Introduction
Bone homeostasis is maintained by repeated cycles of
bone formation and bone resorption in a well-balanced process
called bone remodeling (1).
However, many bone metabolism diseases can disturb the balance in
the bone remodeling process. Postmenopausal osteoporosis is one
such systemic skeletal disease characterized by low bone density
and micro architectural deterioration that lead to increased bone
fragility thus making the bone susceptible to fracture (2,3).
Moreover, lack of functional ovaries leads to a decline in estrogen
levels that increases bone formation and, to a much higher extent,
increases bone resorption, leading to net bone loss (4). Estrogen deficiency may also result in
the over expression of receptor activator of nuclear factor-κB
(NF-κB) ligand (RANKL) by B lymphocytes (5), which may in part explain the
excessive osteoclasts formation and trabecular bone loss in
postmenopausal osteoporosis patients.
Osteoclasts are multinucleated giant cells
differentiated from monocyte-macrophage lineage precursor cells,
and possess a unique ability to resorb bones. Osteoclast
differentiation is primarily governed by 2 key cytokines,
macrophage colony-stimulating factor (M-CSF) and RANKL. M-CSF
supports cell survival and proliferation while RANKL serves as a
signal for osteoclastogenesis (6,7). The
binding of RANKL and its receptor RANK on osteoclast precursor
cells activates downstream pathways including peroxisome
proliferator-activated receptor-γ (PPAR-γ) and c-Fos (8), which increase the expression of
nuclear factor of activated T cells c1 (NFATc1) (9). NFATc1 is a master factor that
activates the expression of osteoclast marker genes and
subsequently results in enhanced differentiation and function of
osteoclasts (10–12). Taking together, interfering with
these pathways may help prevent pathologically enhanced osteoclasts
formation and bone loss.
Iguratimod (T-614), an efficacious and safe
anti-rheumatoid arthritis drug, is reported to exert its
therapeutic effect by reducing the production of inflammatory
cytokines such as interleukin (IL)-1β, IL-6, IL-8 and tumor
necrosis factor (TNF)-α (13,14).
In type II collagen-induced arthritis and spontaneous arthritis
models, iguratimod can reduce joint destruction and bone resorption
(15). However, whether iguratimod
could suppress osteoclasts formation and bone loss in
postmenopausal osteoporosis animal models has not been verified.
Furthermore, in vivo and in vitro studies showed that
PPAR-γ is essential in RANKL-induced osteoclast differentiation
through direct regulation of c-Fos expression (8,16).
Whether PPAR-γ is a target of iguratimod in osteoclastogenesis
should be further explored. Therefore, in the present study, we
investigated the effects of iguratimod on bone loss and osteoclasts
formation in ovariectomized mice models and in primary bone marrow
mononuclear cells (BMMCs) models, and elucidated the underlying
molecular mechanisms.
Materials and methods
Reagents and antibodies
Iguratimod was provided by Simcere Pharmaceutical
Research Co., Ltd. (Jiangsu, China). The drug was suspended in 0.5%
methylcellulose solution for in vivo use, and dissolved in
DMSO (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for in
vitro use. Recombinant soluble mouse M-CSF and RANKL were
obtained from PeproTech (Rocky Hill, NJ, USA). Rabbit antibody
against NFATc1 (no. 8032, dilution 1:1,000) was purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Rabbit antibody
against c-Fos (no. sc-52, dilution 1:200) was purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz Biotechnology, Inc., Dallas,
TX, USA). Rabbit antibody against PPAR-γ (no. BA1693-2, dilution
1:200) and mouse antibody against GAPDH (no. BM1623, dilution
1:200) were purchased from Wuhan Boster Biological Technology, Ltd.
(Wuhan, China). Rosiglitazone was purchased from Abcam (Cambridge,
MA, USA).
Animals
Twelve-weeks-old C57/BL6 female mice (29±2 g)
(Experimental Animal Center of Tongji Hospital, Wuhan, China) were
maintained at a constant temperature of 25°C under a 12-h
light/12-h dark cycle with free access to food and water. All
experimental protocols were approved by the Medical Ethics
Committee of Huazhong University of Science and Technology and were
performed according to the ethical guidelines of the National
Institutes of Health Guide for Care and Use of Laboratory
Animals.
Animals were divided randomly into 3 groups (n=10
mice/group): Sham-operated mice treated with 0.5% methylcellulose
solution (vehicle) (SHAM), bilateral ovariectomized mice treated
with vehicle (OVX) and bilateral ovariectomized mice treated with
iguratimod (30 mg/kg/day) (OVX+T-614) (17). Ovariectomy was performed as
previously described (18).
Briefly, mice were anesthetized by intraperitoneally injecting
pentobarbital sodium at a dose of 50 mg/kg body weight. Bilateral
ovaries were removed through a dorsal approach. All treatments
began on day 1 after operation and were administered orally. After
6 weeks, mice were sacrificed by an overdose of anesthesia to
isolate the femurs and uterus for use in the following
experiments.
Bone structure analysis
The distal femoral bone structure was analysed with
a micro-computed tomography (µ-CT) system (µ-CT50; Scanco Medical,
Bassersdorf, Switzerland). Scans were obtained at 100 kV and 98 µA;
the resolution was set to 10.5 µm. 3D reconstruction were analyzed
using the built-in software in the µ-CT system. Trabecular
structural parameters including bone volume/tissue volume (BV/TV),
structure model index (SMI), trabecular number (Tb.N) and
trabecular separation (Tb. Sp) were also evaluated.
Histological analysis
For histological analysis, femur samples were fixed
in 4% paraformaldehyde for 24 h, decalcified in 10%
ethylenediaminetetraacetic acid (EDTA) solution for 3 weeks and
embedded in paraffin wax. Hematoxylin and eosin (H&E) staining
was performed to observe the trabecular structure.
Tartrate-resistant acid phosphatase (TRAP) staining (Sigma-Aldrich;
Merck KGaA) was performed following standard protocols and the
numbers of osteoclasts near femoral metaphysis were counted
(19). Images were obtained using
Leica Microsystems (Wetzlar, Germany).
Serum biochemistry
For serum biochemical analysis, a retro-orbital
puncture was performed immediately prior to euthanasia to collect
blood. Blood was collected from each mouse and plasma was separated
by centrifugating for 15 min at 1,000 × g in room temperature.
Serum levels of type 1 collagen cross-linked C-terminal telopeptide
(CTX-I) were measured with ELISA kits (Nordic Bioscience
Diagnostics A/S, Herlev, Denmark) according to the manufacturer's
instructions.
Cell cultures
BMMCs were obtained from 6-weeks-old C57BL/6 mice as
previously described (20,21). Briefly, marrow cavities of isolated
femurs and tibias were exposed and flushed with α-minimum essential
medium (α-MEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Cells were then collected and cultured in α-MEM with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin, 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) and M-CSF (30 ng/ml). After 24 h, non-adherent
cells were collected and supplemented with M-CSF (30 ng/ml). After
3 days, adherent cells were seeded in different plates for use in
the following experiments.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was assessed using a CCK-8 assay
(Boster Biological Technology, Ltd.) according to the
manufacturer's instructions. Briefly, BMMCs were seeded at a
density of 5,000 cells/well in 96-well plates. After 24 h, BMMCs
were treated with phosphate-buffered saline (PBS), 0 (vehicle),
0.3, 3 or 30 µg/ml iguratimod in the presence of M-CSF (30 ng/ml).
After 1, 3 and 5 days, medium containing 10% CCK-8 was added to
each well and then incubated in darkness at 37°C for 1 h. The
absorbance was measured on an ELX800 absorbance microplate reader
(Bio-Tek Instruments Inc., Winooski, VT, USA) at a wavelength of
450 nm.
