similar results were observed for CDK4 protein level in the
SPS group. The comparison at 4 days (n=5, F=17.45, P<0.05), 7
days (n=5, F=18.82, P<0.05), and 14 days (n=5, F=19.38,
P<0.05) after SPS and controls was statistically significant
(P<0.05; Fig. 4B).
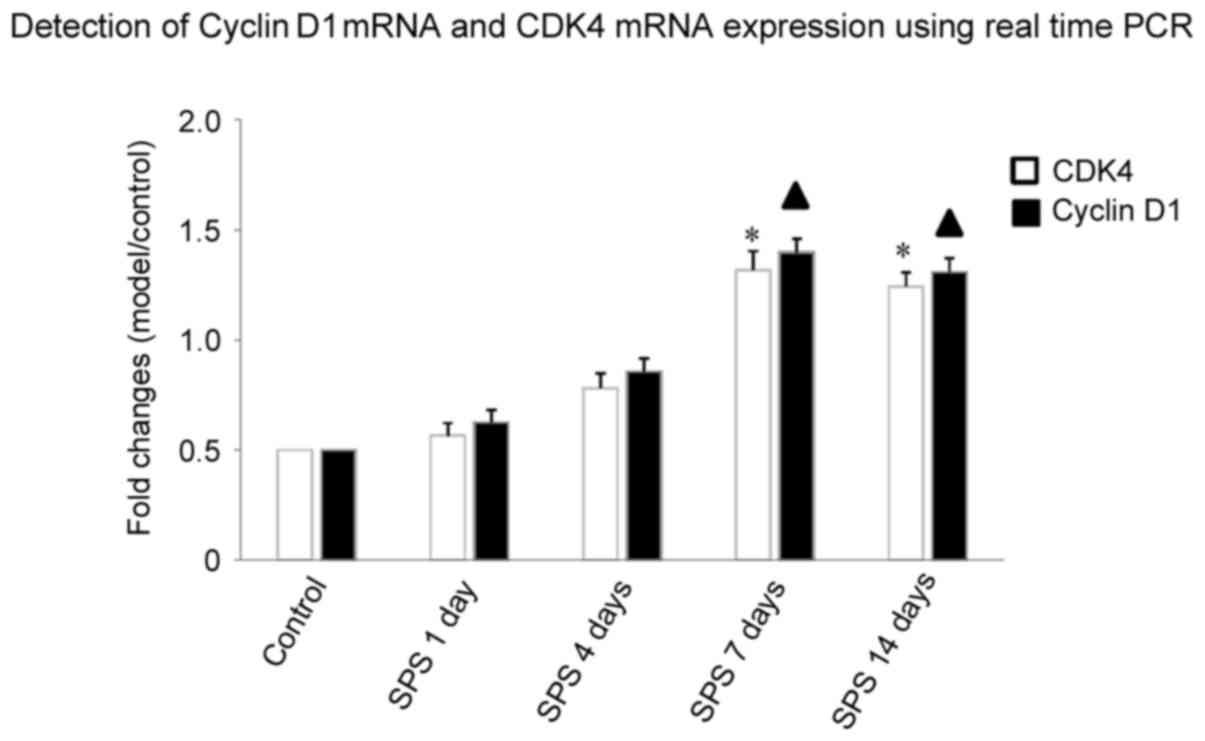
Detection of cyclin D1 mRNA and CDK4
mRNA expression using qPCR
qPCR results showed that after SPS stimulation, at
1, 4, 7 and 14 days, the expression of cyclin D1 mRNA and CDK4 mRNA
in amygdala neurons increased more than in the control group. This
increasing trend of CDK4 mRNA was observed with the extension of
stimulation time, peaking at 7 days (n=5, F=13.28, P<0.05) and
maintaining a high level at 14 days (n=5, F=19.36, P<0.05). The
relative expression of cyclin D1 mRNA in SPS rats at 7 days (n=5,
F=25.39, P<0.05) and 14 days (n=5, F=19.68, P<0.05) after
initial stimulation was statistically significant compared with
control group (P<0.05; Fig.
