Introduction
Acute myocardial infarction (AMI) results from a
disruption of coronary blood flow to the myocardial region that it
supplies. AMI remains the leading cause of mortality and is
associated with a heavy financial burden worldwide (1). Prolonged cardiac ischemia can provoke
tissue damage due to a lack of oxygen and nutrients since the heart
demands high energy to function. The continuous deficiency of
oxygen and nutrients alters ion homeostasis and metabolism,
reducing cardiac contractility and structural organization,
initiating cell death via necrosis and apoptosis (2). Cell death via necrosis is
characterized by cell membrane rupture and the consequent release
of cellular components (e.g., creatine kinase, troponin), which
subsequently provokes an inflammatory response (3). The molecular mechanisms regarding
ischemic injury are multifactorial. A growing number of studies
have investigated the role that reactive oxygen species (ROS) serve
in hypoxia, ranging from beneficial to damaging (4,5). At
the basal level, ROS functions as a mediator for multiple cellular
signaling cascades including cell growth and stress adaptation. In
myocardial ischemia, excess ROS can damage tissues by oxidizing
important cellular components such as proteins, lipids and DNA
(4).
More recently, the role of autophagy in the
pathogenesis of myocardial ischemic injury has been investigated.
Autophagy is an evolutionarily conserved catabolic process that
targets dysfunctional or damaged cytoplasmic constituents to the
lysosome for degradation and recycling (6). Autophagy is essential for the
survival of newborn mice between birth and the onset of suckling
(7,8). Previous research has demonstrated
that HL-1 cells overexpressing green fluorescent protein
(GFP)-microtubule-associated proteins 1A/1B light chain 3 (LC3)
exposed to 2 h of simulated ischemia in the absence of oxygen
exhibited a low level of autophagy (9). Hamacher-Brady et al (9) reported that autophagy was completely
blocked in HL-1 cells exposed to 2 h of simulated ischemia in the
absence of oxygen. Inhibition of autophagy was achieved by
treatments with either 3-methyladenine or wortmannin,
downregulation of Beclin 1 or overexpression of autophagy related 5
(Atg5) K130R; all these interventions sensitized HL-1 cardiac cells
to apoptosis induced by ischemia reperfusion. These indicate the
cardioprotective effect of autophagy against ischemia injury.
Considerable advances have been made in recognizing the molecular
mechanisms that determine the protection afforded by these
strategies. Pharmacological agents targeting key signaling
effectors involved in the ischemic and reperfusion cascades have
also been developed (10), while
most of them have yielded disappointing results in clinical trials
(11). Despite these setbacks,
identifying novel potential ‘druggable’ targets for the clinical
management of AMI is remains imminent.
Sanggenon C, a flavanone Diel-Alder adduct compound,
is isolated from the root bark of Morus cathayana. A
previous study has reported that by inhibiting proteasome function,
Sanggenon C inhibits tumor cell viability via induction of cell
cycle arrest and cell death (12).
Sanggenon C also protected against lipopolysaccharide
(LPS)-stimulated RAW264.7-cell inflammation by inhibiting nitric
oxide (NO) production and inducible nitric oxide synthase
expression via suppressing nuclear factor-κB (NF-κB) activity and
NF-κB inhibitor α activation (13). By suppressing the activation of
NF-κB, Sanggenon C inhibited tumor necrosis factor α (TNFα)
stimulated human polymorphonuclear leukocyte adhesion to human
synovial cell and expression of vascular cell adhesion molecule 1
(14). Since Sanggenon C possesses
antioxidant and anti-inflammatory activities, it may serve as a
cardioprotective agent. The purpose of our study was to determine
the effects of Sanggenon C on cardiomyocyte hypoxia injury.
Materials and methods
Reagents
The primary antibodies included phosphorylated
AMP-activated protein kinase α (p-AMPKα; cat. no. 2535), total
(T)-AMPKα (cat. no. 2603P), p-mechanistic target of rapamycin
(mTOR; cat. no. 2971), T-mTOR (cat. no. 2983), p-forkhead box O3a
(FOXO3a; cat. no. 9465P), T-FOXO3a (cat. no. 2497P), Bcl-2
associated X apoptosis regulator (Bax; cat. no. 2722), Bcl-2
apoptosis regulator (Bcl-2; cat. no. 2870), and GAPDH (cat. no.
2118; all purchased from Cell Signaling Technology, Inc., Danvers,
MA, USA). Sanggenon C (98% purity as determined by high-performance
liquid chromatography analysis) was purchased from Shanghai Winherb
Medical S&T Development Co., Ltd. (Shanghai, China). Fetal
bovine serum was ordered from Gibco (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The cell culture reagents were purchased
from Gibco (Thermo Fisher Scientific, Inc.).