In vitro osteoclastogenesis assay
BMMCs were plated in 96-well plates at a density of
10,000 cells/well and cultured with M-CSF (30 ng/ml) and RANKL (50
ng/ml) in the presence of vehicle or various concentrations of
iguratimod. After 5 days, TRAP staining was performed according to
the manufacturer's instructions. Images were obtained and
TRAP-positive multinucleated (>3 nuclei) cells were counted as
osteoclasts.
Bone pit formation by osteoclasts
BMMCs were seeded at a density of 20,000 cells/well
in a Corning Osteo Assay Surface plate (Corning Inc., Corning, NY,
USA). Cells were cultured with M-CSF (30 ng/ml) and RANKL (100
ng/ml) for 7 days, then treated with vehicle or various
concentrations of iguratimod for an additional 5 days. Then, the
plate was washed with 5% sodium hypochlorite for 5 min. Images of
bone resorption were captured and quantified.
RNA extraction, reverse transcription
and real-time quantitative PCR
BMMCs were seeded at a density of 1×105
cells/mm2 in 6-well plates. Cells were cultured with
M-CSF (30 ng/ml) and RANKL (50 ng/ml) in the presence of vehicle or
3 µg/ml iguratimod for 5 days. Then total RNA was extracted from
BMMCs using TRIzol reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA) as previously described (21). First-strand cDNA was synthesized
using ReverTra Ace qPCR RT kit (Toyobo Co., Ltd., Osaka, Japan) to
perform RT-qPCR using the Thunderbird SYBR qPCR Mix (Toyobo Co.,
Ltd.) and a Bio-Rad Q5 instrument (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). All reactions were performed according to the
manufacturer's instructions, and target gene expression was
normalized to the reference gene glyceraldehyde 3-phosphate
dehydrogenase (GAPDH). The relative expression levels of each gene
were calculated using the comparative 2−ΔΔCt method
(22). The primers used for
RT-qPCR are listed in Table I.
 | Table I.Primers used in RT-qPCR. |
Table I.
Primers used in RT-qPCR.
| Genes | Primers
(5′-3′) |
|---|
| NFATc1 |
|
| F |
CAACGCCCTGACCACCGATAG |
| R |
GGGAAGTCAGAAGTGGGTGGA |
| TRAP |
|
| F |
TACCTGTGTGGACATGACC |
| R |
CAGATCCATAGTGAAACCGC |
| Cathepsin K |
|
| F |
TGTATAACGCCACGGCAAA |
| R |
GGTTCACATTATCACGGTCACA |
| MMP-9 |
|
| F |
TCCAGTACCAAGACAAAGCCTA |
| R |
TTGCACTGCACGGTTGAA |
| c-Fos |
|
| F |
GGTGAAGACCGTGTCAGGAG |
| R |
TATTCCGTTCCCTTCGGATT |
| GAPDH |
|
| F |
CTCCCACTCTTCCACCTTCG |
| R |
TTGCTGTAGCCGTATTCATT |
Western blot analysis
BMMCs were seeded at a density of 1×105
cells/well in 6-well plates. To detect effect of iguratimod on
c-Fos and NFATc1 expression, cells were cultured with M-CSF (30
ng/ml) and RANKL (50 ng/ml) in the presence of vehicle or 3 µg/ml
iguratimod for 2 days and 5 days. To detect the crosstalk between
iguratimod and PPAR-γ, cells were cultured with M-CSF and RANKL in
the presence of vehicle, 3 µg/ml iguratimod or 1 µM rosiglitazone
for 5 days.
Cell lysates were prepared with the RIPA Lysis
Buffer (Boster Biological Technology, Ltd.) containing 1 mM
phenylmethanesulfonyl fluoride (PMSF; Boster Biological Technology,
Ltd.). The lysates were centrifuged for 20 min at 12,000 × g. Then
supernatants were collected. Protein concentration of each sample
was detected using BCA protein assay (no. AR0146; Boster Biological
Technology, Ltd.) according to the standard protocol. Western blot
analysis was then performed as described (21,23).
In brief, total cell proteins were separated on 10% SDS-PAGE and
transferred to polyvinylidene fluoride (PVDF) membranes (Millipore,
Billerica, MA, USA). Subsequently, membranes were blocked with 5%
bovine serum albumin (BSA) and immunoblotted with corresponding
primary antibody overnight at 4°C. Then, the membranes were
incubated with appropriate horseradish peroxidase-labelled
secondary antibody (nos. BA1001 or BA1003, dilution 1:2,000; Boster
Biological Technology, Ltd.) for 1 h at room temperature.
Immunoreactivity was detected with enhanced chemiluminescence
(Boster Biological Technology, Ltd.) and images were taken by
ChemiDoc™ XRS+ System with Image Lab™ software (Bio-Rad
Laboratories, Inc.).
Statistical analysis
All quantitative data are expressed as means ± SD.
Statistical analysis between 2 groups was performed using Student's
t-test. Statistical comparison of more than 2 groups was performed
using one-way analysis of variance (ANOVA) followed by a Tukey's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of iguratimod on OVX-induced
bone loss
Six weeks after operation, mice in three groups were
sacrificed. We used µ-CT scanning to analyze the trabecular bone
changes in distal femoral metaphyses of mice. The results
demonstrated significant decrease in trabecular BV/TV and Tb. N and
increase in SMI and Tb. Sp in the OVX group when compared with the
SHAM group. Compared with the OVX group, treatment of OVX mice with
iguratimod significantly attenuated trabecular bone loss revealed
by changes in histomorphometric parameters (Fig. 1A and B). Furthermore, mice in the
OVX and OVX+T-614 groups exhibited a marked decrease in the wet
weight of uterus (Fig. 1C)
compared with the SHAM group, suggesting the success of
ovariectomy.