5).
Discussion
PTSD is an anxiety-related neurological disorder and
mental illness caused by severe stimulation by stressors. About 10%
of people will experience PTSD after exposure to a serious
stressful event (37). One of the
features of PTSD is an exaggerated fear response that does not
resolve with the passage of time after the stimulating event, even
in the absence of continuous stimuli. PTSD patients react strongly
to fearful situations and may experience a strong sense of
helplessness (38). Amygdalae are
involved in the regulation of emotion and play a key role in fear
memory. Magnetic resource imaging (MRI) studies reveal significant
volume reductions in the amygdalae of adult patients with PTSD
(39,40). Our previous study noted a higher
apoptosis rate in the amygdalae of SPS rats with TdT-mediated dUTP
nick-end labeling (TUNEL)-staining and double-labeled flow
cytometry methods. Alterations in apoptosis-related proteins (Bcl-2
and Bax) also occurred in the amygdala of the SPS rats (23). These studies suggest that abnormal
structure and function in the amygdala is involved in PTSD.
Our previous studies used TUNEL and flow cytometry
to show that SPS induced enhanced apoptosis in cells in the
amygdala, hippocampus, and medial prefrontal cortex (23,41,42).
In this experiment, we observed through TEM that apoptosis occurred
after SPS stimulation. Specifically, it was represented by a
declining presence of cell bodies, cytoplasm concentration, and a
frontier set of the chromatin. DAPI fluorescence was employed to
detect nuclear pycnosis. After SPS stimulation, the fluorescence of
cell nuclei in the amygdala increased gradually as a consequence of
the pycnotic changes. These results prove that SPS increased neural
death in the amygdala, which may contribute to the pathogenesis of
PTSD. Functional changes of amygdala cells caused by PTSD may be
the neural structural underpinning of PTSD symptoms.
Research has shown that in damaged axons, certain
regulatory factors of the cell cycle, especially cyclin Dl and
CDK4, are highly expressed (43,44),
and may play a significant role in the induction of cell apoptosis
after central lesions.
The location of CDK4 expression in neurons of the
amygdala of PTSD-like rats was observed using immunofluorescence,
and quantitative analysis of cyclin D1 and CDK4 was performed by
western blotting and qPCR. Cyclin D1 and CDK4 expression was
gradually upregulated after SPS stimulation, peaking after 7 d.
This high expression of cyclin D1 and CDK4 suggests that PTSD may
activate the neural cell cycle, and the high expression of cyclin
D1 and CDK4 fails to play a regulating and controlling role in the
cycle. Such unusually high activation of the cell cycle may be one
of the main causes of neural cell apoptosis. The following reasons
are possible: Mature neural cells are completely differentiated,
and will not enter the cell cycle in normal and non-pathological
conditions, and the expression time and trend of cyclin Dl and CDK4
matches that of the neural cell apoptosis. Using
immunofluorescence, we discovered that CDK4 only exists in
non-apoptotic cytoplasm, suggesting that it may play a role in the
cell apoptosis process, as a significant factor influencing or
inducing cell apoptosis.
We discovered that amygdala neurons went into
apoptosis in PTSD-like SPS rats and consider that the high
expression of cyclin Dl and CDK4 in amygdala neurons may accelerate
the apoptosis of these cells. Possible mechanisms include the
following factors: (1) Due to SPS
stimulation, cyclin Dl and CDK4 are highly expressed in neural
cells. The proliferation signal sent by cyclin Dl and CDK4 to cells
is contradictory and incorrect, which may promote the expression of
another apoptotic factor or directly triggers apoptosis. (2) Following SPS stimulation, high
expression of cyclin Dl activates the cell cycle, but a lack of
necessary enzymes for processing from the G1 to S phase l occurs,
which may result in slowing of the cell cycle and cause apoptosis.