Cell culture
Rat cardiac H9c2 cells (Cell Bank of the Chinese
Academy of Sciences, Shanghai, China) were cultured in Dulbecco's
modified Eagle's medium (DMEM; cat. no. C11885; Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(FBS; cat. no. 10099-133; Gibco; Thermo Fisher Scientific, Inc.,),
100 U/ml penicillin/100 mg/ml streptomycin (cat. no. 15140; Gibco;
Thermo Fisher Scientific, Inc.) and 5% CO2 at 37°C. The
media was changed every 1–2 days and subcultured to 70–80%
confluency. Cells were plated at an appropriate density according
to each experimental design. Cells were seeded at a density of
1×106/well onto 6-well culture plates for mRNA
extraction, 5×103/well in 24-well plates for cell
surface area examination and 10×107/well onto 10-cm
diameter culture plates for protein extraction. Following 24 h
adherence, the culture medium was changed to serum-free DMEM 12 h
prior to the experiment. Cells were pretreated with Sanggenon C (1,
10 and 100 µM) and/or Compound C (CpC; 20 µM) for 12 h in
serum-free DMEM at 37°C and then cells were maintained at 37°C,
under a hypoxic atmosphere of 95% N2 and 5%
CO2 for 24 h.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from frozen H9c2 cells using
TRIzol (cat. no. 15596-026; Thermo Fisher Scientific, Inc.). RNA
yield and purity were evaluated using a SmartSpec Plus
Spectrophotometer (Bio-Rad Laboratories Inc., Hercules, CA, USA),
comparing the absorbance (A) 260/A280 and A230/260 ratios. RT was
performed on RNA (2 µg of each sample) to produce cDNA using oligo
(dT) primers and the Transcriptor First Strand cDNA Synthesis kit
(cat. no. 04896866001; Roche Diagnostics, Basel Switzerland). The
PCR products were quantified using a LightCycler 480 SYBR-Green 1
Master mix (cat. no. 04707516001; Roche Diagnostics). Following an
initial 5 min denaturation step at 95°C, a total of 42
primer-extension cycles were carried out. Each cycle consisted of a
10 sec denaturation step at 95°C, a 20 sec annealing step at 60°C,
and a 20 sec incubation at 72°C for extension. Then a final
extension step was performed at 72°C for 10 min. The double
standard curve was used to quantify the PCR results. Calibrator
normalized ratio = (concentration of sample target/concentrations
of sample reference)/(concentration of calibrator
target/concentration of calibrator reference) (15). The results were normalized against
GAPDH gene expression. The sequences of the oligonucleotide primers
(Sangon Biotech Co., Ltd., Shanghai, China) were as follows: TNFα
forward, 5′-AGCATGATCCGAGATGTGGAA-3′ and reverse,
5′-TAGACAGAAGAGCGTGGTGGC-3′; interleukin-1 (IL-1) forward,
5′-GGGATGATGACGACCTGCTAG-3′ and reverse,
5′-ACCACTTGTTGGCTTATGTTCTG-3′; IL-6 forward,
5′-GTTGCCTTCTTGGGACTGATG-3′ and reverse,
5′-ATACTGGTCTGTTGTGGGTGGT-3′; Beclin-1 forward,
5′-AGCTTTTCTGGACTGTGTGC-3′ and reverse, 5′-TGAACTTGAGCGCCTTTGTC-3′;
Atg5 forward, 5′-CAAGGATGCAGTTGAGGCTC-3′ and reverse,
5′-AGTTTCCGGTTGATGGTCCA-3′; p62 forward, 5′-AAGAGGCTCCATCACCAGAG-3′
and reverse, 5′-CCCCTTGACTCTGGCTGTAA-3′.
ROS measurement
The level of intracellular ROS generation was
assessed using the fluorescent dye 2′,7′-dichlorofluorescin
diacetate (DCFH-DA). Following the indicated treatments, cells were
washed twice with PBS and then incubated with serum-free DMEM and
1×10−5 mol/l DCFH-DA in a 37°C incubator for 30 min.
Subsequently, cells were washed with PBS for three times to
eliminate the residual DCFH-DA. A fluorescence microscope (BX51;
Olympus Corporation, Tokyo, Japan) was also used to evaluate the
DCFH florescence of cells on coverslips.
Antioxidative assessments
Antioxidative capacity was assessed by the release
of NO and the activity of SOD according to the protocol of the NO
detection kit, SOD detection kit, respectively (Beyotime Institute
of Biotechnology, Haimen, China). For NO detection, the supernatant
was collected and transferred to another 96-well plate. The NO
released from H9c2 cells was measured at 540 nm using
spectrophotometer according to the manufacturer's instructions
(Beyotime Institute of Biotechnology). For SOD assessment, proteins
were extracted and quantified before measuring the activity of SOD
at 450 nm using the commercial kit (Beyotime Institute of
Biotechnology).
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick-end labelling
(TUNEL) staining
Cells were cultured on cover slips in a 24-well
plate, fixed in 4% paraformaldehyde for 5 min at room temperature
and then permeabilized in 0.1% Triton X-100 for 5 min in room
temperature following treatment. TUNEL staining according to the
protocol of ApopTag® Plus Fluorescein in situ
Apoptosis Detection kit (EMD Millipore, Billerica, MA, USA) for 1 h
at 37°C. Nuclei were labeled with DAPI and DNA fragmentation was
quantified under fluorescence microscope (magnification, ×200;
BX51TRF; Olympus Corporation). The percentages of TUNEL-positive
cells relative to DAPI-positive cells were calculated by an
investigator in a blinded manner.
Western blot analysis
Cultured cardiac H9c2 cells were lysed in
radioimmunoprecipitation (RIPA) lysis buffer [720 µl RIPA, 20 µl
phenylmethylsulfonyl fluoride (1 mM), 100 µl cOmplete™
protease inhibitor cocktail (cat. no. 04693124001; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany)], 100 µl phosSTOP (cat. no.
04906837001; Roche Diagnostics), 50 µl NaF (1 mM), 10 µl
Na3VO4/ml and the protein concentration was
measured by the bincinchoninic assay method. A total of 30 µg cell
lysate was used for protein separation using SDS-PAGE on a 10% gel.
The proteins were then transferred to polyvinylidene difluoride
(PVDF) membranes (EMD Millipore). Specific protein expression
levels were normalized to the GAPDH protein levels of the total
cell lysate and cytosolic proteins on the same PVDF membranes,
which were blocked with 5% non-fat milk at room temperature for 2
h. The following primary antibodies were used: p-AMPKα, T-AMPKα,
p-mTOR, T-mTOR, p-FOXO3a, T-FOXO3a, Bax, Bcl-2, and GAPDH. The
primary antibodies were diluted at 1:1,000. Antibody incubation was
performed overnight with gentle shaking at 4°C. Quantification of
the western blots was performed using an Odyssey infrared imaging
system (LI-COR, Lincoln, NE, USA). The secondary antibodies, goat
anti-rabbit IRdye® 800 CW (cat. no. 926-32211; LI-COR)
IgG and goat anti-mouse IRdye 800 CW (cat. no. 926-32210; LI-COR),
were used at a 1:10,000 dilution at 37°C in Odyssey blocking for 1
h. The blots were scanned using an infrared LI-COR scanner,
allowing for simultaneous detection of two targets (phosphorylated
and total protein) within the same experiment.