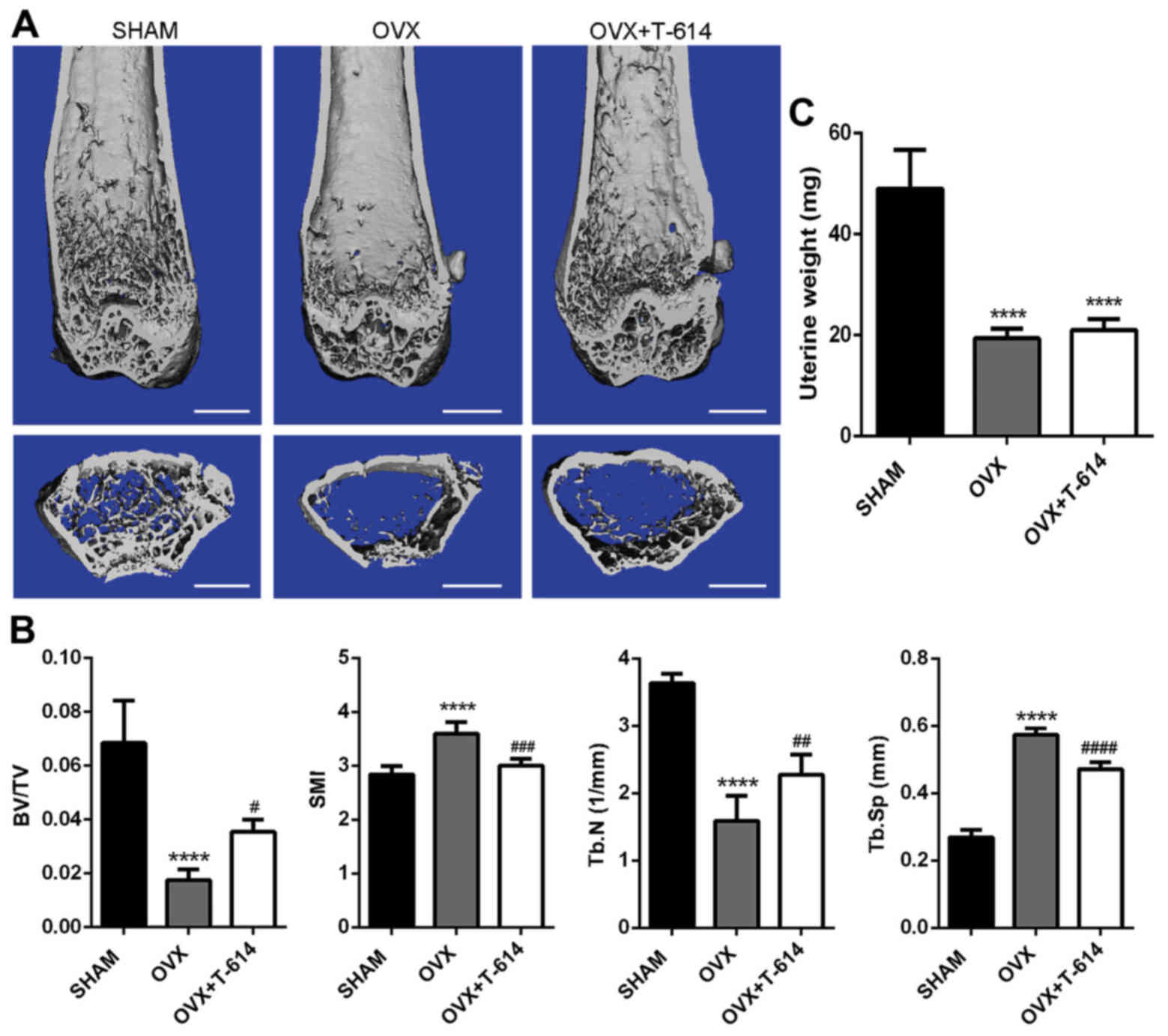 | Figure 1.Iguratimod alleviates bone loss in
ovariectomized mice. (A) µ-CT images of distal femurs from
representative specimens from the SHAM, OVX and OVX+T-614 groups.
Scale bar, 1 mm. (B) Histograms represent the 3D trabecular
structural parameters of the distal femur: Trabecular BV/TV, SMI,
Tb.N and Tb.Sp. (C) Mice uterus was isolated and weighed. Data are
presented as means ± SD. n=10. ****P<0.0001 vs. SHAM;
#P<0.05, ##P<0.01,
###P<0.001, ####P<0.0001 vs. OVX. SHAM,
sham operated and vehicle treated mice; OVX,
bilateral-ovariectomized and vehicle treated mice; T-614,
bilateral-ovariectomized and iguratimod treated mice; BV/TV, bone
volume/tissue volume; SMI, structure model index; Tb. N, trabecular
number; Tb. Sp, trabecular separation. |
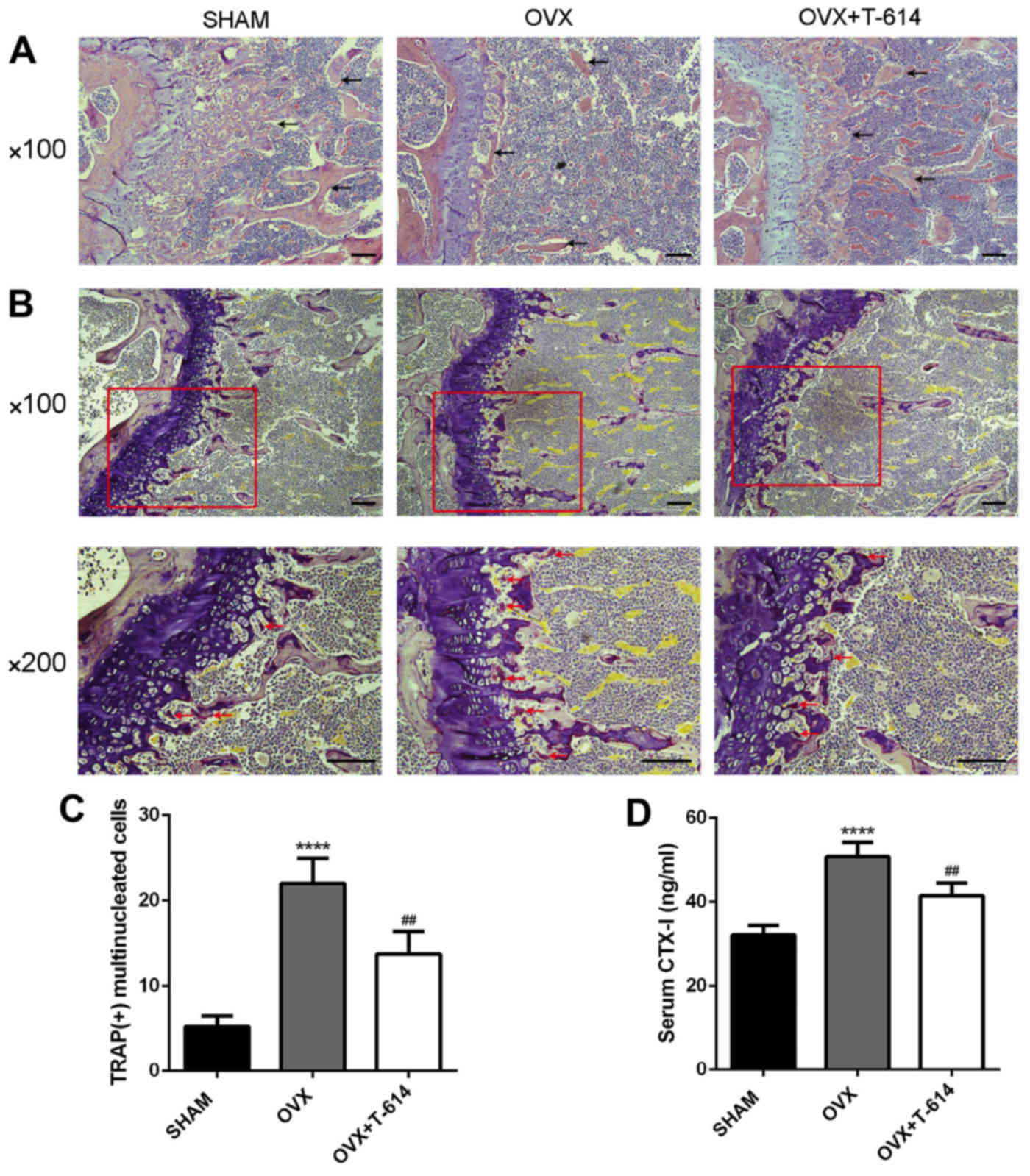
H&E staining of the femoral sections were then
used to further corroborate these results. Trabeculae in the OVX
group were rare and thin in regions proximal and distal to the
growth plate. Treatment with iguratimod significantly increased
trabecular density and thickness when compared to the OVX group
(Fig. 2A).
We then stained femoral sections with TRAP to
investigate the effects of iguratimod on osteoclasts
differentiation. Mice in the OVX+T-614 group had reduced numbers of
TRAP-positive multinucleated cells, analyzed by the number of
osteoclasts per 200× version, compared with mice in the OVX group
(Fig. 2B and C).