(3) After SPS simulation, the
CDK4/cyclin D1-retinoblastoma protein (pRB)-E2F signal transduction
pathway is activated, and phosphorylated pRB is highly expressed,
releasing free E2F1, which may be related to cell apoptosis
(45).
In our study, SPS is used to establish the PTSD.
Evidences from behavioral and neuroendocrinological studies
examined that 7 days exposed to SPS is needed to develop PTSD. In
the present study, we focus on the expression of CDK4 and cyclin D1
at 1, 4, 7 and 14 days after SPS exposure to explore different
change of both factors during PTSD development (including acute
period: 1 day and 4 days after SPS exposure). We did not find
significant change in CDK 4 and cyclin D1 in the amygdala.
In conclusion, the experimental results show that
changes in the expression of cyclin D1 and CDK4 may accelerate
apoptosis of amygdala neurons in PTSD-like rats. Treatments
interfering with the protein expression of the cell cycle,
administered soon after trauma-induced stress, may lower the
apoptosis of neural cells. This new hypothesis leads to a potential
strategy for treatment of PTSD.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 31140060), the Science and
Technology Planning Project of Shenyang (no. F16-205-1-35), and the
Natural Science Foundation of Liaoning Province, China (no.
.201602274).
References
|
1
|
Bremner JD: The relationship between
cognitive and brain changes in posttraumatic stress disorder. Ann N
Y Acad Sci. 1071:80–86. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Francati V, Vermetten E and Bremner JD:
Functional neuroimaging studies in posttraumatic stress disorder:
Review of current methods and findings. Depress Anxiety.
24:202–218. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brown VM, LaBar KS, Haswell CC, Gold AL;
Mid-Atlantic MIRECC Workgroup, ; McCarthy G and Morey RA: Altered
resting-state functional connectivity of basolateral and
centromedial amygdala complexes in posttraumatic stress disorder.
Neuropsychopharmacology. 39:351–359. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bahraini NH, Breshears RE, Hernández TD,
Schneider AL, Forster JE and Brenner LA: Traumatic brain injury and
posttraumatic stress disorder. Psychiatr Clin North Am. 37:55–75.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klumpers F, Morgan B, Terburg D, Stein DJ
and van Honk J: Impaired acquisition of classically conditioned
fear-potentiated startle reflexes in humans with focal bilateral
basolateral amygdala damage. Soc Cogn Affect Neurosci.
10:1161–1168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Harding AJ, Stimson E, Henderson JM and
Halliday GM: Clinical correlates of selective pathology in the
amygdala of patients with Parkinson's disease. Brain. 11:2431–2445.
2002. View Article : Google Scholar
|
|
7
|
Vyas A, Mitra R, Rao BS Shankaranarayana
and Chattarji S: Chronic stress induces contrasting patterns of
dendritic remodeling in hippocampal and amygdaloid neurons. J
Neurosci. 22:6810–6818. 2002.PubMed/NCBI
|
|
8
|
Miller MM and McEwen BS: Establishing an
agenda for translational research on PTSD. Ann N Y Acad Sci.
1071:294–312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sarro EC, Sullivan RM and Barr G:
Unpredictable neonatal stress enhances adult anxiety and alters
amygdala gene expression related to serotonin and GABA.
Neuroscience. 258:147–161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
El K, houry-Malhame M, Reynaud E, Soriano
A, Michael K, Salgado-Pineda P, Zendjidjian X, Gellato C, Eric F,
Lefebvre MN, Rouby F, et al: Amygdala activity correlates with
attentional bias in PTSD. Neuropsychologia. 49:1969–1973. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hayes JP, Hayes SM and Mikedis AM:
Quantitative meta-analysis of neural activity in posttraumatic
stress disorder. Biol Mood Anxiety Disord. 2:92012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kühn S and Gallinat J: Gray matter
correlates of posttraumatic stress disorder: A quantitative
meta-analysis. Biol Psychiatry. 73:70–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Morey RA, Gold AL, LaBar KS, Beall SK,
Brown VM, Haswell CC, Nasser JD, Wagner HR and McCarthy G;
Mid-Atlantic MIRECC Workgroup, : Amygdala volume changes in
posttraumatic stress disorder in a large case-controlled veterans
group. Arch Gen Psychiatry. 69:1169–1178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brenner LA: Neuropsychological and
neuroimaging findings in traumatic brain injury and post-traumatic
stress disorder. Dialogues Clin Neurosci. 13:311–323.