Statistical analysis
Data is expressed as the mean ± standard error of
the mean. Differences among groups were determined by a two-way
analysis of variance followed by Tukey's post hoc test. Statistical
analyses were conducted using SPSS software (version 19.0; IBM
Corp., Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
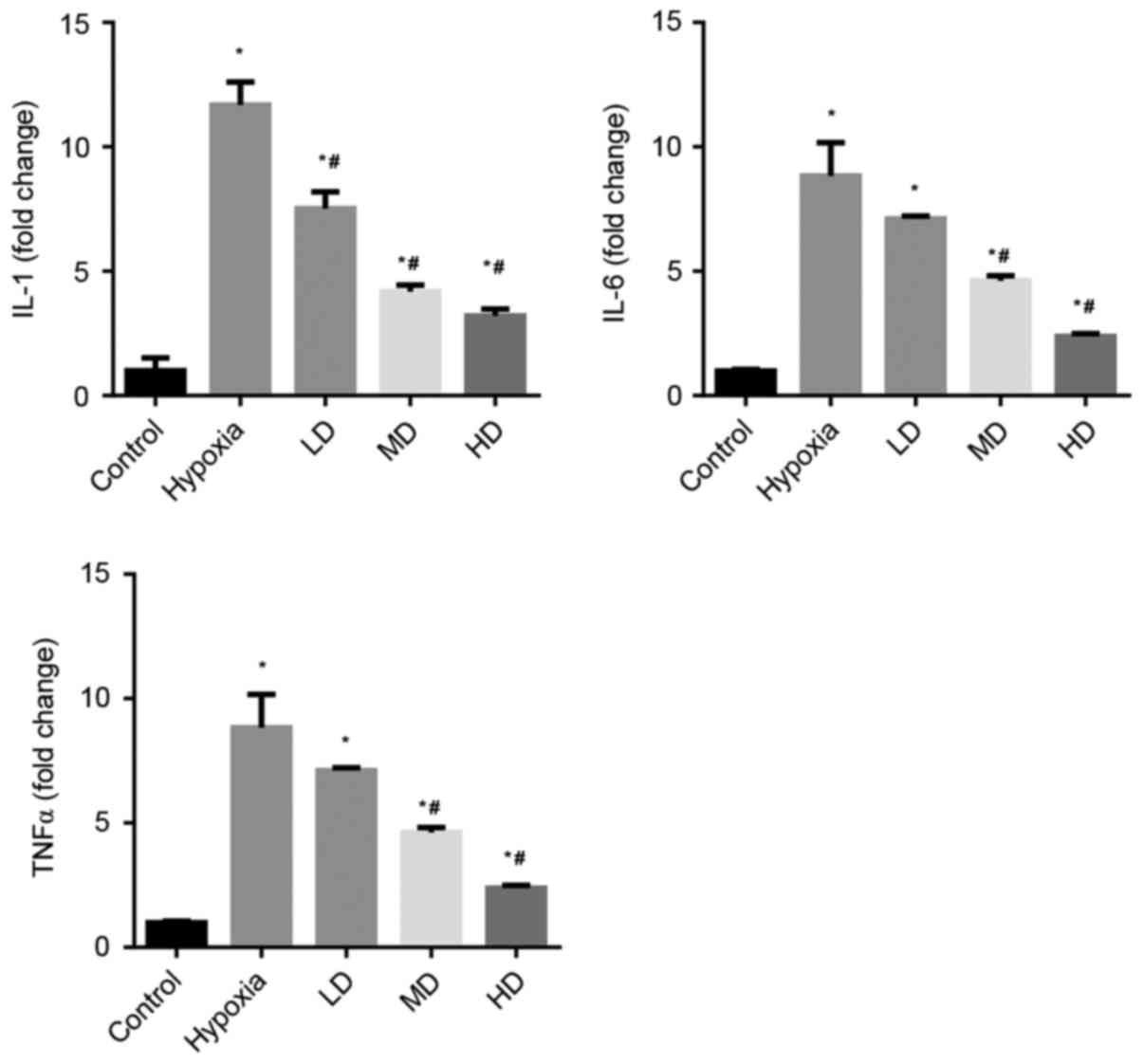
Sanggenon C suppresses hypoxia-induced
inflammation in cardiomyocytes
The effects of Sanggenon C on the induction of TNFα,
IL-1β and IL-6 in cardiomyocytes exposed to hypoxia were measured
by RT-qPCR. The expression levels of pro-inflammatory cytokines
TNFα, IL-1β and IL-6 were significantly increased in the hypoxia
group. Sanggenon C treatment significantly attenuated this increase
in a concentration-dependent manner (Fig. 1).