Moreover, compared with the OVX group, mice in the
OVX+T-614 group also displayed decreased serum levels of CTX-I,
which is a biomarker of bone resorption (Fig. 2D). These results together suggest
that treatment with iguratimod can attenuate OVX-induced bone loss
by inhibiting the differentiation of osteoclasts.
Effect of iguratimod on RANKL-mediated
osteoclastogenesis and osteoclasts function in vitro. To
further explore the impact of iguratimod on osteoclastogenesis, we
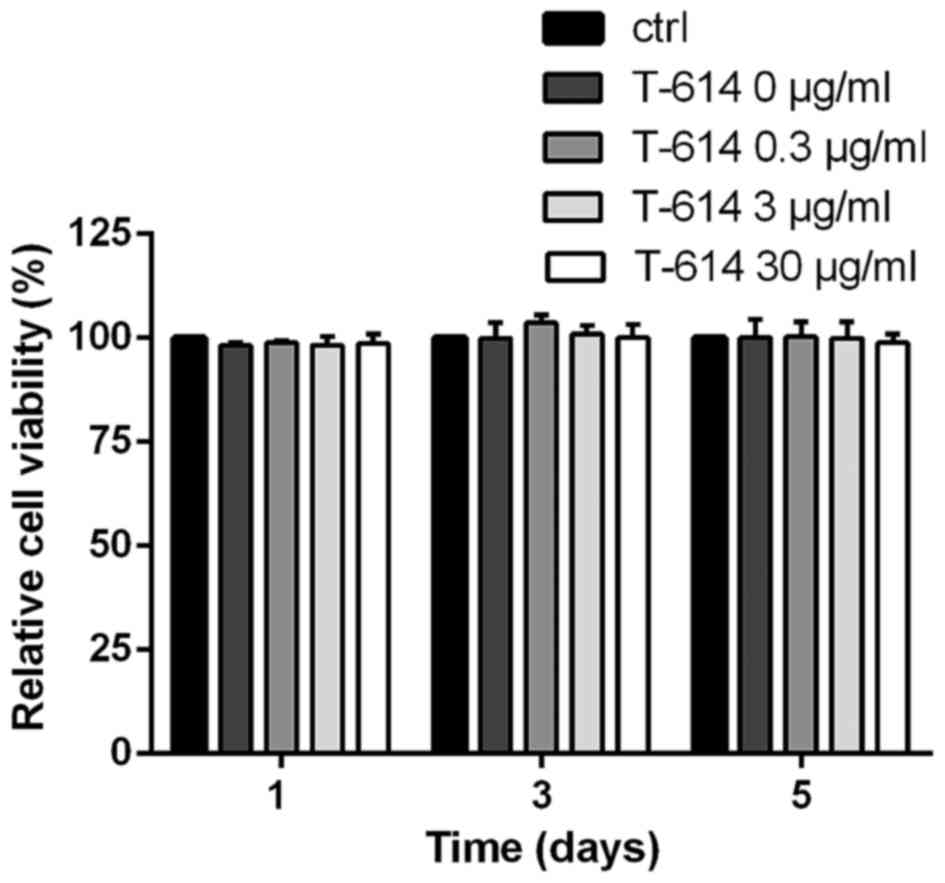
tested its effect on BMMCs. We first detected the potential
cytotoxicity of iguratimod using the CCK-8. As shown in Fig. 3, even at 30 µg/ml concentration,
iguratimod did not influence the viability and proliferation of
BMMCs.
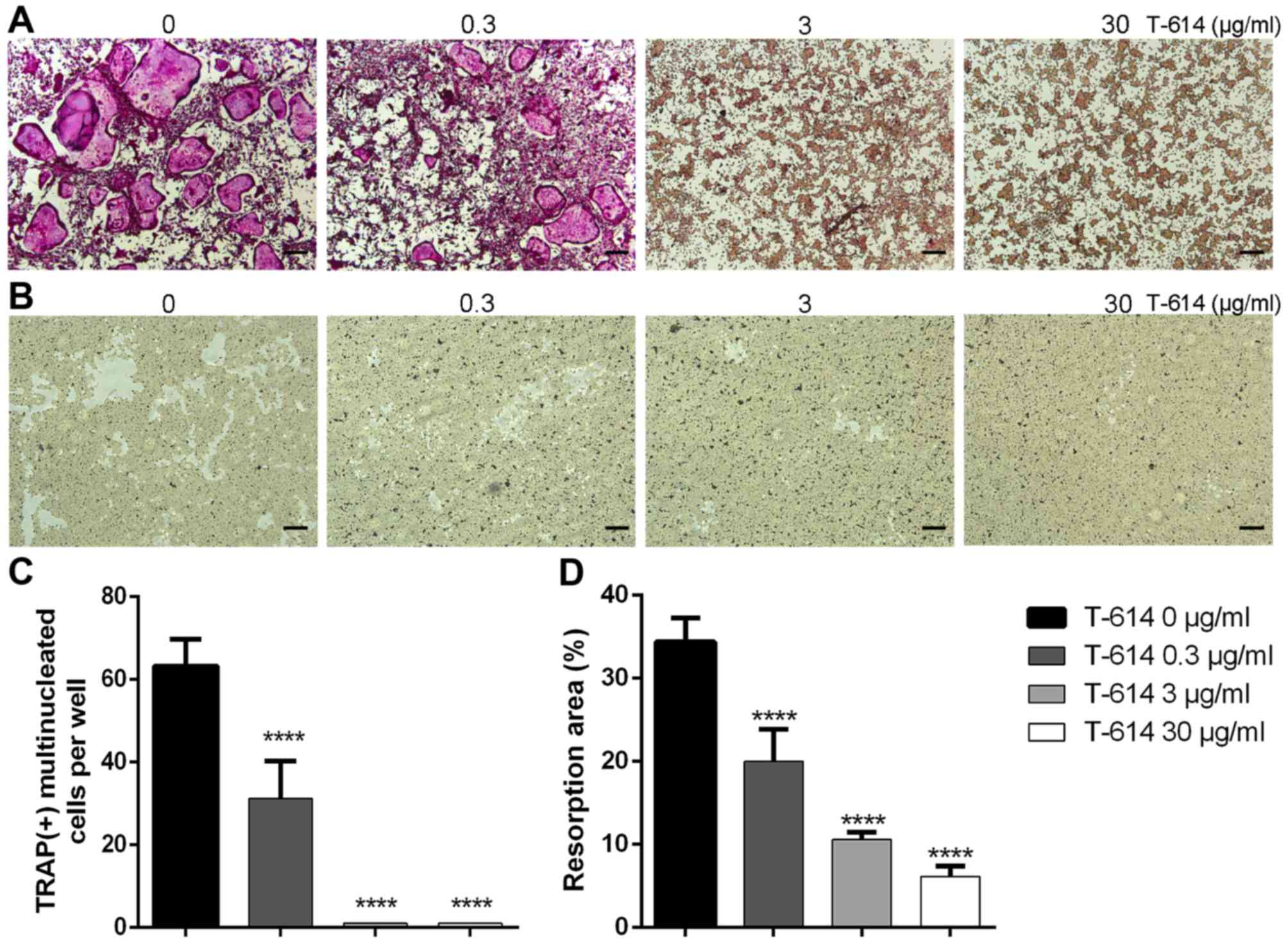
We then treated BMMCs with different concentrations
of iguratimod (0, 0.3, 3 or 30 µg/ml) in the presence of RANKL (50
ng/ml) and M-CSF (30 ng/ml) for 5 days. As shown in Fig. 4, iguratimod strongly inhibited
RANKL-mediated osteoclastogenesis in a dose-dependent manner. At
the concentration of 3 µg/ml iguratimod, there were no visible
TRAP-positive multinucleated cells.