2011.PubMed/NCBI
|
|
15
|
Fu XL and Gao DS: Endoplasmic reticulum
proteins quality control and the unfolded protein response: The
regulative mechanism of organisms against stress injuries.
Biofactors. 40:569–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu H, Wang HT, Han F and Shi YX: Activity
of 5-HT1A receptor is involved in neuronal apoptosis of the
amygdala in a rat model of post-traumatic stress disorder. Mol Med
Rep. 4:291–295. 2011.PubMed/NCBI
|
|
17
|
Wimalawansa SJ: Mechanisms of developing
post-traumatic stress disorder: New targets for drug development
and other potential interventions. CNS Neurol Disord Drug Targets.
13:807–816. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kohda K, Harada K, Kato K, Hoshino A,
Motohashi J, Yamaji T, Morinobu S, Matsuoka N and Kato N:
Glucocorticoid receptor activation is involved in producing
abnormal phenotypes of single prolonged stress rats: A putative
post-traumatic stress disorder model. Neuroscience. 148:22–33.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li XM, Han F, Liu DJ and Shi YX:
Single-prolonged stress induced mitochondrial-dependent apoptosis
in hippocampus in the rat model of post-traumatic stress disorder.
J Chem Neuroanat. 40:248–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu H, Li H, Xu A, Kan Q and Liu B: Role
of phosphorylated ERK in amygdale neuronal apoptosis in
single-prolonged stress rats. Mol Med Rep. 3:1059–1063.
2010.PubMed/NCBI
|
|
21
|
Xiao B, Han F and Shi YX: Dysfunction of
Ca2+/CaM kinase IIalpha cascades in the amygdala in post-traumatic
stress disorder. Int J Mol Med. 24:795–799. 2009.PubMed/NCBI
|
|
22
|
Xiao B, Yu B, Wang HT, Han F and Shi YX:
Single-prolonged stress induces apoptosis by activating cytochrome
C/caspase-9 pathway in a rat model of post-traumatic stress
disorder. Cell Mol Neurobiol. 31:37–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ding J, Han F and Shi Y: Single-prolonged
stress induces apoptosis in the amygdala in a rat model of
post-traumatic stress disorder. J Psychiatr Res. 44:48–55. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Milewska M, Grabiec K and
Grzelkowska-Kowalczyk K: Interactions of proliferation and
differentiation signaling pathways in myogenesis. Postepy Hig Med
Dosw (Online). 68:516–526. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moschovi M, Alexiou GA, Patereli A, Siozos
G, Sfakianos G, Prodromou N and Stefanaki K: Immunohistochemical
expression of cell-cycle regulators in pediatric embryonal brain
tumors. J Neurooncol. 109:529–534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Buendía-Monreal M, Rentería-Canett I,
Guerrero-Andrade O, Bravo-Alberto CE, Martínez-Castilla LP, García
E and Vázquez-Ramos JM: The family of maize D-type cyclins: Genomic
organization, phylogeny and expression patterns. Physiol Plant.
143:297–308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Casimiro MC, Velasco-Velázquez M,
Aguirre-Alvarado C and Pestell RG: Overview of cyclins D1 function
in cancer and the CDK inhibitor landscape: Past and present. Expert
Opin Invest Drugs. 23:295–304. 2014. View Article : Google Scholar
|
|
28
|
Lee Y, Dominy JE, Choi YJ, Jurczak M,
Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al:
Cyclin D1-Cdk4 controls glucose metabolism independently of cell
cycle progression. Nature. 510:547–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kabadi SV and Faden AI: Selective CDK
inhibitors: Promising candidates for future clinical traumatic
brain injury trials. Neural Regen Res. 9:1578–1580. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu J, Stoica BA, Dinizo M, Pajoohesh-Ganji
A, Piao C and Faden AI: Delayed cell cycle pathway modulation
facilitates recovery after spinal cord injury. Cell Cycle.