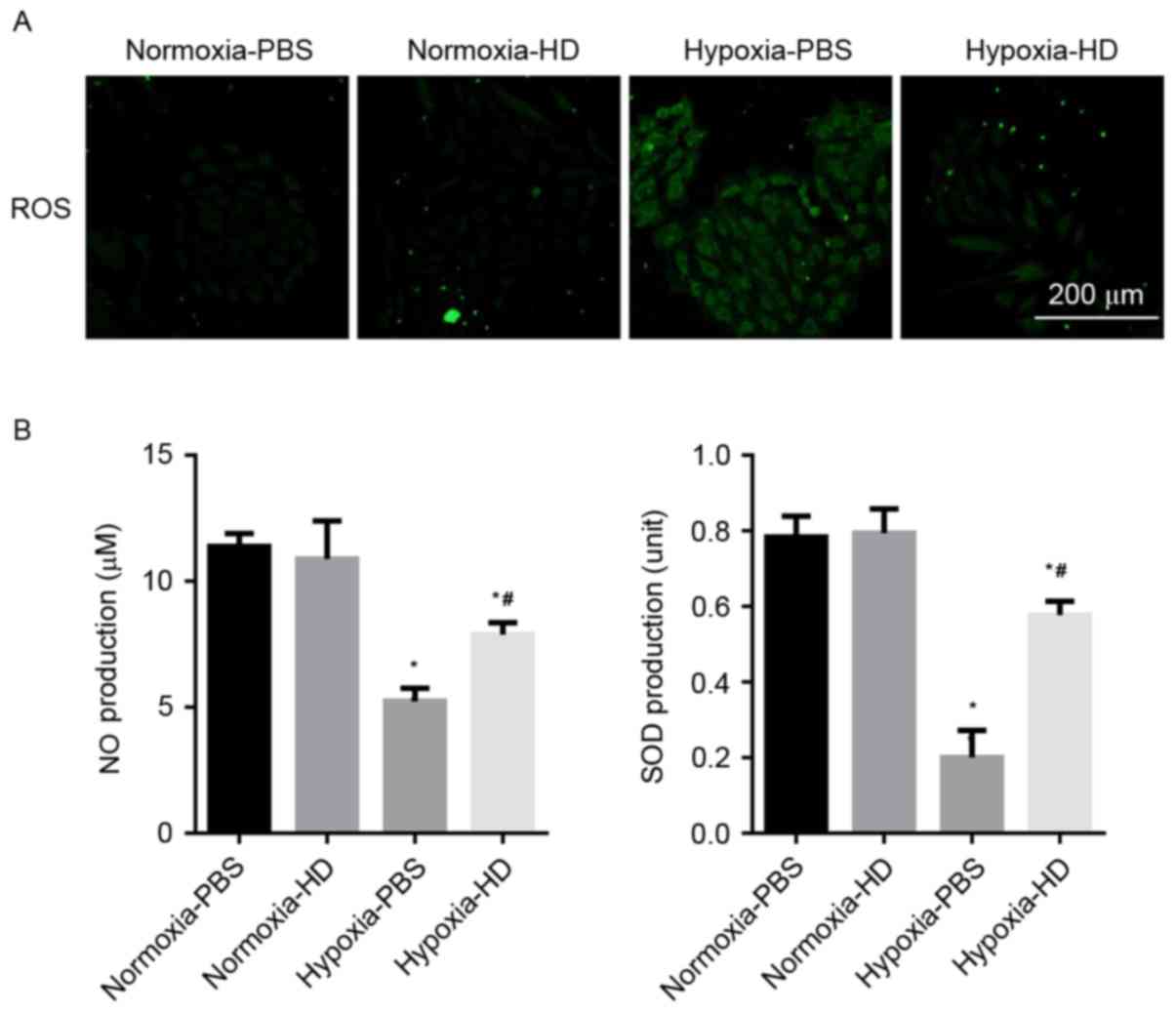
Sanggenon C inhibits the oxidative
stress induced by hypoxia in cardiomyocytes
Cells incubated with DCFH-DA were used to determine
the ROS production. A marked increase of ROS was observed in H9c2
cells exposed to hypoxia, while 100 µM Sanggenon C suppressed
hypoxia-induced ROS generation. The effect of Sanggenon C on the
release of antioxidants was also measured. Exposure to hypoxia for
24 h significantly decreased the release of NO and SOD activity in
H9c2 cells and 100 µM Sanggenon C significantly attenuated this
decrease (Fig. 2A and B).
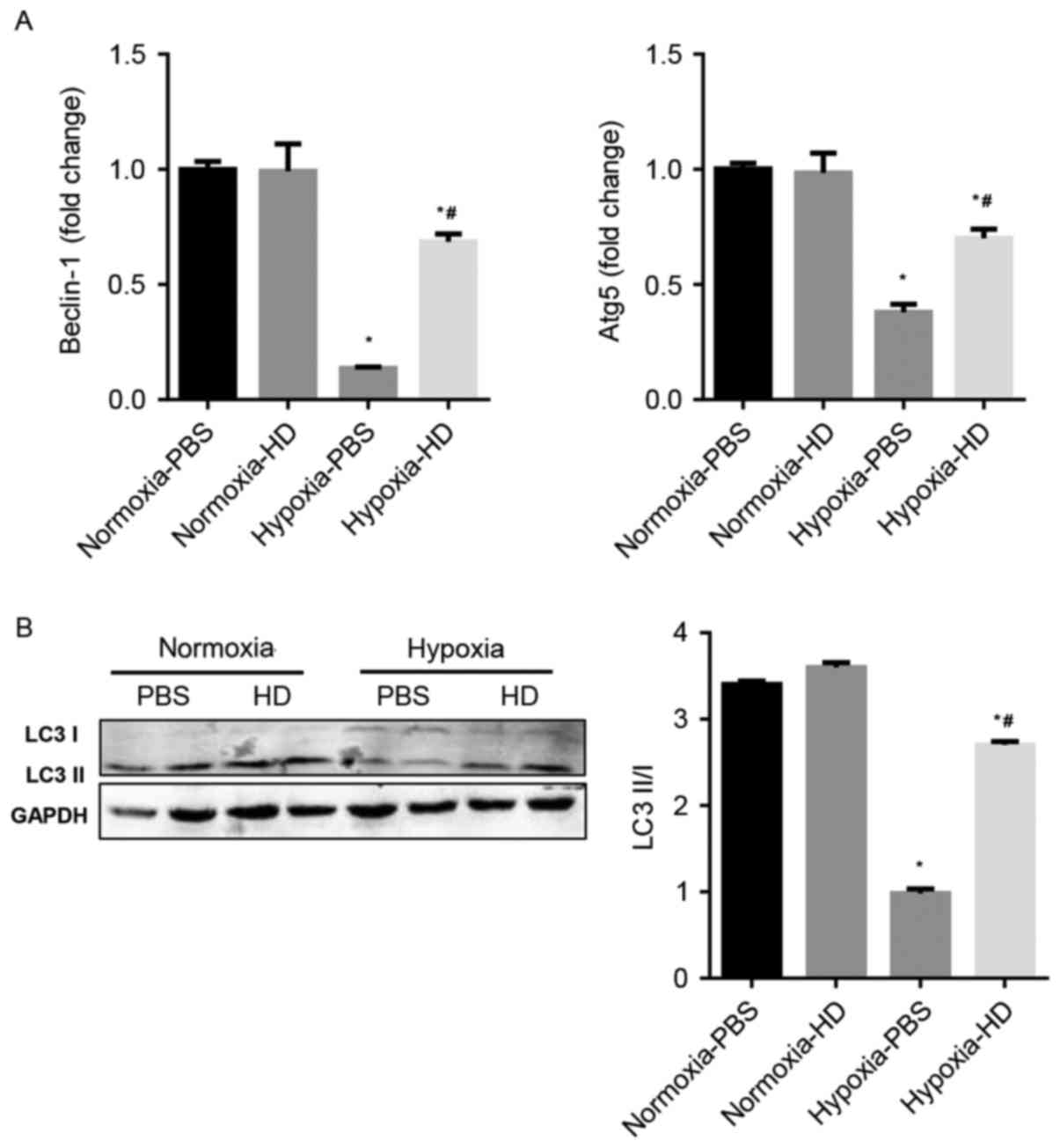
Sanggenon C activates autophagy in
response to hypoxia in cardiomyocytes
The effect of Sanggenon C on cardiomyocyte autophagy
in response to hypoxia was explored. Previous studies have
demonstrated that Beclin-1, p62, Atg5 and LC3 are