To evaluate the effect of iguratimod on the bone
resorption function of osteoclasts, an osteo assay surface plate
was used. After seeding onto a bone slice, BMMCs were cultured with
RANKL (100 ng/ml) and M-CSF (30 ng/ml) for 7 days, and then
additional 5 days in the presence of different concentrations of
iguratimod. As shown in Fig. 4B and
D, iguratimod significantly suppressed the bone resorption
function of osteoclasts.
Effect of iguratimod on RANKL-induced
c-Fos, NFATc1 and osteoclast marker gene expression
Stimulation of RANKL may activate a variety of
transcription factors including c-Fos and downstream NFATc1.
Subsequently, activated NFATc1 can increase the expression of
osteoclast marker genes such as TRAP, cathepsin K and matrix
metalloprotein-9 (MMP-9). We then examined the effect of iguratimod
on these genes. As shown in Fig.
5A, RT-qPCR showed that incubation with RANKL significantly
increased the mRNA expression of c-Fos, NFATc1, TRAP, cathepsin K
and MMP-9 in BMMCs on day 5. Iguratimod drastically suppressed
RANKL-induced upregulation of all these genes at the concentration
of 3 µg/ml which is close to the plasma concentration of iguratimod
in rheumatoid arthritis patients treated with 50–100 mg/day
(13). The effects of iguratimod
on the expression of c-Fos and NFATc1 were corroborated by
immunoblotting (Fig. 5B and
C).
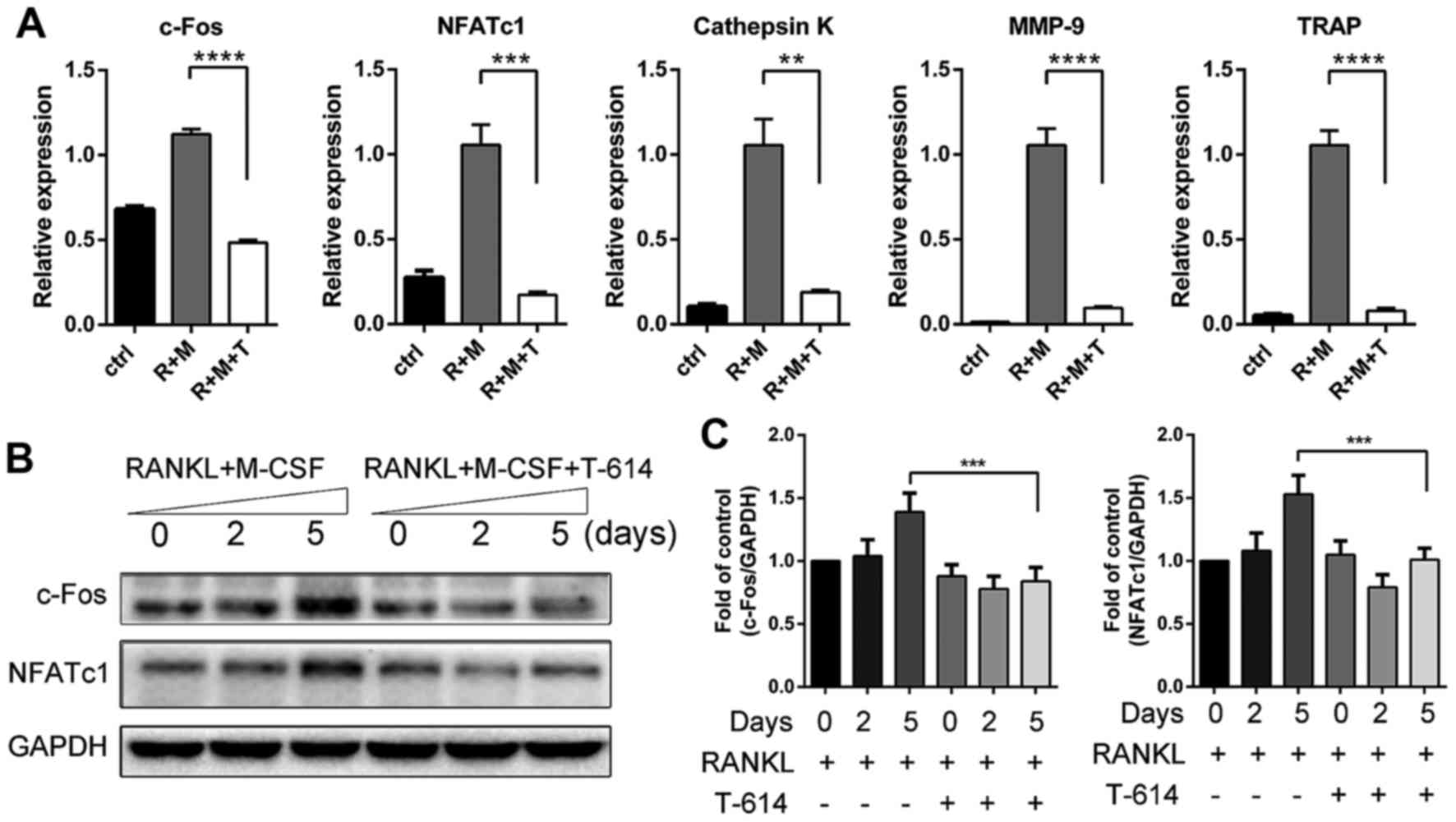 | Figure 5.Iguratimod inhibits the expression of
c-Fos, NFATc1 and osteoclast marker genes. (A) The mRNA levels of
c-Fos, NFATc1 and osteoclast marker genes were detected using
RT-qPCR. Data are presented as means ± SD. (B and C) Proteins were
extracted at indicated times and protein expression levels of c-Fos
and NFATc1 were detected by western blotting (B) and quantified
(C). The experiments were repeated 3 times independently. Data are
presented as means ± SD. **P<0.01, ***P<0.001,
****P<0.0001. NFATc1, nuclear factor of activated T cells c1;
MMP-9, matrix metalloproteinase-9; TRAP, tartrate-resistant acid
phosphatase; RANKL, receptor activator of nuclear factor-κB ligand;
M-CSF, macrophage colony-stimulating factor; GAPDH, glyceraldehyde