11:1782–1795. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Folch J, Junyent F, Verdaguer E, Auladell
C, Pizarro JG, Beas-Zarate C, Pallàs M and Camins A: Role of cell
cycle re-entry in neurons: A common apoptotic mechanism of neuronal
cell death. Neurotox Res. 22:195–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang W, Bu B, Xie M, Zhang M, Yu Z and Tao
D: Neural cell cycle dysregulation and central nervous system
diseases. Prog Neurobiol. 89:1–17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pei L, Zhang Y, Zhang Y, Chu X, Zhang J,
Wang R, Liu M, Zhu X and Yu W: Peroxisome proliferator-activated
receptor gamma promotes neuroprotection by modulating cyclin D1
expression after focal cerebral ischemia. Can J Physiol Pharmacol.
88:716–723. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee CH, Yoo KY, Choi JH, Park OK, Hwang
IK, Choi SY, Kim DH and Won MH: Cyclin D1 immunoreactivity changes
in CA1 pyramidal neurons and dentate granule cells in the gerbil
hippocampus after transient forebrain ischemia. Neurol Res.
33:93–100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liberzon I, Krstov M and Young EA:
Stress-restress: Effects on ACTH and fast feedback.
Psychoneuroendocrinology. 22:443–453. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Paxinos G and Watson C: The rat brain in
stereotaxic coordinates. 4th edition. Academic Press; San Diego.
CA: 1998
|
|
37
|
Koch SB, van Zuiden M, Nawijn L, Frijling
JL, Veltman DJ and Olff M: Intranasal oxytocin as strategy for
medication-enhanced psychotherapy of PTSD: Salience processing and
fear inhibition processes. Psychoneuroendocrinology. 40:242–256.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Amos T, Stein DJ and Ipser JC:
Pharmacological interventions for preventing post-traumatic stress
disorder (PTSD). Cochrane Database Syst Rev. 7:CD0062392014.
|
|
39
|
Karl A, Schaefer M, Malta LS, Dörfel D,
Rohleder N and Werner A: A meta-analysis of structural brain
abnormalities in PTSD. Neurosci Biobehav Rev. 30:1004–1031. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Driessen M, Beblo T, Mertens M, Piefke M,
Rullkoetter N, Silva-Saavedra A, Reddemann L, Rau H, Markowitsch
HJ, Wulff H, et al: Posttraumatic stress disorder and fMRI
activation patterns of traumatic memory in patients with borderline
personality disorder. Biol Psychiatry. 55:603–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li X, Han F, Liu D and Shi Y: Changes of
Bax, Bcl-2 and apoptosis in hippocampus in the rat model of
post-traumatic stress disorder. Neurol Res. 32:579–586. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Y, Han F and Shi Y: Increased neuronal
apoptosis in medial prefrontal cortex is accompanied with changes
of Bcl-2 and Bax in a rat model of post-traumatic stress disorder.
J Mol Neurosci. 51:127–137. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nobs L, Nestel S, Kulik A, Nitsch C and
Atanasoski S: Cyclin D1 is required for proliferation of
Olig2-expressing progenitor cells in the injured cerebral cortex.
Glia. 61:1443–1455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kabadi SV, Stoica BA, Loane DJ, Byrnes KR,
Hanscom M, Cabatbat RM, Tan MT and Faden AI: Cyclin D1 gene
ablation confers neuroprotection in traumatic brain injury. J
Neurotrauma. 29:813–827. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Carnevale J, Palander O, Seifried LA and
Dick FA: DNA damage signals through differentially modified E2F1
molecules to induce apoptosis. Mol Cell Biol. 32:900–912. 2012.
View Article : Google Scholar : PubMed/NCBI
|