autophagy-associated proteins and enhanced autophagy is accompanied
by an increased ratio of LC3 II/LC3 I and Atg5 and Beclin-1
expression (16). The results
demonstrated that autophagy significantly decreased in
hypoxia-exposed cardiomyocytes, however increased in 100 µM
Sanggenon C pre-treated cardiomyocytes (Fig. 3A and B).
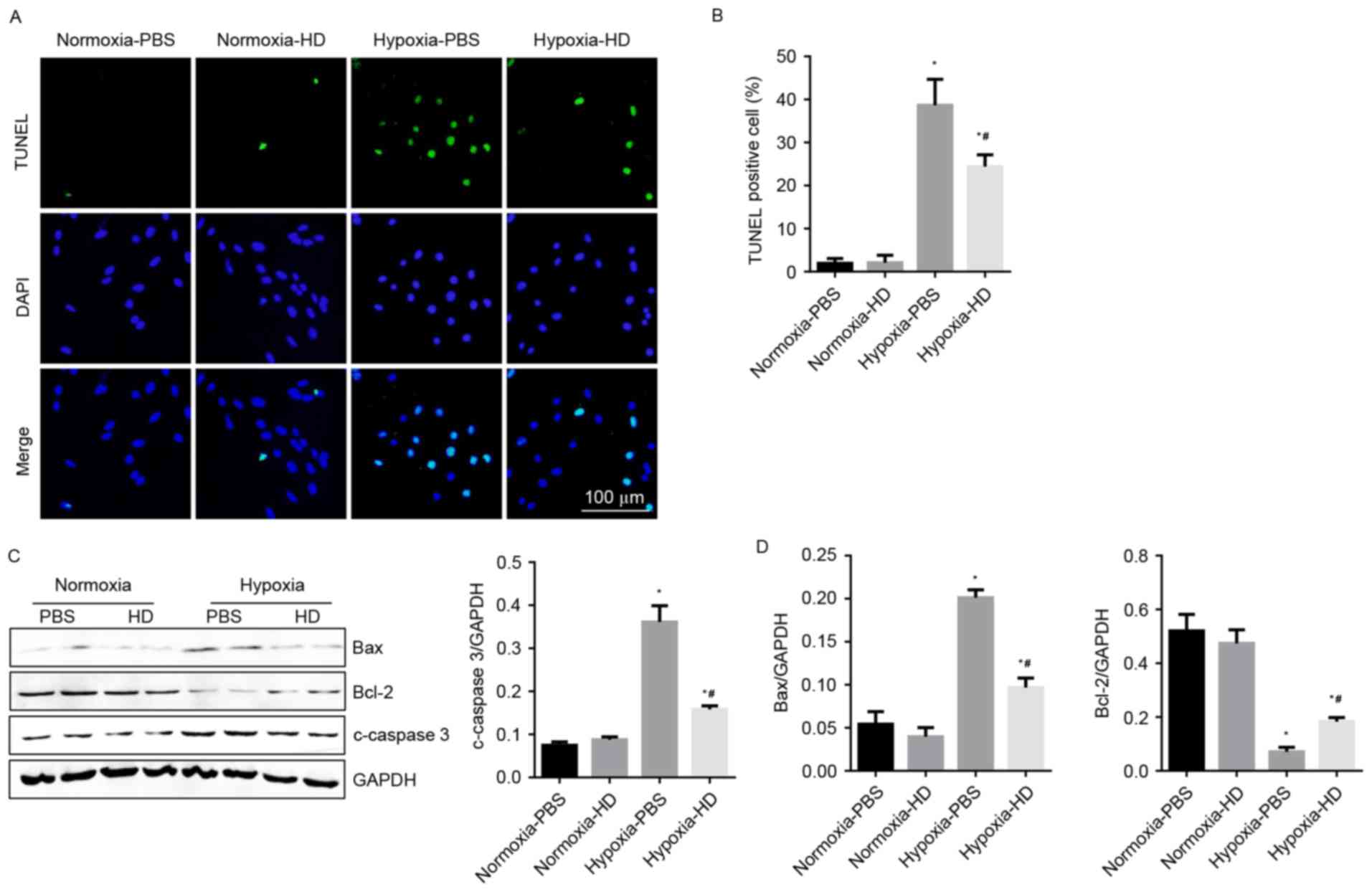
Sanggenon C attenuates hypoxia-induced
apoptosis in cardiomyocytes
To demonstrate the effect of Sanggenon C on
cardiomyocyte apoptosis in response to hypoxia, TUNEL staining and
western blotting were performed to detect cell apoptosis. Results
revealed that the rate of apoptotic cells increased significantly
after 24 h of hypoxia. Sanggenon C significantly reduced
hypoxia-induced apoptosis (Fig. 4A and
B). As expected, changes in expression of apoptosis-associated
proteins were also consistent with the results above (Fig. 4C and D).