3-phosphate dehydrogenase; ctrl, M-CSF treated controls; R+M,
RANKL+M-CSF; R+M+T, RANKL+M-CSF+T-614 (iguratimod). |
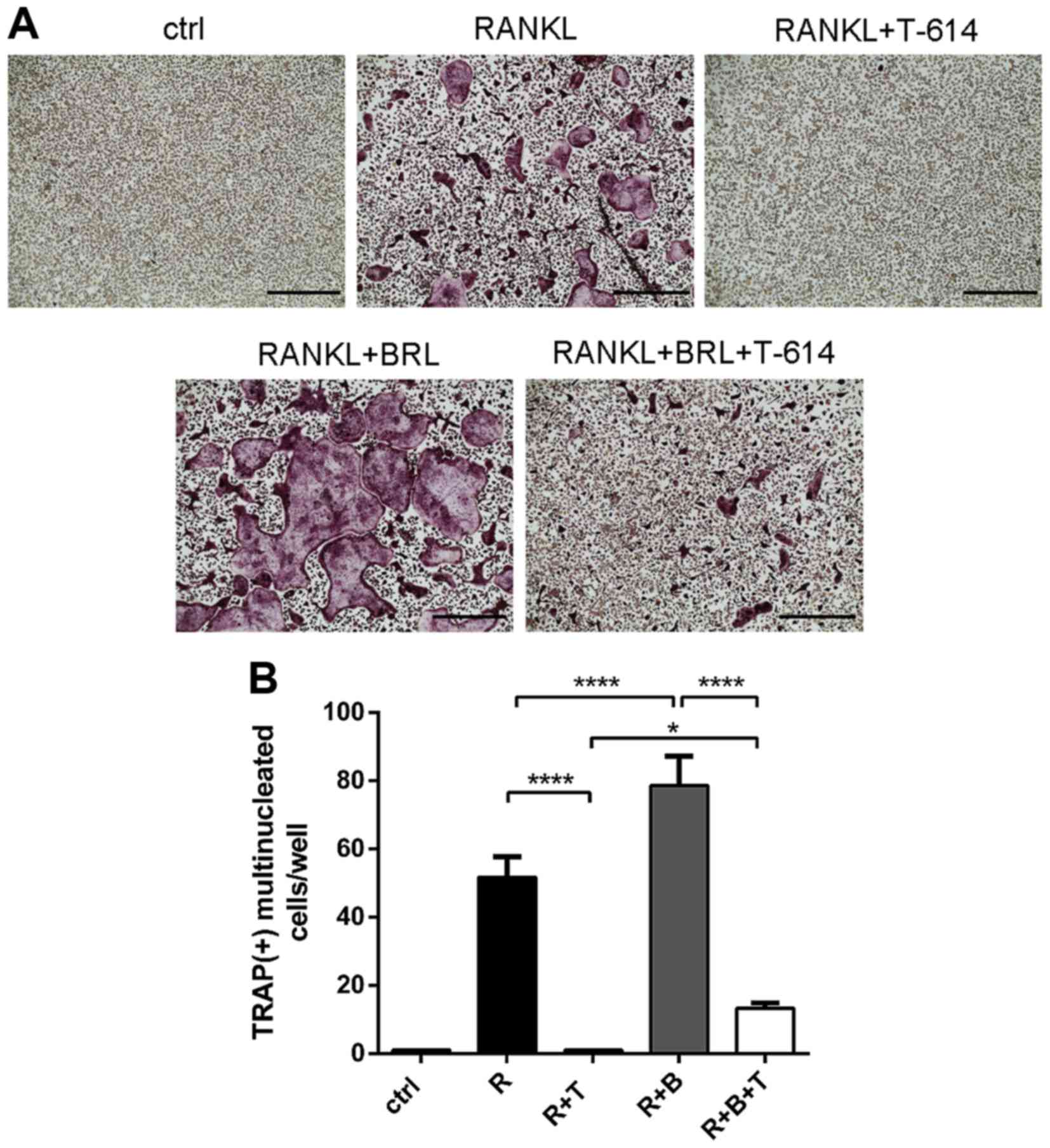
Effect of iguratimod on PPAR-γ
signaling
PPAR-γ plays an essential role in osteoclastogenesis
though directly regulating c-Fos. Therefore, we then explored
whether iguratimod suppresses osteoclastogenesis though targeting
PPAR-γ signaling. BMMCs were treated with 3 µg/ml iguratimod and/or
1 µM rosiglitazone (BRL, an agonist of PPAR-γ) in the presence of
RANKL and M-CSF for 5 days. As shown in Fig. 6, RANKL-induced osteoclasts
formation is further promoted by rosiglitazone. Treatment of
rosiglitazone could partly reverse the inhibitory effect of
iguratimod.
To further validate the effect of iguratimod on the
PPAR-γ signaling, proteins were extracted and subjected to
immunoblotting. Consistently, rosiglitazone partly reversed the
inhibitory effect of iguratimod on the expression of PPAR-γ
(Fig. 7). The expression of
downstream c-Fos and NFATc1 were also suppressed by iguratimod and
partly retrieved by rosiglitazone.
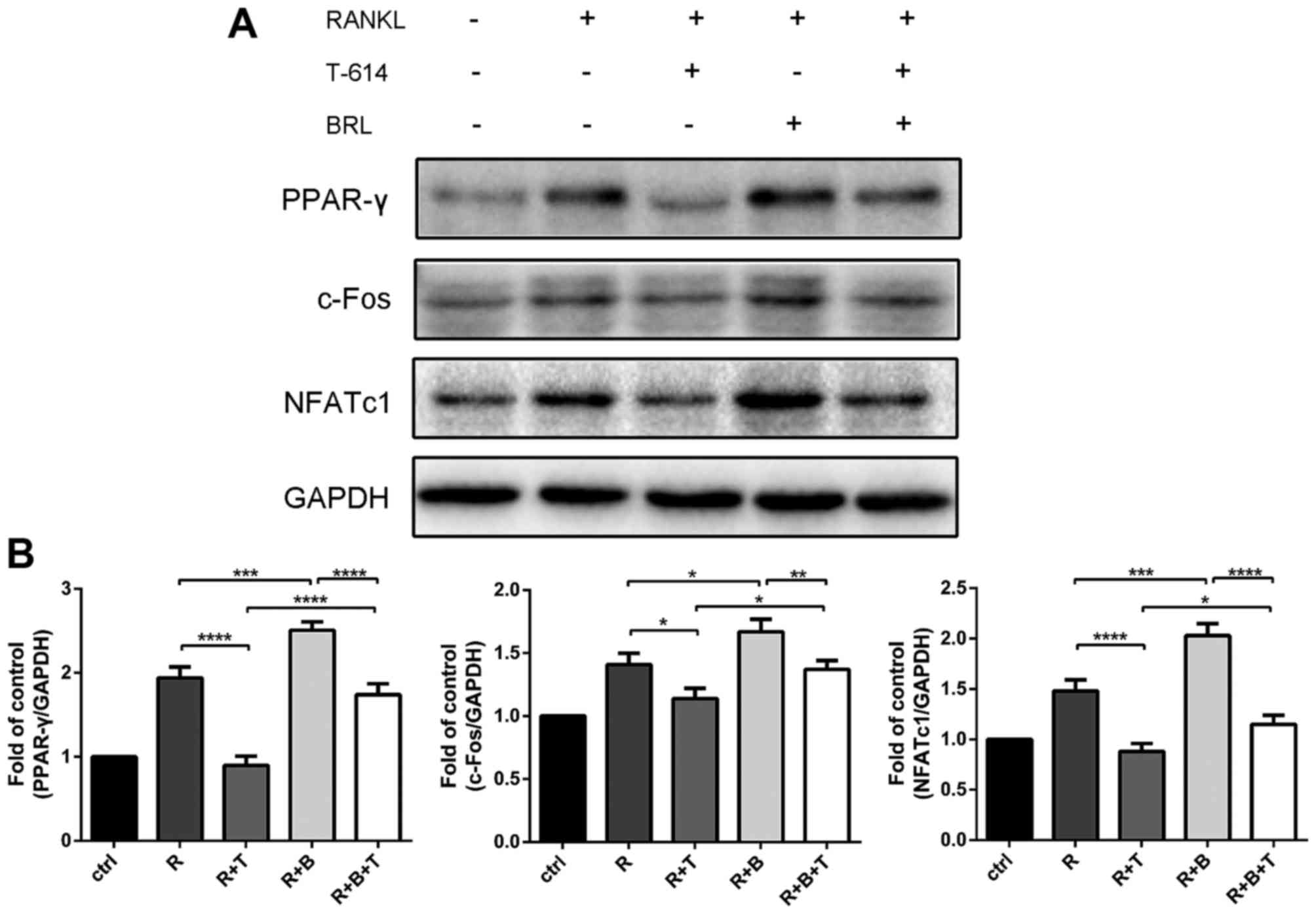 | Figure 7.Iguratimod blocks PPAR-γ/c-Fos
signaling. Proteins were extracted, and the protein expression
levels of PPAR-γ, c-Fos and NFATc1 were detected (A) and quantified
(B). The experiments were repeated 3 times independently. Data are
presented as means ± SD. *P<0.05, **P<0.01, ***P<0.001,
****P<0.0001. PPAR-γ, peroxisome proliferator-activated
receptor-γ; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; ctrl,
M-CSF treated controls; R, RANKL; T, iguratimod (T-614); B,
rosiglitazone (BRL). |
Discussion
Postmenopausal osteoporosis is an important clinical
issue, which affects 50% of women over age 45 years (24). Exploring an agent to mitigate this
problem effectively and safely will meet the needs of
postmenopausal osteoporosis patients. In our previous study we
observed that iguratimod reduced osteoclasts formation and bone
destruction in the Walker 256 rat mammary gland carcinoma cells
induced bone cancer pain model (25). Other studies showed that mammary
carcinoma cells are not bone-resorbing cells, but they stimulate
osteocytes to express RANKL (26,27).