Effect of Sanggenon C on
AMPKα/mTOR/FOXO3a signaling
Cellular energy levels are associated with the
autophagy machinery by means of three major energy sensing
pathways: AMPK, mTOR, and FOXO3a (16). In the current study, activated
AMPKα was decreased, and phosphorylated mTOR and FOXO3a were
increased in cardiomyocytes under hypoxia. Sanggenon C
pre-treatment significantly increased the activation of AMPKα, and
reduced the phosphorylation of mTOR and FOXO3a (Fig. 5A and B). To confirm the protective
effects of Sanggenon C on hypoxia-induced cardiomyocyte injury were
mediated by AMPKα signaling, an AMPKα inhibitor, CpC (20 µM) was
used. The anti-apoptotic and pro-autophagy effects of Sanggenon C
in response to hypoxia were abolished by CpC (Fig. 5C-E). The data indicated that
enhanced autophagy and reduced inflammation, oxidative stress,
apoptosis in Sanggenon C-pretreated cardiomyocytes exposed to
hypoxia may be mediated through the regulation of the
AMPKα/mTOR/FOXO3a signaling pathway.
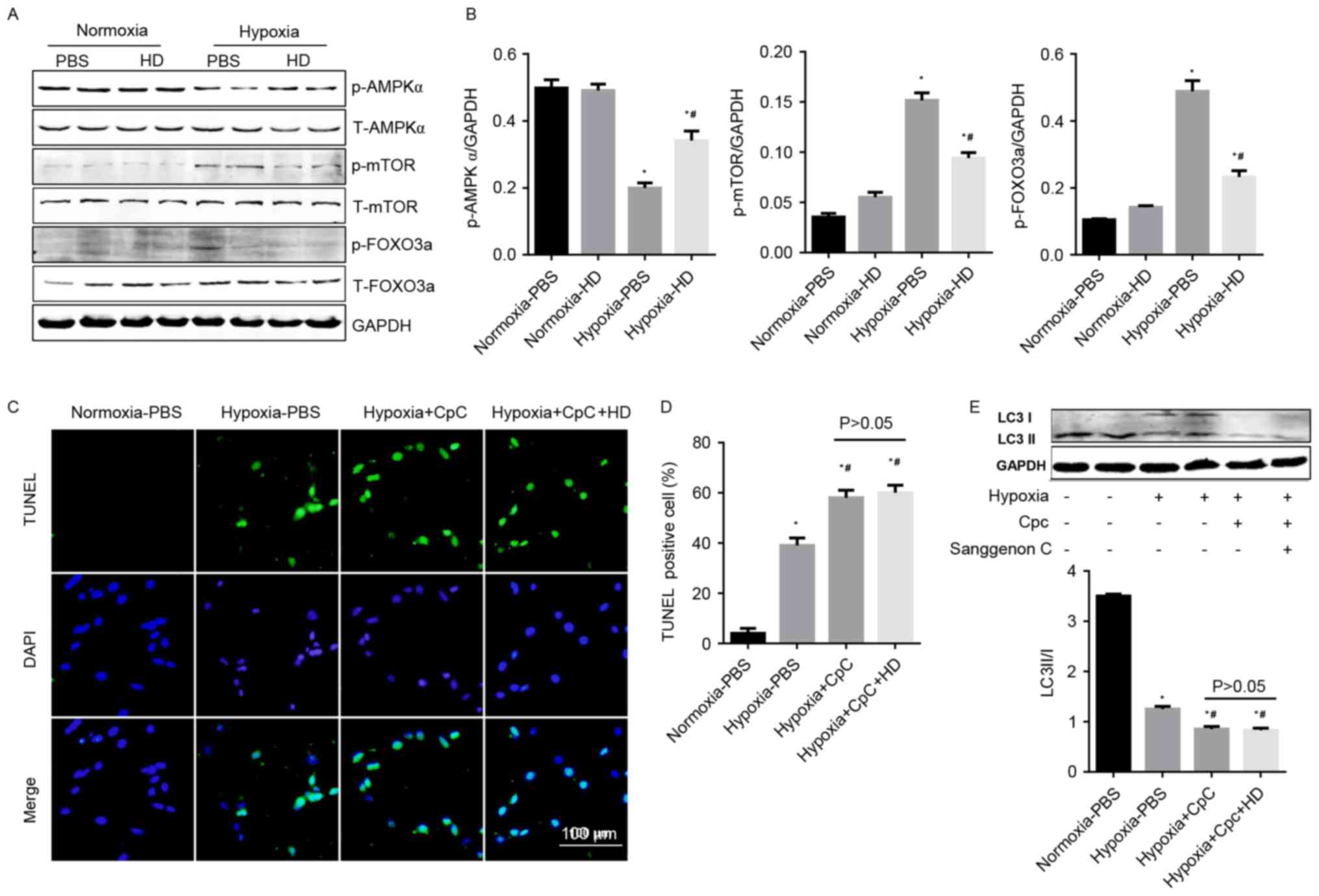 | Figure 5.Effect of Sanggenon C on
AMPKα/mTOR/FOXO3a signaling. (A) Western blot analysis the effect
of Sanggenon C (100 µM) on the activation of AMPKα pathways
including phosphorylated (p-) and total (T-) AMPKα, mTOR and FOXO3a
with (B) densitometry analysis (n=6). (C) TUNEL staining of
cardiomyocytes pretreated with Sanggenon C (100 µM) or AMPKa
inhibitor, CpC (20 µM) and exposed to hypoxia for 24 h (n=3) and
(D) quantification of TUNEL-positive cells. (E) Western blot
analysis the effect of Sanggenon C (100 µM) and CpC on the
transformation of LC3 I to LC3 II (n=6). *P<0.05 vs.
normoxia-PBS; #P<0.05 vs. hypoxia-PBS. HD, high dose
100 µM Sanggenon C; p-, phospho; T-, total; AMPKα, AMP-activated
protein kinase α; mTOR, mechanistic target of rapamycin; FOXO3a,
forkhead box O3a; TUNEL, terminal
deoxynucleotidyl-transferase-mediated dUTP nick-end labeling; LC3,
microtubule-associated proteins 1A/1B light chain 3; CpC, compound
C. |
Discussion
In the present study, it was demonstrated that
Sanggenon C could increase cardiomyocyte autophagy in hypoxic
conditions and prevent myocyte apoptosis. The increase in autophagy
was associated with ROS clearance and the inhibition of
inflammation, which would lead to cell death. The
cardiac-protective effect of Sanggenon C was mediated by the
modulation of AMPKα/mTOR/FOXO3a signaling.
In myocardial infarction and cardiomyocyte hypoxia,
necrotic cardiomyocytes are capable of releasing a wide range of
damage-associated molecular signals that activate innate immune
pathways triggering an inflammatory response (17). Although inflammation is required
for the phagocytotic removal of dead cells and for the activation
of reparative mesenchymal cells, overactive, dysregulated,
temporally-prolonged, or spatially expanded inflammatory responses
may cause death of viable cardiomyocytes. This may also enhance
matrix degradation (thus promoting dilative remodeling) and extend
fibrosis (18). The oxidant and
antioxidant imbalance within cardiomyocytes, which favors the
accumulation of oxidants, can lead to cellular damage that
constitutes oxidative stress (19). In the healthy myocardium, ROS is an
unintended byproduct of mitochondrial respiration, where its
concentration is tightly controlled to a low steady state level by
antioxidants. However, in the ischemic heart, NO•,
O2•−, and NO3−
formation are elevated. The electron leakage from complexes I and
III of the electron transport chain is primarily responsible for
O2•− generation in mitochondria. This in turn
damages the inner mitochondrial membrane, leading to decreased ATP
production and then subsequently cellular damage (5). Previous studies have reported that
Sanggenon C possesses antioxidant and anti-inflammatory activities
(12,13). In addition to this, the present
study demonstrated that Sanggenon C reduced the expression of
pro-inflammatory cytokines, decreased ROS generation and increased
the antioxidant production in cardiomyocytes in response to
hypoxia.
Autophagy involves many actions that are essential
for cell survival. It preserves energy availability and removes
damaged organelles through limited cellular catabolism. Removal of
damaged mitochondria is particularly important since these
organelles produce ROS and contribute to cell stress and damage.
Autophagy reduces pro-inflammatory signals by eliminating
intracellular organisms, degrading pro-inflammatory signaling
platforms, and by controlling cytokine production and release.
Studies have demonstrated that cells exposed to 2 h of simulated
ischemia in the absence of oxygen exhibited a low level of
autophagy (9,20). It is widely agreed that autophagy
elicited by myocardial infarction protects the heart from ischemic
injury. Due to the abrupt interruption of exogenous nutrient
supply, metabolites from autophagic digestion become a major source
for energy production (21).
Stimulation of autophagy in mice using TAT-p27 fusion protein
reduces infarct size and improves cardiac performance following
myocardial infarction (22).
Furthermore, elevated basal levels of autophagy in communities that
live at high altitudes are associated with diminished
ischemia-reperfusion injury (23).
The results of the current showed that autophagy reduced sharply
after 24 h of hypoxia, whereas Sanggenon C-pretreatment induced
cardiomyocyte autophagy. Increased autophagy in the Sanggenon
C-treated cardiomyocyte may contribute to the protection against
hypoxia.
To investigate the mechanism of induced autophagy
following Sanggenon C treatment, the signal pathway associated with
autophagy was detected. Autophagy is regulated by multiple
signaling pathways, involving nutrients, stress, hormones, growth
factors and intracellular energy information (8). AMPK, a sensor of the intracellular
AMP/ATP ratio, is activated in response to elevated intracellular
AMP by ATP hydrolysis. In conditions when ATP is depleted, AMPK is
activated and subsequently phosphorylates eukaryotic elongation
factor-2 kinase. This leads to a balance between the induction of
autophagy and the inhibition of peptide elongation (24). AMPK also suppresses mTOR activity
by interfering with GTPase activity, leading to the activation of
autophagy (8). mTOR, a sensor of
nutrients, is repressed under conditions of nutrient deprivation
and hypoxia. Repression of mTOR promotes increased autophagic
activity (25). FOXO3a, an
evolutionarily conserved subfamily of transcription factors
involved in regulation of energy metabolism, is also modulated by
AMPK. Activation of the FOXO3a transcriptional program initially
induces autophagy as an attempt to retain energy for cell survival
(26). Treatment with Sanggenon C
significantly increased the activation of AMPKα and FOXO3a, but
suppressed the activation of mTOR in hypoxia cardiomyocytes. These
results suggested that the regulation of AMPKα/mTOR/FOXO3a signal
pathway by Sanggenon C is the compensatory mechanisms that induce
autophagy and attenuate hypoxia injury in cardiomyocytes.