Over expression of RANKL may enhance osteoclasts formation and bone
destruction. Considering the similar roles osteoclasts play in
malignant and benign bone resorption, we hypothesized that
iguratimod may also have therapeutic effects in benign bone
metabolism diseases such as postmenopause osteoporosis.
In postmenopausal osteoporosis patients, dysfuction
of ovaries induces estrogen deficiency and subsequently leads to
the over expression of RANKL, which contributes to excessive
osteoclastogenesis and trabecular bone loss (5). Our rationale to use ovariectomized
mice as animal models in this study is based on reports that all
major characteristics of bone loss associated with estrogen
deficiency in humans can be mimicked in ovariectomized mice
(4,28). Our data showed that ovariectomy in
mice led to trabecular bone loss, characterized by decreased BV/TV,
Tb.N and increased SMI, Tb.Sp. Treatment of iguratimod
significantly mitigated increased osteoclasts formation and
increased serum levels of CTX-I, whereas these osteoporotic effects
were significantly alleviated by treatment with iguratimod. These
results suggested that iguratimod may be a therapeutic agent for
OVX-induced bone loss. Taking into account the vital role of RANKL
in postmenopausal bone loss, we presumed that targeting
RANKL-induced osteoclastogenesis may be a reasonable explanation
for the therapeutic effects of iguratimod in OVX mice.
Stimulation of RANKL facilitates the activation of
c-Fos, which subsequently contributes to the induction and
auto-amplification of NFATc1 (9).
A previous report showed that stimulation of RANKL failed to
increase NFATc1 levels in c-Fos deficient cells (29). Finally, NFATc1 activates osteoclast
marker genes and promotes the formation and function of osteoclasts
(30). Consistent with the in
vivo study, our data showed that iguratimod could inhibit
RANKL-induced osteoclasts formation and bone resorption activity of
BMMCs in a dose-dependent manner. In addition, iguratimod
drastically suppressed RANKL-mediated expression of NFATc1 and
subsequently suppressed the activation of a number of osteoclast
marker genes, including MMP-9 and cathepsin K, which can directly
degrade collagens in demineralized hard tissues (11). Taken together, targeting
RANKL-induced osteoclastogenesis may be a reasonable explanation
for the therapeutic effects of iguratimod in OVX mice.
Considering the increased expression of PPAR-γ in
bone marrow of postmenopausal osteoporosis patients and
ovariectomized animals (31,32),
the association between iguratmod and PPAR-γ should be noticed.
Although known as a key regulator of adipogenesis, the role of
PPAR-γ in osteoclastogenesis is also well characterized. A previous
study showed that thiazolidinediones (PPAR-γ agonists) may cause
increased bone resorption in mice and rats (33). PPAR-γ-deficient mice suffer from
impaired osteoclasts function and osteopetrosis caused by a direct
reduction of c-Fos activation (16). In the present study, we revealed
that iguratimod suppressed RANKL-induced osteoclast formation and
treatment with rosiglitazone could partly reverse this inhibitory
effect. Consistently, iguratimod also suppressed RANKL-induced
expression of PPAR-γ, c-Fos and NFATc1. Treatment of rosiglitazone
partly reversed the inhibitory effect of iguratimod. Thus, the
prevention of RANKL-mediated bone resorption by iguratimod could
also be attributed to the suppression of NFATc1 expression via
blocking the PPAR-γ/c-Fos pathways.
Previously, Gan et al reported that
iguratimod suppresses RANKL-induced osteoclasts differentiation and
migration in RAW264.7 cells via NF-κB and MAPK pathways (34). Wang et al also showed the
effect of iguratimod on RANKL and OPG expression in serum and
IL-1β-induced fibroblast-like synoviocytes from patients with
rheumatoid arthritis (35). These
studies both implied the relationship between iguratimod and
osteoclastogenesis in rheumatoid arthritis. Differently, our study
verified that iguratimod could suppress osteoclasts formation and
bone loss in postmenopausal osteoporosis animal models. In
addition, RAW264.7 cells were derived from leukemia cells, our
study further confirmed the effect of iguratimod on RANKL-induced
osteoclastogenesis and the underlying mechanisms in primary BMMCs.
Though NF-κB and MAPK pathways play important roles in
RANKL-induced osteoclasts differentiation, PPAR-γ is also essential
in RANKL-induced osteoclastogenesis through direct regulation of
c-Fos expression (8,16). Considering the increased expression
of PPAR-γ in bone marrow of postmenopausal osteoporosis patients
and ovariectomized animals (31,32),
inhibition of osteoclastogenesis via blocking the PPAR-γ/c-Fos
pathway may contribute to the therapeutic effects of iguratimod in
postmenopausal osteoporosis animal models.
In conclusion, the present study, to our knowledge,
is the first to demonstrate that iguratimod can prevent
ovariectomy-induced bone loss and that iguratimod can inhibit
PPAR-γ/c-Fos pathway in RANKL-induced osteoclastogenesis.
Considering the lifelong need to treat osteoporosis and that
iguratimod is well-tolerated in long-term use (36), the anti-osteoclastogenic activity
of iguratimod under clinical settings should be addressed in
future.
Acknowledgements
This study was supported by grants from the National
Nature Science Foundation of China (nos. 81572094 and
81371915).