In conclusion, the current results supported the
theory that Sanggenon C, administered as a pre-treatment prior to
cardiomyocyte hypoxia, attenuates the inflammatory response and ROS
production provoked during hypoxia. Notably, Sanggenon C was
demonstrated to promote autophagy and render the cardiomyocyte
resistant to hypoxic injury. The modulation of AMPKα/mTOR/FOXO3a
signaling pathway restored autophagy as a reflex reaction.
Acknowledgements
This study was supported by the Applied and
Technologic Research Program of Huai'an (grant no. HAS2014009) and
the Research Fund for the Technology Development Project of Nanjing
Medical University (grant no. 2013NJMU226).
References
|
1
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heusch G and Gersh BJ: The pathophysiology
of acute myocardial infarction and strategies of protection beyond
reperfusion: A continual challenge. Eur Heart J. 38:774–784.
2017.PubMed/NCBI
|
|
3
|
Westman PC, Lipinski MJ, Luger D, Waksman
R, Bonow RO, Wu E and Epstein SE: Inflammation as a driver of
adverse left ventricular remodeling after acute myocardial
infarction. J Am Coll Cardiol. 67:2050–2060. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou T, Chuang CC and Zuo L: Molecular
characterization of reactive oxygen species in myocardial
ischemia-reperfusion injury. Biomed Res Int. 2015:8649462015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurian GA, Rajagopal R, Vedantham S and
Rajesh M: The role of oxidative stress in myocardial ischemia and
reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev.
2016:16564502016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lippai M and Szatmári Z: Autophagy-from
molecular mechanisms to clinical relevance. Cell Biol Toxicol.
33:145–168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nishida K and Otsu K: Autophagy during
cardiac remodeling. J Mol Cell Cardiol. 95:11–18. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen-Scarabelli C, Agrawal PR, Saravolatz
L, Abuniat C, Scarabelli G, Stephanou A, Loomba L, Narula J,
Scarabelli TM and Knight R: The role and modulation of autophagy in
experimental models of myocardial ischemia-reperfusion injury. J
Geriatr Cardiol. 11:338–348. 2014.PubMed/NCBI
|
|
9
|
Hamacher-Brady A, Brady NR, Logue SE,
Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA and Gustafsson AB:
Response to myocardial ischemia/reperfusion injury involves Bnip3
and autophagy. Cell Death Differ. 14:146–157. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kharbanda RK: Cardiac conditioning: A
review of evolving strategies to reduce ischaemia-reperfusion
injury. Heart. 96:1179–1186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Downey JM and Cohen MV: Why do we still
not have cardioprotective drugs? Circ J. 73:1171–1177. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang H, Liu N, Zhao K, Zhu C, Lu X, Li S,
Lian W, Zhou P, Dong X, Zhao C, et al: Sanggenon C decreases tumor
cell viability associated with proteasome inhibition. Front Biosci
(Elite Ed). 3:1315–1325. 2011.PubMed/NCBI
|
|
13
|
Dat NT, Binh PT, le TP Quynh, Huong HT and
Minh CV: Sanggenon C and O inhibit NO production, iNOS expression
and NF-κB activation in LPS-induced RAW264.7 cells. Immunopharmacol
Immunotoxicol. 34:84–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li LC, Shen F, Hou Q and Cheng GF:
Inhibitory effect and mechanism of action of Sanggenon C on human
polymorphonuclear leukocyte adhesion to human synovial cells. Acta
Pharmacol Sin. 23:138–142. 2002.PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nishida K, Kyoi S, Yamaguchi O, Sadoshima
J and Otsu K: The role of autophagy in the heart. Cell Death
Differ. 16:31–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rienks M and Papageorgiou AP: Novel
regulators of cardiac inflammation: Matricellular proteins expand
their repertoire. J Mol Cell Cardiol. 91:172–178. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frangogiannis NG: Inflammation in cardiac
injury, repair and regeneration. Curr Opin Cardiol. 30:240–245.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Y: Oxidative stress and cardiac
repair/remodeling following infarction. Am J Med Sci. 334:197–205.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang ZV and Hill JA: Protein quality
control and metabolism: Bidirectional control in the heart. Cell
Metab. 21:215–226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun X, Momen A, Wu J, Noyan H, Li R, von
Harsdorf R and Husain M: p27 protein protects metabolically
stressed cardiomyocytes from apoptosis by promoting autophagy. J
Biol Chem. 289:16924–16935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu Y, Sun Q, Li Z, Chen J, Shen C, Song Y
and Zhong Q: High basal level of autophagy in high-altitude
residents attenuates myocardial ischemia-reperfusion injury. J
Thorac Cardiovasc Surg. 148:1674–1680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bairwa SC, Parajuli N and Dyck JR: The
role of AMPK in cardiomyocyte health and survival. Biochim Biophys
Acta. 1862:2199–2210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gallagher LE, Williamson LE and Chan EY:
Advances in autophagy regulatory mechanisms. Cells. 5:pii: E242016.
View Article : Google Scholar
|
|
26
|
Chiacchiera F and Simone C: The
AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle.
9:1091–1096. 2010. View Article : Google Scholar : PubMed/NCBI
|