References
|
1
|
Boyle WJ, Simonet WS and Lacey DL:
Osteoclast differentiation and activation. Nature. 423:337–342.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seeman E and Delmas PD: Bone quality-the
material and structural basis of bone strength and fragility. N
Engl J Med. 354:2250–2261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
An J, Yang H, Zhang Q, Liu C, Zhao J,
Zhang L and Chen B: Natural products for treatment of osteoporosis:
The effects and mechanisms on promoting osteoblast-mediated bone
formation. Life Sci. 147:46–58. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manolagas SC, O'Brien CA and Almeida M:
The role of estrogen and androgen receptors in bone health and
disease. Nat Rev Endocrinol. 9:699–712. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Onal M, Xiong J, Chen X, Thostenson JD,
Almeida M, Manolagas SC and O'Brien CA: Receptor activator of
nuclear factor κB ligand (RANKL) protein expression by B
lymphocytes contributes to ovariectomy-induced bone loss. J Biol
Chem. 287:29851–29860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arai F, Miyamoto T, Ohneda O, Inada T,
Sudo T, Brasel K, Miyata T, Anderson DM and Suda T: Commitment and
differentiation of osteoclast precursor cells by the sequential
expression of c-Fms and receptor activator of nuclear factor kappaB
(RANK) receptors. J Exp Med. 190:1741–1754. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lacey DL, Timms E, Tan HL, Kelley MJ,
Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S,
et al: Osteoprotegerin ligand is a cytokine that regulates
osteoclast differentiation and activation. Cell. 93:165–176. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gu DR, Lee JN, Oh GS, Kim HJ, Kim MS and
Lee SH: The inhibitory effect of beta-lapachone on RANKL-induced
osteoclastogenesis. Biochem Biophys Res Commun. 482:1073–1079.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wagner EF and Eferl R: Fos/AP-1 proteins
in bone and the immune system. Immunol Rev. 208:126–140. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li C, Yang Z, Li Z, Ma Y, Zhang L, Zheng
C, Qiu W, Wu X, Wang X, Li H, et al: Maslinic acid suppresses
osteoclastogenesis and prevents ovariectomy-induced bone loss by
regulating RANKL-mediated NF-κB and MAPK signaling pathways. J Bone
Miner Res. 26:644–656. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takayanagi H: The role of NFAT in
osteoclast formation. Ann N Y Acad Sci. 1116:227–237. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Conaway HH, Henning P, Lie A, Tuckermann J
and Lerner UH: Activation of dimeric glucocorticoid receptors in
osteoclast progenitors potentiates RANKL induced mature osteoclast
bone resorbing activity. Bone. 93:43–54. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kohno M, Aikawa Y, Tsubouchi Y,
Hashiramoto A, Yamada R, Kawahito Y, Inoue K, Kusaka Y, Kondo M and
Sano H: Inhibitory effect of T-614 on tumor necrosis factor-alpha
induced cytokine production and nuclear factor-kappaB activation in
cultured human synovial cells. J Rheumatol. 28:2591–2596.
2001.PubMed/NCBI
|
|
14
|
Kawakami A, Tsuboi M, Urayama S, Matsuoka
N, Yamasaki S, Hida A, Aoyagi T, Furuichi I, Nakashima T, Migita K,
et al: Inhibitory effect of a new anti-rheumatic drug T-614 on
costimulatory molecule expression, cytokine production, and antigen
presentation by synovial cells. J Lab Clin Med. 133:566–574. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Du F, Lü LJ, Fu Q, Dai M, Teng JL, Fan W,
Chen SL, Ye P, Shen N, Huang XF, et al: T-614, a novel
immunomodulator, attenuates joint inflammation and articular damage
in collagen-induced arthritis. Arthritis Res Ther. 10:R1362008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wan Y, Chong LW and Evans RM: PPAR-gamma
regulates osteoclastogenesis in mice. Nat Med. 13:1496–1503. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo Q, Sun Y, Liu W, Qian C, Jin B, Tao F,
Gu Y, Wu X, Shen Y and Xu Q: A novel disease-modifying
antirheumatic drug, iguratimod, ameliorates murine arthritis by
blocking IL-17 signaling, distinct from methotrexate and
leflunomide. J Immunol. 191:4969–4978. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fan H, Ji F, Lin Y, Zhang M, Qin W, Zhou Q
and Wu Q: Electroacupuncture stimulation at CV4 prevents
ovariectomy-induced osteoporosis in rats via Wnt-β-catenin
signaling. Mol Med Rep. 13:2485–2491. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Y, Wang XX, Zhao BJ, Bu J, Su YR and
Zhang J: Effects of icariin on orthodontic tooth movement in rats.
Int J Clin Exp Med. 8:8608–8616. 2015.PubMed/NCBI
|
|
20
|
Koga T, Inui M, Inoue K, Kim S, Suematsu
A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, et al:
Costimulatory signals mediated by the ITAM motif cooperate with
RANKL for bone homeostasis. Nature. 428:758–763. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Guan H, Li J, Fang Z, Chen W and
Li F: Amlexanox suppresses osteoclastogenesis and prevents
ovariectomy-induced bone loss. Sci Rep. 5:135752015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Zhu Y, Zheng S, Ni C, Zhao L, Liu
C, Chen A and Xiao J: Amiloride inhibits osteoclastogenesis by
suppressing nuclear factor-κB and mitogen-activated protein kinase
activity in receptor activator of nuclear factor-κB-induced
RAW264.7 cells. Mol Med Rep. 11:3451–3456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guan H, Zhao L, Cao H, Chen A and Xiao J:
Epoxyeicosanoids suppress osteoclastogenesis and prevent
ovariectomy-induced bone loss. FASEB J. 29:1092–1101. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cline-Smith A, Gibbs J, Shashkova E,
Buchwald ZS, Novack DV and Aurora R: Pulsed low-dose RANKL as a
potential therapeutic for postmenopausal osteoporosis. JCI Insight.
1:e888392016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun Y, Ye DW, Zhang P, Wu YX, Wang BY,
Peng G and Yu SY: Anti-rheumatic drug iguratimod (T-614) alleviates
cancer-induced bone destruction via down-regulating interleukin-6
production in a nuclear factor-κB-dependent manner. J Huazhong Univ
Sci Technolog Med Sci. 36:691–699. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Käkönen SM and Mundy GR: Mechanisms of
osteolytic bone metastases in breast carcinoma. Cancer. 97 Suppl
3:S834–S839. 2003. View Article : Google Scholar
|
|
27
|
Roodman GD: Genes associate with abnormal
bone cell activity in bone metastasis. Cancer Metastasis Rev.
31:569–578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jilka RL, Hangoc G, Girasole G, Passeri G,
Williams DC, Abrams JS, Boyce B, Broxmeyer H and Manolagas SC:
Increased osteoclast development after estrogen loss: Mediation by
interleukin-6. Science. 257:88–91. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Asagiri M and Takayanagi H: The molecular
understanding of osteoclast differentiation. Bone. 40:251–264.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ortega N, Behonick D, Stickens D and Werb
Z: How proteases regulate bone morphogenesis. Ann N Y Acad Sci.
995:109–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bu S, Chen Y, Wang S, Zhang F and Ji G:
Treadmill training regulates β-catenin signaling through
phosphorylation of GSK-3β in lumbar vertebrae of ovariectomized
rats. Eur J Appl Physiol. 112:3295–3304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li GW, Xu Z, Chang SX, Nian H, Wang XY and
Qin LD: Icariin prevents ovariectomy-induced bone loss and lowers
marrow adipogenesis. Menopause. 21:1007–1016. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sottile V, Seuwen K and Kneissel M:
Enhanced marrow adipogenesis and bone resorption in
estrogen-deprived rats treated with the PPARgamma agonist BRL49653
(rosiglitazone). Calcif Tissue Int. 75:329–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gan K, Yang L, Xu L, Feng X, Zhang Q, Wang
F, Tan W and Zhang M: Iguratimod (T-614) suppresses RANKL-induced
osteoclast differentiation and migration in RAW264.7 cells via
NF-κB and MAPK pathways. Int Immunopharmacol. 35:294–300. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang XT, Li P, Xu TS, Ding R, Zhang X and
Bi LQ: Effect of iguratimod and methotrexate on RANKL and OPG
expression in serum and IL-1β-induced fibroblast-like synoviocytes
from patients with rheumatoid arthritis. Cell Mol Biol
(Noisy-le-grand). 62:44–50. 2016. View Article : Google Scholar
|
|
36
|
Okamura K, Yonemoto Y, Suto T, Okura C and
Takagishi K: Efficacy at 52 weeks of daily clinical use of
iguratimod in patients with rheumatoid arthritis. Mod Rheumatol.
25:534–539. 2015. View Article : Google Scholar : PubMed/NCBI
|